Mitochondrial FAD-linked Glycerol-3-phosphate Dehydrogenase: A Target for Cancer Therapeutics

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Experimental
2.1. Cell Lines and Growth Conditions
2.2. Screening Library
2.3. High-Throughput Screening for Small Molecules Altering H2O2 Production
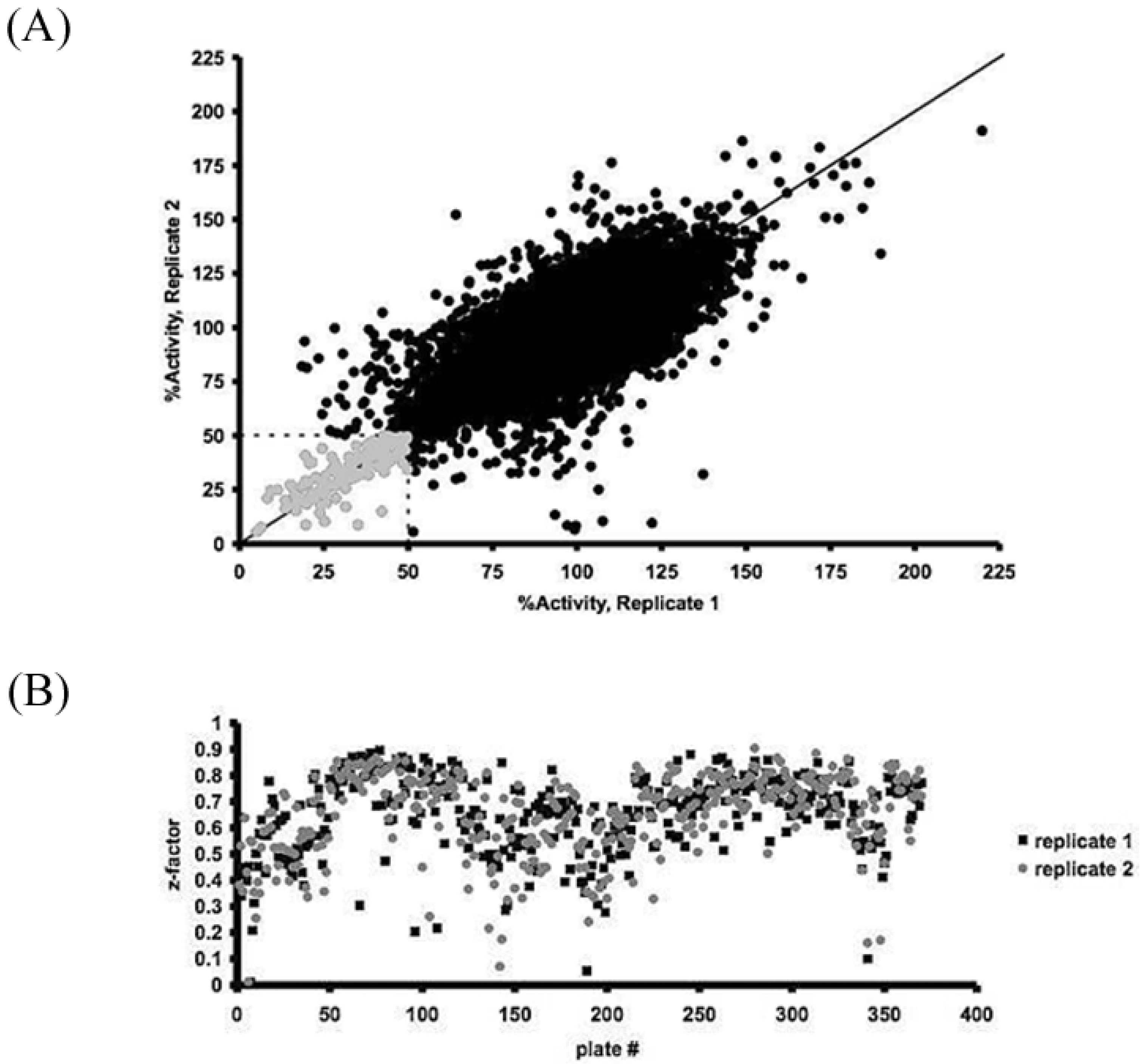
2.4. Assay Optimization and Validation for High-Throughput Screening
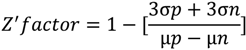
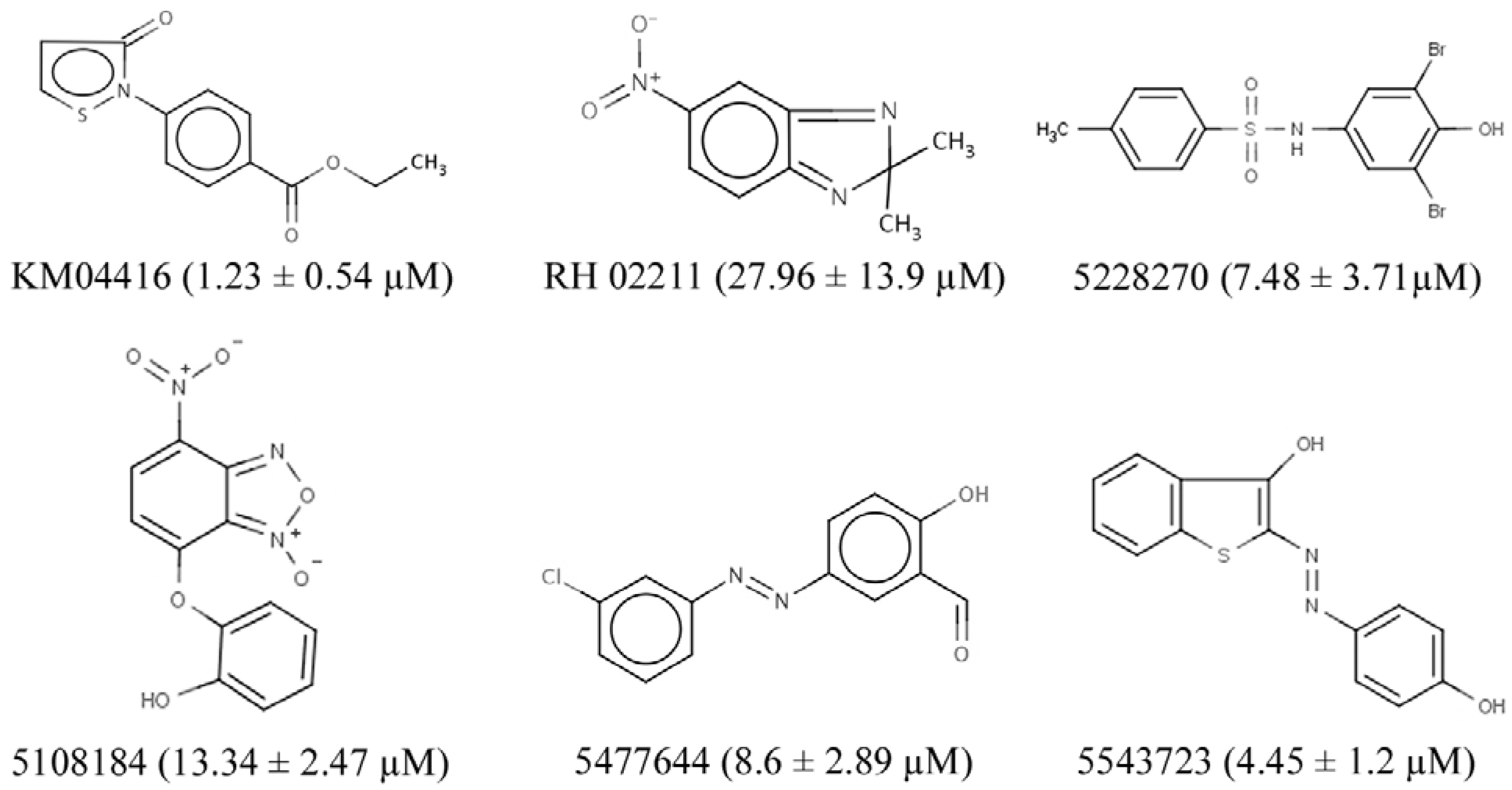
2.5. Identification of GPD2-Specific Inhibitors
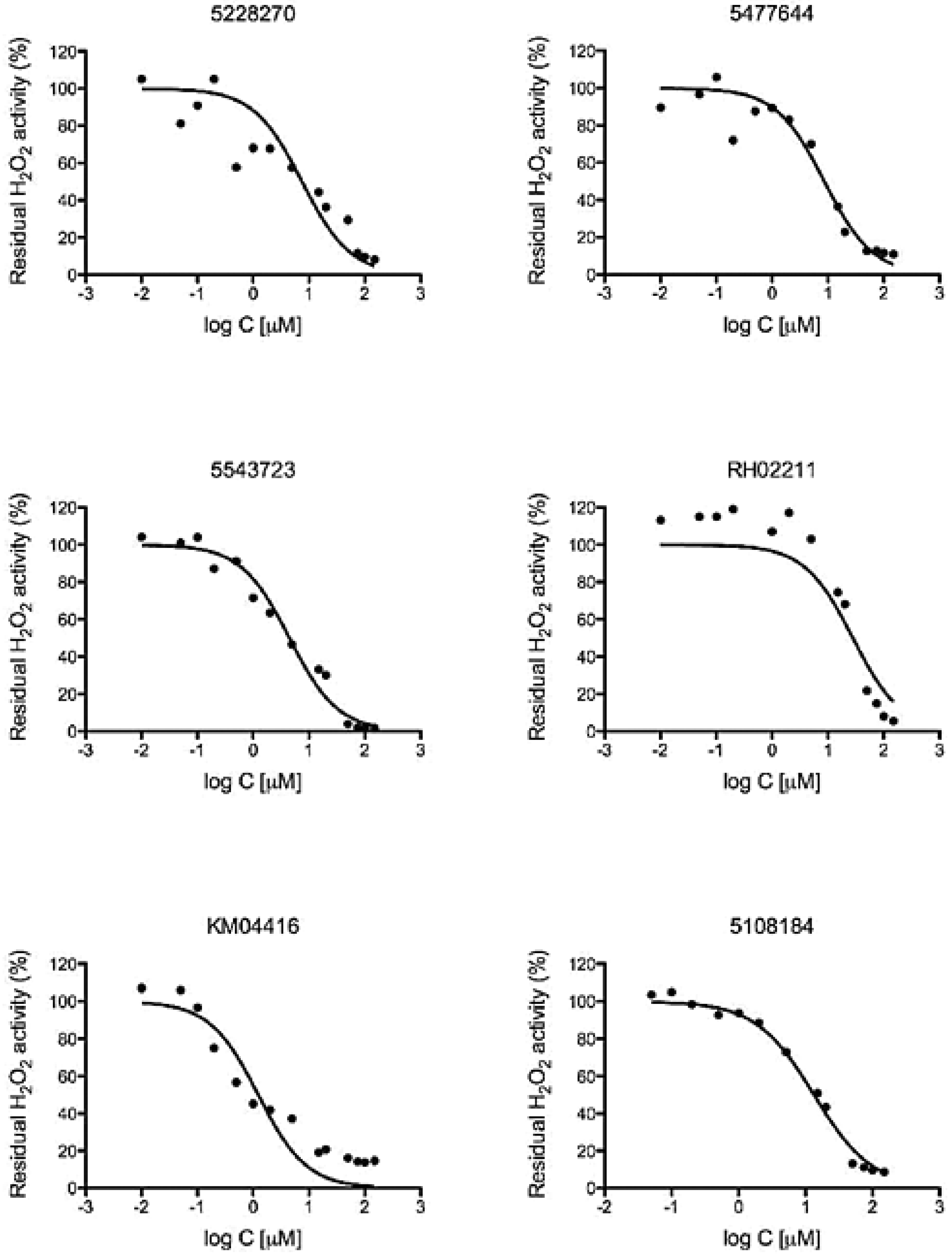
2.6. Determination of EC50 Values
2.7. Proliferation Assay
2.8. Cytotoxicity Assays
2.9. Statistical Analysis
3. Results and Discussion
3.1. High-Throughput Screening for the iSolation of Compounds Altering H2O2 Production
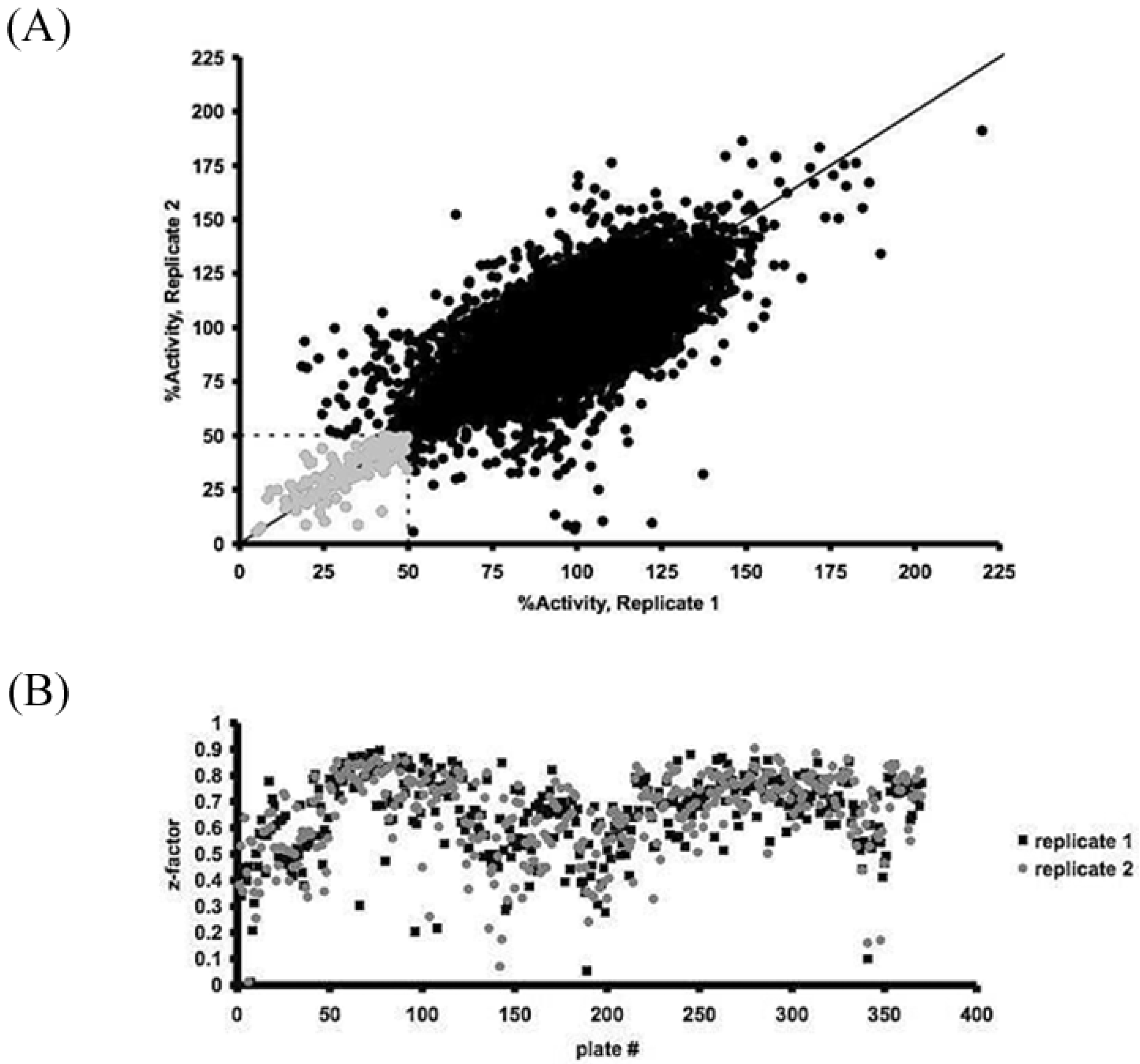
3.2. GPD2 Specificity
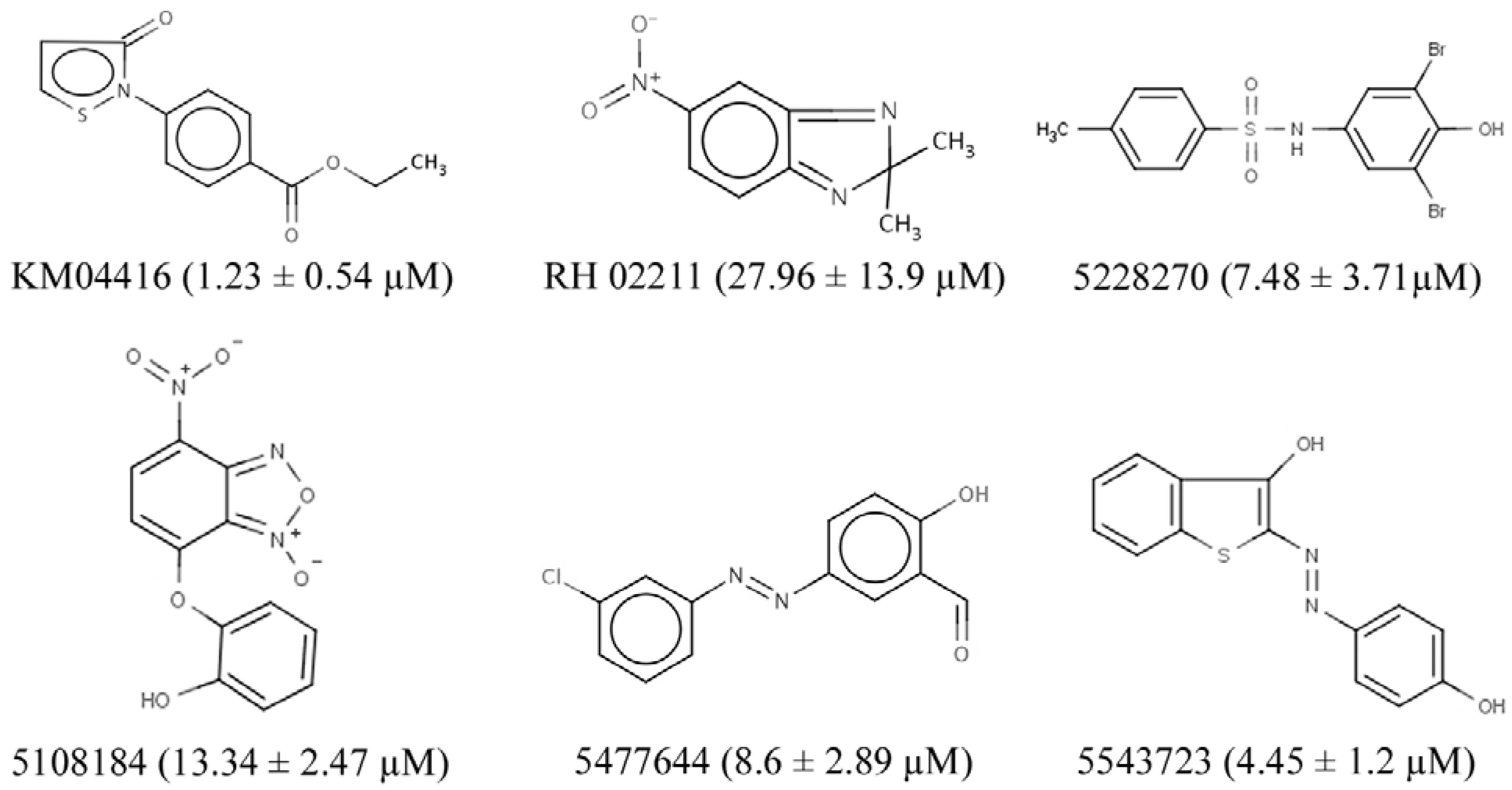
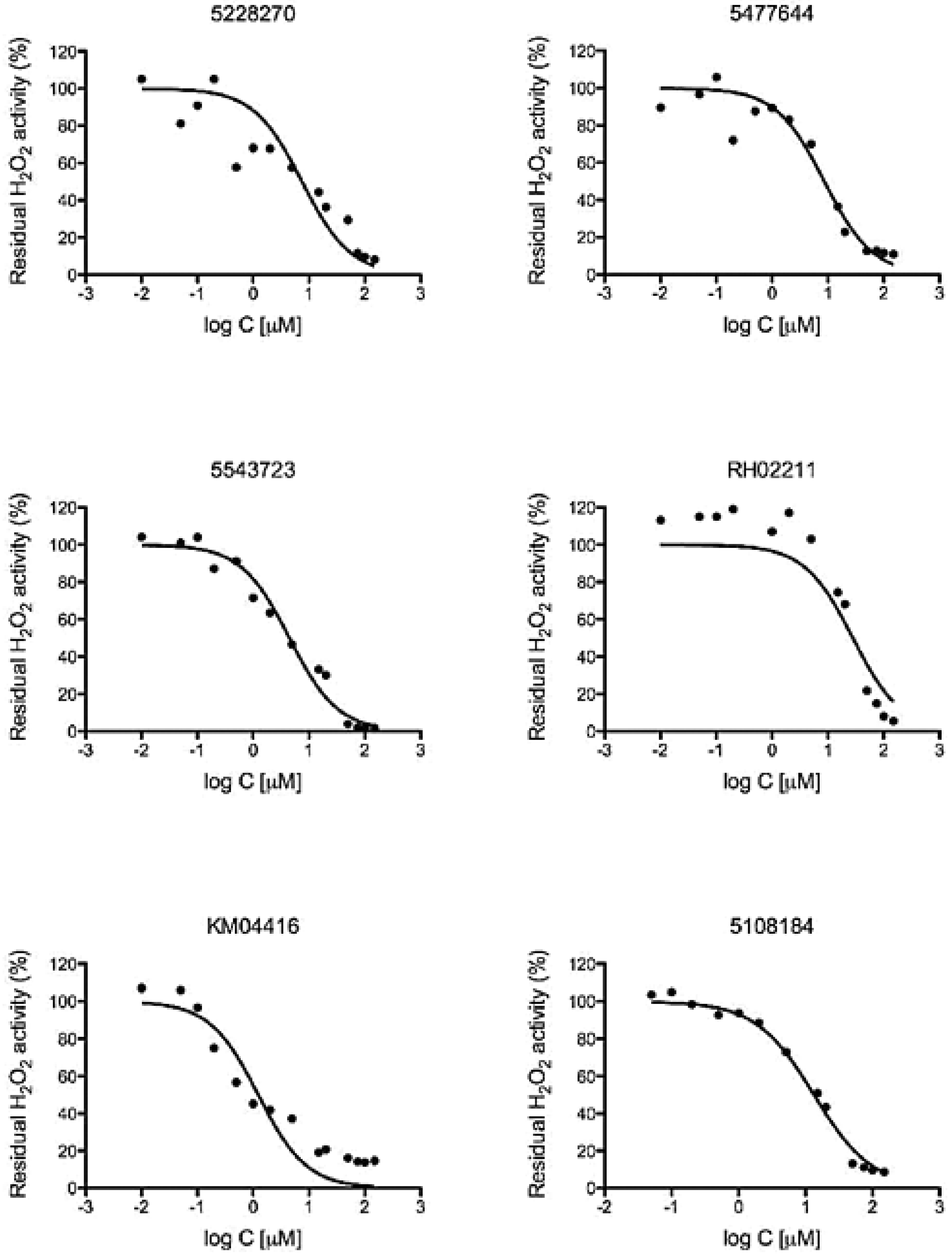
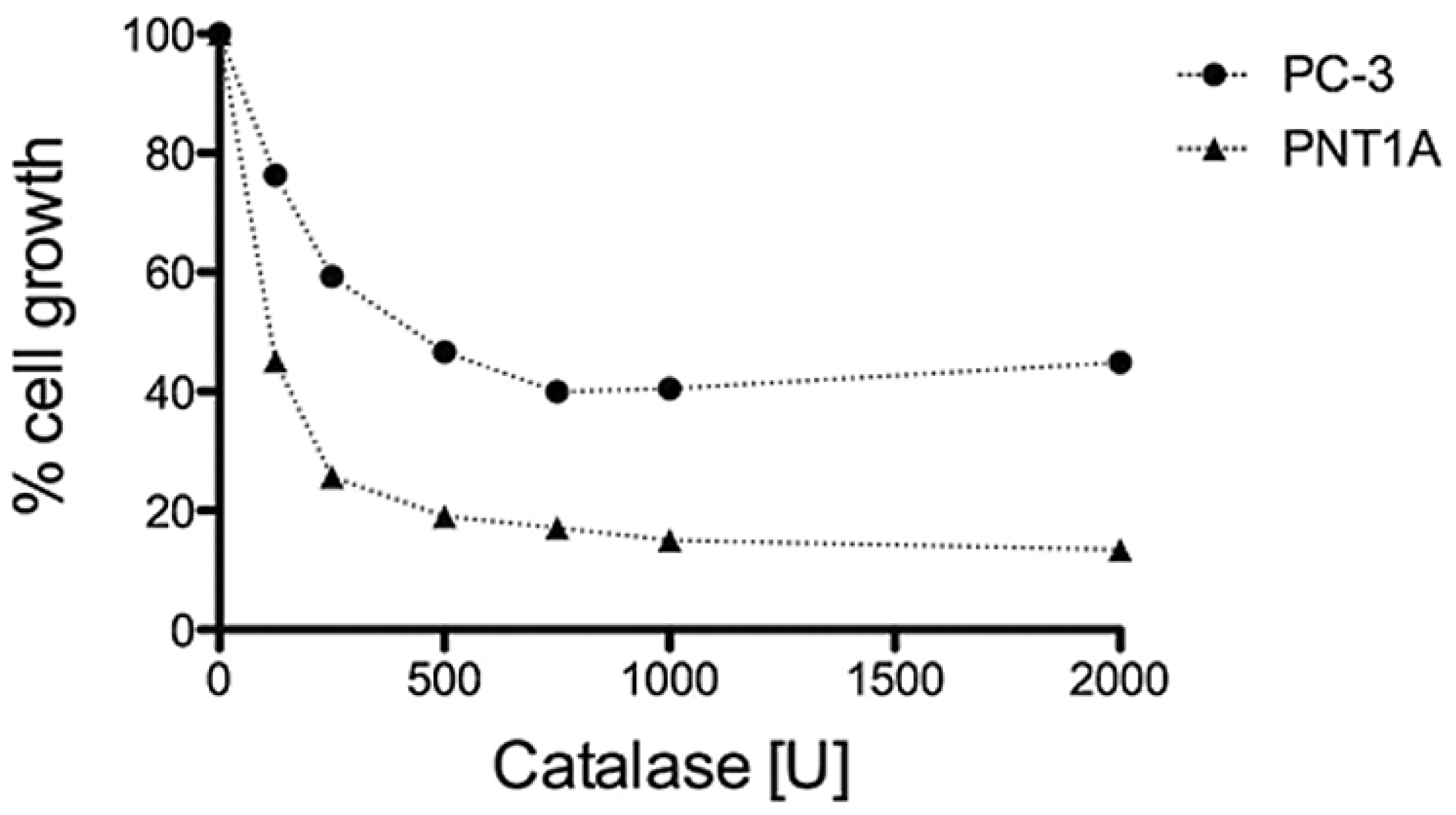
3.3. H2O2 is Required for PC-3 Cell Growth
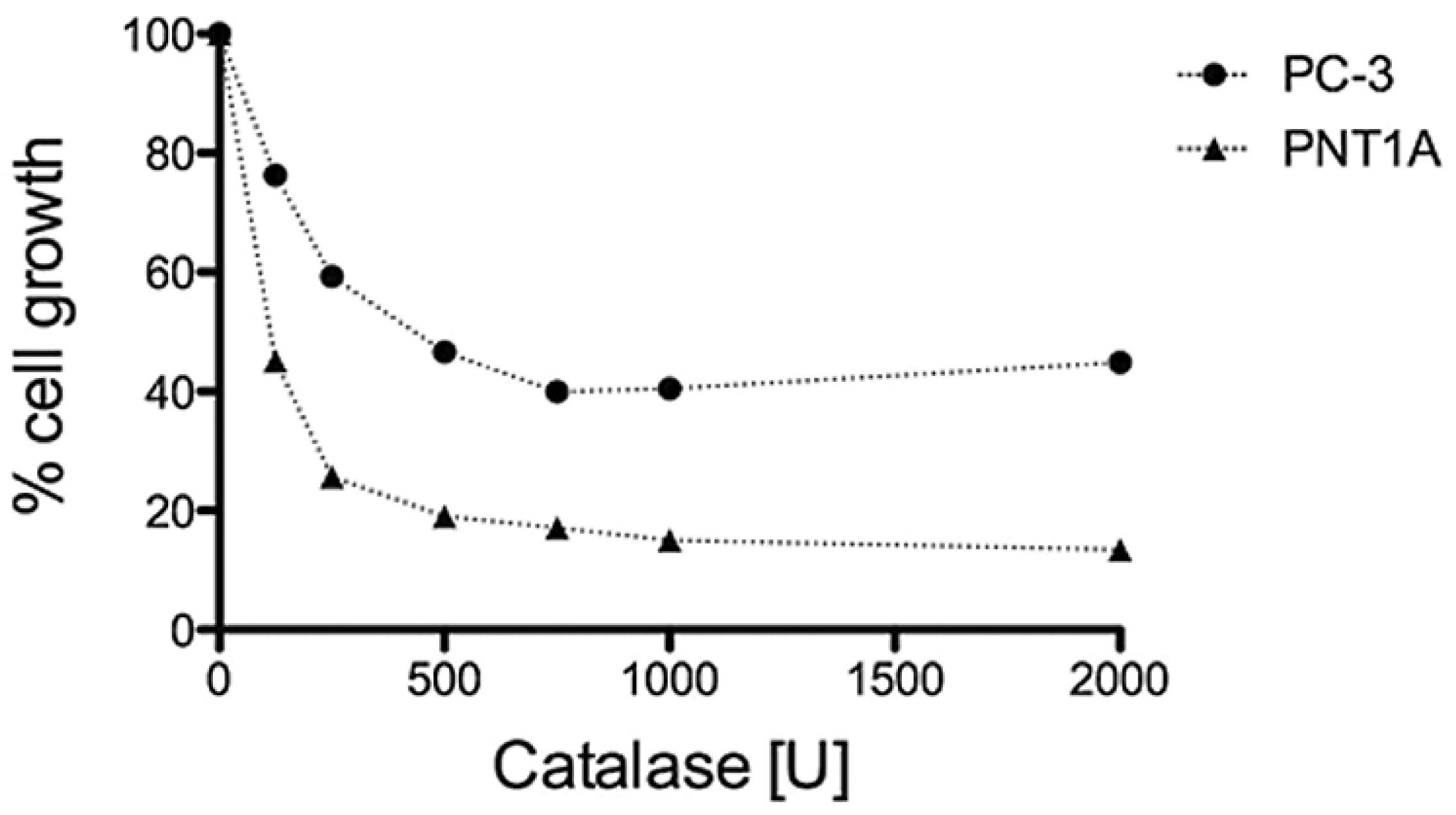
3.4. GPD2 Activity Inhibitors Impact Survival of Prostate Cancer Cells
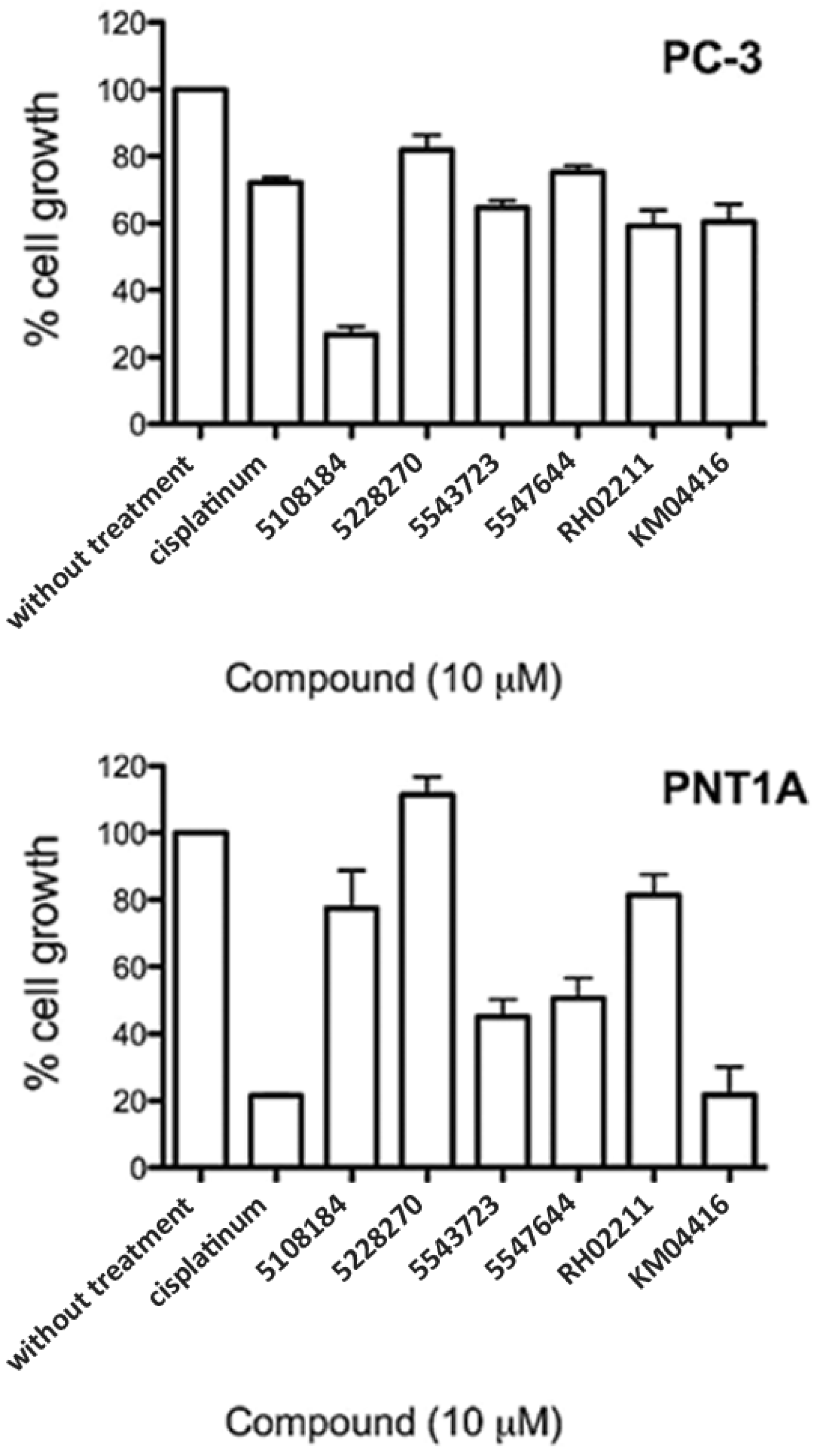
4. Conclusions
Acknowledgments
Conflicts of Interest
References
- Liou, G.Y.; Storz, P. Reactive oxygen species in cancer. Free Radic. Res. 2010, 44, 479–496. [Google Scholar]
- Kumar, B.; Koul, S.; Khandrika, L.; Meacham, R.B.; Koul, H.K. Oxidative stress is inherent in prostate cancer cells and is required for aggressive phenotype. Cancer Res. 2008, 68, 1777–1785. [Google Scholar] [CrossRef]
- Finkel, T. Reactive oxygen species and signal transduction. IUBMB Life 2001, 52, 3–6. [Google Scholar] [CrossRef]
