Structural Bioinformatics and Protein Docking Analysis of the Molecular Chaperone-Kinase Interactions: Towards Allosteric Inhibition of Protein Kinases by Targeting the Hsp90-Cdc37 Chaperone Machinery


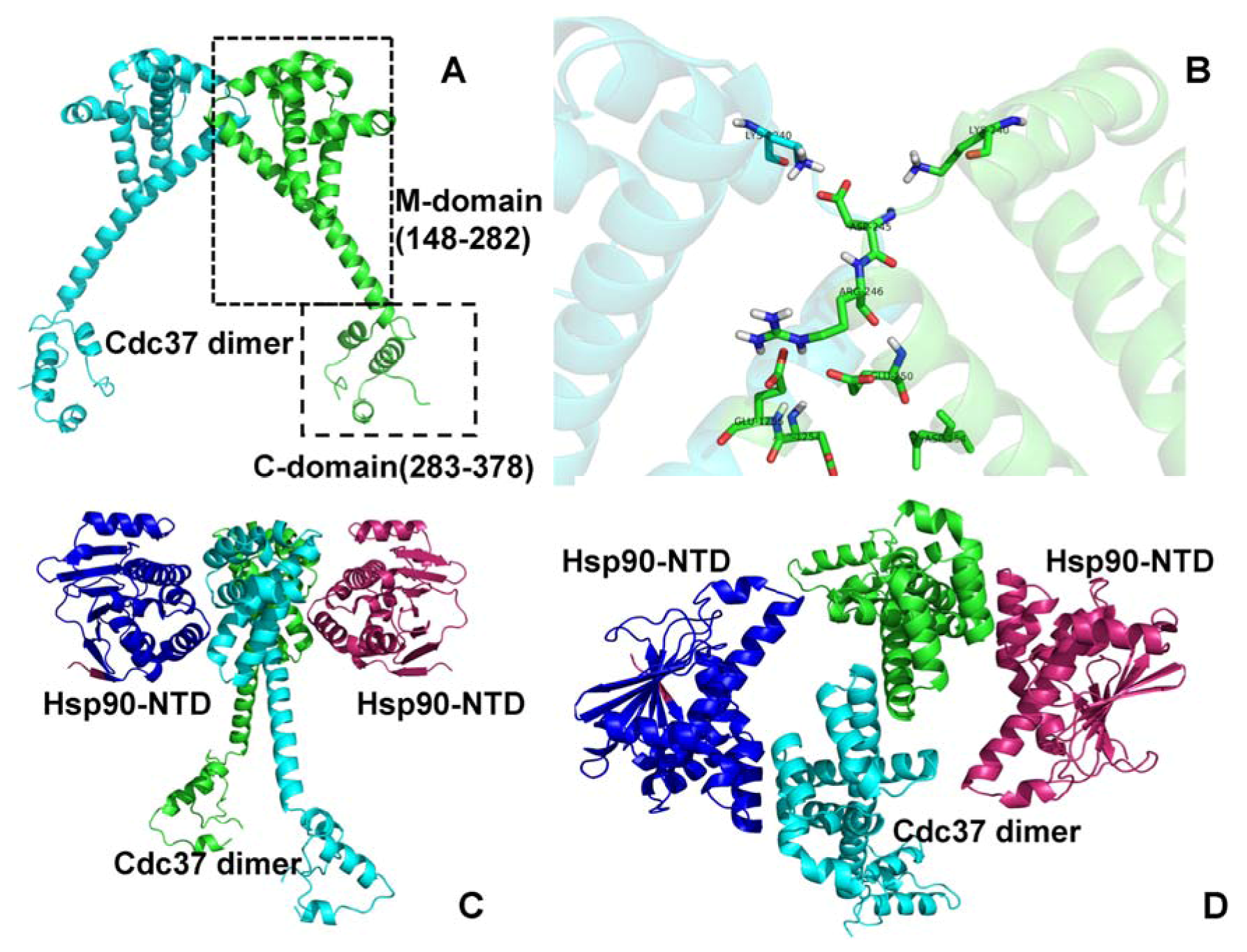{kind=link}
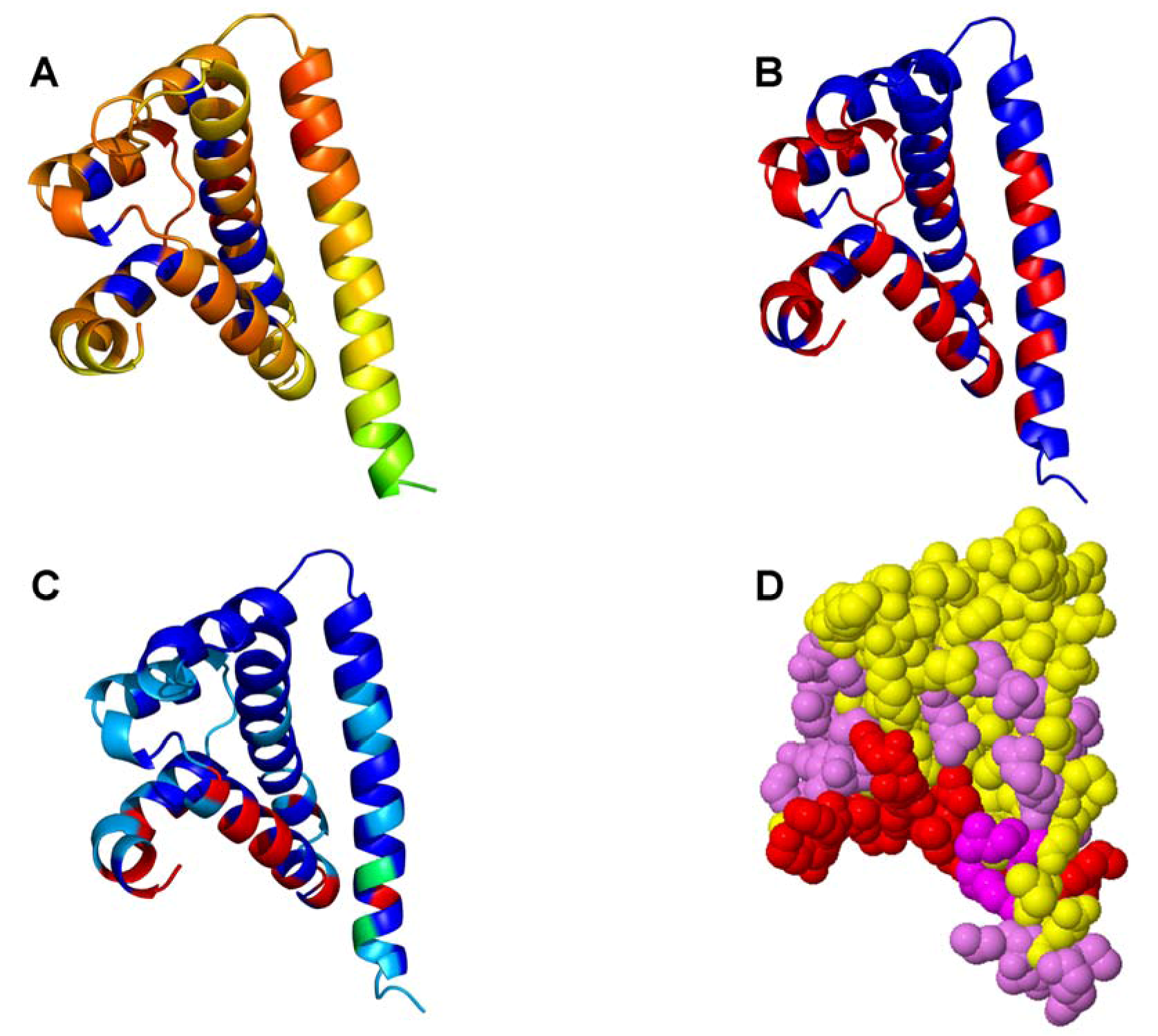{kind=link}
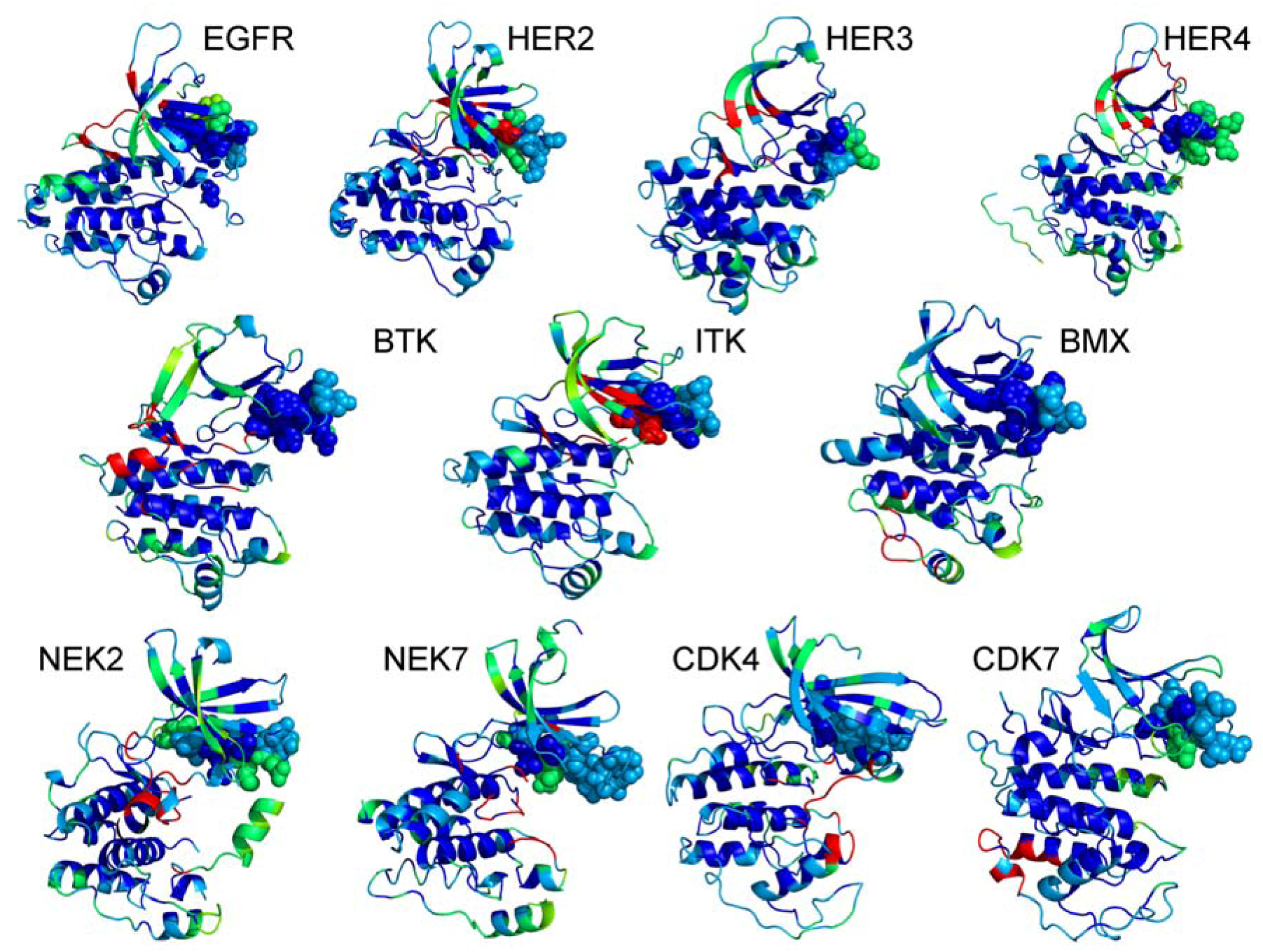