- Irani, K.; Xia, Y.; Zweier, J.L.; Sollott, S.J.; Der, C.J.; Fearon, E.R.; Sundaresan, M.; Finkel, T.; Goldschmidt-Clermont, P.J. Mitogenic signaling mediated by oxidants in Ras-transformed fibroblasts. Science 1997, 275, 1649–1652. [Google Scholar] [CrossRef]
- Irani, K. Oxidant signaling in vascular cell growth, death, and survival: A review of the roles of reactive oxygen species in smooth muscle and endothelial cell mitogenic and apoptotic signaling. Circ. Res. 2000, 87, 179–183. [Google Scholar]
- Polytarchou, C.; Hatziapostolou, M.; Papadimitriou, E. Hydrogen peroxide stimulates proliferation and migration of human prostate cancer cells through activation of activator protein-1 and up-regulation of the heparin affin regulatory peptide gene. J. Biol. Chem. 2005, 280, 40428–40435. [Google Scholar]
- Andreyev, A.Y.; Kushnareva, Y.E.; Starkov, A.A. Mitochondrial metabolism of reactive oxygen species. Biochemistry 2005, 70, 200–214. [Google Scholar]
- Chowdhury, S.K.; Gemin, A.; Singh, G. High activity of mitochondrial glycerophosphate dehydrogenase and glycerophosphate-dependent ROS production in prostate cancer cell lines. Biochem. Biophys. Res. Commun. 2005, 333, 1139–1145. [Google Scholar] [CrossRef]
- Kwong, L.K.; Sohal, R.S. Substrate and site specificity of hydrogen peroxide generation in mouse mitochondria. Arch. Biochem. Biophys. 1998, 350, 118–126. [Google Scholar] [CrossRef]
- Miwa, S.; St-Pierre, J.; Partridge, L.; Brand, M.D. Superoxide and hydrogen peroxide production by Drosophila mitochondria. Free Radic. Biol. Med. 2003, 35, 938–948. [Google Scholar] [CrossRef]
- Mracek, T.; Pecinova, A.; Vrbacky, M.; Drahota, Z.; Houstek, J. High efficiency of ROS production by glycerophosphate dehydrogenase in mammalian mitochondria. Arch. Biochem. Biophys. 2009, 481, 30–36. [Google Scholar]
- Kaighn, M.E.; Narayan, K.S.; Ohnuki, Y.; Lechner, J.F.; Jones, L.W. Establishment and characterization of a human prostatic carcinoma cell line (PC-3). Invest. Urol. 1979, 17, 16–23. [Google Scholar]
- Klingenberg, M. Localization of the glycerol-phosphate dehydrogenase in the outer phase of the mitochondrial inner membrane. Eur. J. Biochem. 1970, 13, 247–252. [Google Scholar] [CrossRef]
- Mracek, T.; Holzerova, E.; Drahota, Z.; Kovarova, N.; Vrbacky, M.; Jesina, P.; Houstek, J. ROS generation and multiple forms of mammalian mitochondrial glycerol-3-phosphate dehydrogenase. Biochim. Biophys. Acta 2014, 1837, 98–111. [Google Scholar] [CrossRef]
- Saheki, T.; Inoue, K.; Ono, H.; Katsura, N.; Yokogawa, M.; Yoshidumi, Y.; Furuie, S.; Kuroda, E.; Ushikai, M.; Asakawa, A.; et al. Effects of supplementation on food intake, body weight and hepatic metabolites in the citrin/mitochondrial glycerol-3-phosphate dehydrogenase double-knockout mouse model of human citrin deficiency. Mol. Genet. Metab 2012, 107, 322–329. [Google Scholar] [CrossRef]
- Alfadda, A.; DosSantos, R.A.; Stepanyan, Z.; Marrif, H.; Silva, J.E. Mice with deletion of the mitochondrial glycerol-3-phosphate dehydrogenase gene exhibit a thrifty phenotype: Effect of gender. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2004, 287, R147–R156. [Google Scholar] [CrossRef]
- DosSantos, R.A.; Alfadda, A.; Eto, K.; Kadowaki, T.; Silva, J.E. Evidence for a compensated thermogenic defect in transgenic mice lacking the mitochondrial glycerol-3-phosphate dehydrogenase gene. Endocrinology 2003, 144, 5469–5479. [Google Scholar] [CrossRef]
- Gudayol, M.; Vidal-Taboada, J.M.; Usac, E.F.; Costa, A.; Cristobal, P.; Dell’Anna, C.; Oliva, R.; Gomis, R. Detection of a new variant of the mitochondrial glycerol-3-phosphate dehydrogenase gene in Spanish type 2 DM patients. Biochem. Biophys. Res.Commun. 1999, 263, 439–445. [Google Scholar] [CrossRef]
- Koike, G.; Van, V.P.; Shiozawa, M.; Galli, J.; Li, L.S.; Glaser, A.; Balasubramanyam, A.; Brown, L.J.; Luthman, H.; Szpirer, C.; et al. Genetic mapping and chromosome localization of the rat mitochondrial glycerol-3-phosphate dehydrogenase gene, a candidate for non-insulin-dependent diabetes mellitus. Genomics 1996, 38, 96–99. [Google Scholar] [CrossRef]
- MacDonald, M.J.; Longacre, M.J.; Langberg, E.C.; Tibell, A.; Kendrick, M.A.; Fukao, T.; Ostenson, C.G. Decreased levels of metabolic enzymes in pancreatic islets of patients with type 2 diabetes. Diabetologia 2009, 52, 1087–1091. [Google Scholar] [CrossRef]
- Rasschaert, J.; Malaisse-Lagae, F.; Sener, A.; Leclercq-Meyer, V.; Herberg, L.; Malaisse, W.J. Impaired FAD-glycerophosphate dehydrogenase activity in islet and liver homogenates of fa/fa rats. Mol. Cell Biochem. 1994, 135, 137–141. [Google Scholar] [CrossRef]
- Novials, A.; Vidal, J.; Franco, C.; Ribera, F.; Sener, A.; Malaisse, W.J.; Gomis, R. Mutation in the calcium-binding domain of the mitochondrial glycerophosphate dehydrogenase gene in a family of diabetic subjects. Biochem. Biophys. Res. Commun. 1997, 231, 570–572. [Google Scholar] [CrossRef]
- Fabregat, M.E.; Benito, C.; Gudayol, M.; Vidal, J.; Gallart, T.; Malaisse, W.J.; Gomis, R. Enzyme-linked immunosorbent assay of autoantibodies against mitochondrial glycerophosphate dehydrogenase in insulin-dependent and non-insulin-dependent diabetic subjects. Biochem. Mol. Med. 1997, 62, 172–177. [Google Scholar] [CrossRef]
- Daoud, H.; Gruchy, N.; Constans, J.M.; Moussaoui, E.; Saumureau, S.; Bayou, N.; Amy, M.; Vedrine, S.; Vu, P.Y.; Rotig, A.; et al. Haploinsufficiency of the GPD2 gene in a patient with nonsyndromic mental retardation. Hum. Genet. 2009, 124, 649–658. [Google Scholar] [CrossRef]
- Barge-Schaapveld, D.Q.; Ofman, R.; Knegt, A.C.; Alders, M.; Hohne, W.; Kemp, S.; Hennekam, R.C. Intellectual disability and hemizygous GPD2 mutation. Am. J. Med. Genet. A 2013, 161A, 1044–1050. [Google Scholar]
- Oliveira, H.C.; Cosso, R.G.; Alberici, L.C.; Maciel, E.N.; Salerno, A.G.; Dorighello, G.G.; Velho, J.A.; de Faria, E.C.; Vercesi, A.E. Oxidative stress in atherosclerosis-prone mouse is due to low antioxidant capacity of mitochondria. FASEB J. 2005, 19, 278–280. [Google Scholar]
- Cussenot, O.; Berthon, P.; Berger, R.; Mowszowicz, I.; Faille, A.; Hojman, F.; Teillac, P.; Le, D.A.; Calvo, F. Immortalization of human adult normal prostatic epithelial cells by liposomes containing large T-SV40 gene. J. Urol. 1991, 146, 881–886. [Google Scholar]
- Zhou, M.; Diwu, Z.; Panchuk-Voloshina, N.; Haugland, R.P. A stable nonfluorescent derivative of resorufin for the fluorometric determination of trace hydrogen peroxide: Applications in detecting the activity of phagocyte NADPH oxidase and other oxidases. Anal. Biochem. 1997, 253, 162–168. [Google Scholar] [CrossRef]
- Benz, R.; McLaughlin, S. The molecular mechanism of action of the proton ionophore FCCP (carbonylcyanide p-trifluoromethoxyphenylhydrazone). Biophys. J. 1983, 41, 381–398. [Google Scholar] [CrossRef]
- Zhang, J.H.; Chung, T.D.; Oldenburg, K.R. A simple statistical parameter for use in evaluation and validation of high throughput screening assays. J. Biomol. Screen. 1999, 4, 67–73. [Google Scholar] [CrossRef]
- Bhandari, P.; Gowrishankar, J. An Escherichia coli host strain useful for efficient overproduction of cloned gene products with NaCl as the inducer. J. Bacteriol. 1997, 179, 4403–4406. [Google Scholar]
- Brown, L.J.; Koza, R.A.; Everett, C.; Reitman, M.L.; Marshall, L.; Fahien, L.A.; Kozak, L.P.; MacDonald, M.J. Normal thyroid thermogenesis but reduced viability and adiposity in mice lacking the mitochondrial glycerol phosphate dehydrogenase. J. Biol. Chem. 2002, 277, 32892–32898. [Google Scholar] [CrossRef]
- Chowdhury, S.K.; Raha, S.; Tarnopolsky, M.A.; Singh, G. Increased expression of mitochondrial glycerophosphate dehydrogenase and antioxidant enzymes in prostate cancer cell lines/cancer. Free Radic. Res. 2007, 41, 1116–1124. [Google Scholar] [CrossRef]
- Colussi, T.; Parsonage, D.; Boles, W.; Matsuoka, T.; Mallett, T.C.; Karplus, P.A.; Claiborne, A. Structure of alpha-glycerophosphate oxidase from Streptococcus sp.: A template for the mitochondrial alpha-glycerophosphate dehydrogenase. Biochemistry 2008, 47, 965–977. [Google Scholar] [CrossRef]
- Cole, E.S.; Lepp, C.A.; Holohan, P.D.; Fondy, T.P. Isolation and characterization of flavin-linked glycerol-3-phosphate dehydrogenase from rabbit skeletal muscle mitochondria and comparison with the enzyme from rabbit brain. J. Biol. Chem. 1978, 253, 7952–7959. [Google Scholar]
- Garrib, A.; McMurray, W.C. Purification and characterization of glycerol-3-phosphate dehydrogenase (flavin-linked) from rat liver mitochondria. J. Biol. Chem. 1986, 261, 8042–8048. [Google Scholar]
- Preston, T.J.; Muller, W.J.; Singh, G. Scavenging of extracellular H2O2 by catalase inhibits the proliferation of HER-2/Neu-transformed rat-1 fibroblasts through the induction of a stress response. J. Biol. Chem. 2001, 276, 9558–9564. [Google Scholar] [CrossRef]
- Ricci, G.; De, M.F.; Antonini, G.; Turella, P.; Bullo, A.; Stella, L.; Filomeni, G.; Federici, G.; Caccuri, A.M. 7-Nitro-2,1,3-benzoxadiazole derivatives, a new class of suicide inhibitors for glutathione S-transferases. Mechanism of action of potential anticancer drugs. J. Biol. Chem. 2005, 280, 26397–26405. [Google Scholar] [CrossRef]
- Seaton, A.; Higgins, C.; Mann, J.; Baron, A.; Bailly, C.; Neidle, S.; van den Berg, H. Mechanistic and anti-proliferative studies of two novel, biologically active bis-benzimidazoles. Eur. J. Cancer 2003, 39, 2548–2555. [Google Scholar] [CrossRef]
- Dionisi, O.; Cittadini, A.; Gelmuzzi, G.; Galeotti, T.; Terranova, T. The role of the alpha-glycerophosphate shuttle in the reoxidation of cytosolic NADH in Ehrlich ascites tumour cells. Biochim. Biophys. Acta 1970, 216, 71–79. [Google Scholar] [CrossRef]
- Hunt, S.M.; Osnos, M.; Rivlin, R.S. Thyroid hormone regulation of mitochondrial alpha-glycerophosphate dehydrogenase in liver and hepatoma. Cancer Res. 1970, 30, 1764–1768. [Google Scholar]
- Karsten, U.; Sydow, G.; Wollenberger, A.; Graffi, A. Rat liver glycerolphosphate dehydrogenases: Activity changes and induction by thyroid hormone of the mitochondrial enzyme in hepatomas and in precancerous and growing liver. Acta Biol. Med. Ger. 1971, 26, 1131–1140. [Google Scholar]
- Peron, F.G.; Haksar, A.; Lin, M.; Kupfer, D.; Robidoux, W., Jr.; Kimmel, G.; Bedigian, E. Studies on respiration and 11 beta-hydroxylation of deoxycorticosterone in mitochondria and intact cells isolated from the Snell adrenocortical carcinoma 494. Cancer Res. 1974, 34, 2711–2719. [Google Scholar]
- Abraham, S.; Bartley, J.; DeOme, K.B.; Faulkin, L.J., Jr.; Medina, D. Enzyme activities in induced and serially transplanted murine mammary adenocarcinomas. J. Natl. Cancer Inst. 1973, 51, 251–256. [Google Scholar]
- Criss, W.E. Control of the adenylate charge in the Morris “minimal-deviation” hepatomas. Cancer Res. 1973, 33, 51–56. [Google Scholar]
- Harding, J.W., Jr.; Pyeritz, E.A.; Morris, H.P.; White, H.B., III. Proportional activities of glycerol kinase and glycerol 3-phosphate dehydrogenase in rat hepatomas. Biochem. J. 1975, 148, 545–550. [Google Scholar]
- Shonk, C.E.; Arison, R.N.; Koven, B.J.; Majima, H.; Boxer, G.E. Enzyme patterns in human tissues. 3. Glycolytic enzymes in normal and malignant tissues of the colon and rectum. Cancer Res. 1965, 25, 206–213. [Google Scholar]
- Shonk, C.E.; Morris, H.P.; Boxer, G.E. Patterns of glycolytic enzymes in rat liver and hepatoma. Cancer Res. 1965, 25, 671–676. [Google Scholar]
- Liu, Y. Fatty acid oxidation is a dominant bioenergetic pathway in prostate cancer. Prostate Cancer Prostatic. Dis. 2006, 9, 230–234. [Google Scholar] [CrossRef]
- Katusic, Z.S.; Vanhoutte, P.M. Superoxide anion and endothelial regulation of arterial tone. Semin. Perinatol. 1991, 15, 30–33. [Google Scholar]
- Glass, C.K.; Witztum, J.L. Atherosclerosis. the road ahead. Cell 2001, 104, 503–516. [Google Scholar] [CrossRef]
- Fulda, S.; Galluzzi, L.; Kroemer, G. Targeting mitochondria for cancer therapy. Nat. Rev. Drug Discov. 2010, 9, 447–464. [Google Scholar] [CrossRef]
- Kroemer, G. Mitochondria in cancer. Oncogene 2006, 25, 4630–4632. [Google Scholar] [CrossRef]
- Trachootham, D.; Alexandre, J.; Huang, P. Targeting cancer cells by ROS-mediated mechanisms: A radical therapeutic approach? Nat. Rev. Drug Discov. 2009, 8, 579–591. [Google Scholar] [CrossRef]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Singh, G. Mitochondrial FAD-linked Glycerol-3-phosphate Dehydrogenase: A Target for Cancer Therapeutics. Pharmaceuticals 2014, 7, 192-206. https://doi.org/10.3390/ph7020192
Singh G. Mitochondrial FAD-linked Glycerol-3-phosphate Dehydrogenase: A Target for Cancer Therapeutics. Pharmaceuticals. 2014; 7(2):192-206. https://doi.org/10.3390/ph7020192
Chicago/Turabian StyleSingh, Gurmit. 2014. "Mitochondrial FAD-linked Glycerol-3-phosphate Dehydrogenase: A Target for Cancer Therapeutics" Pharmaceuticals 7, no. 2: 192-206. https://doi.org/10.3390/ph7020192