{kind=link}
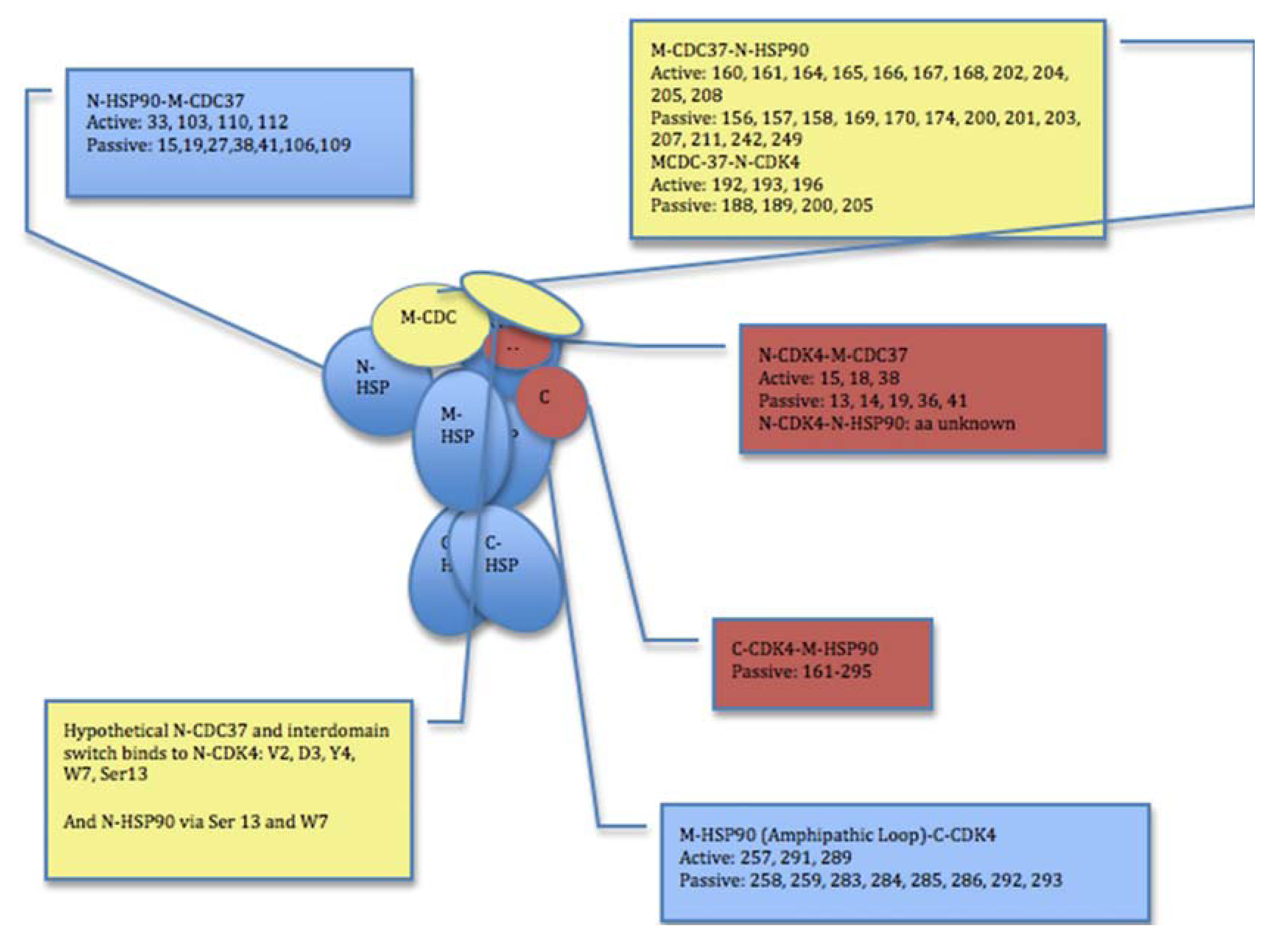{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
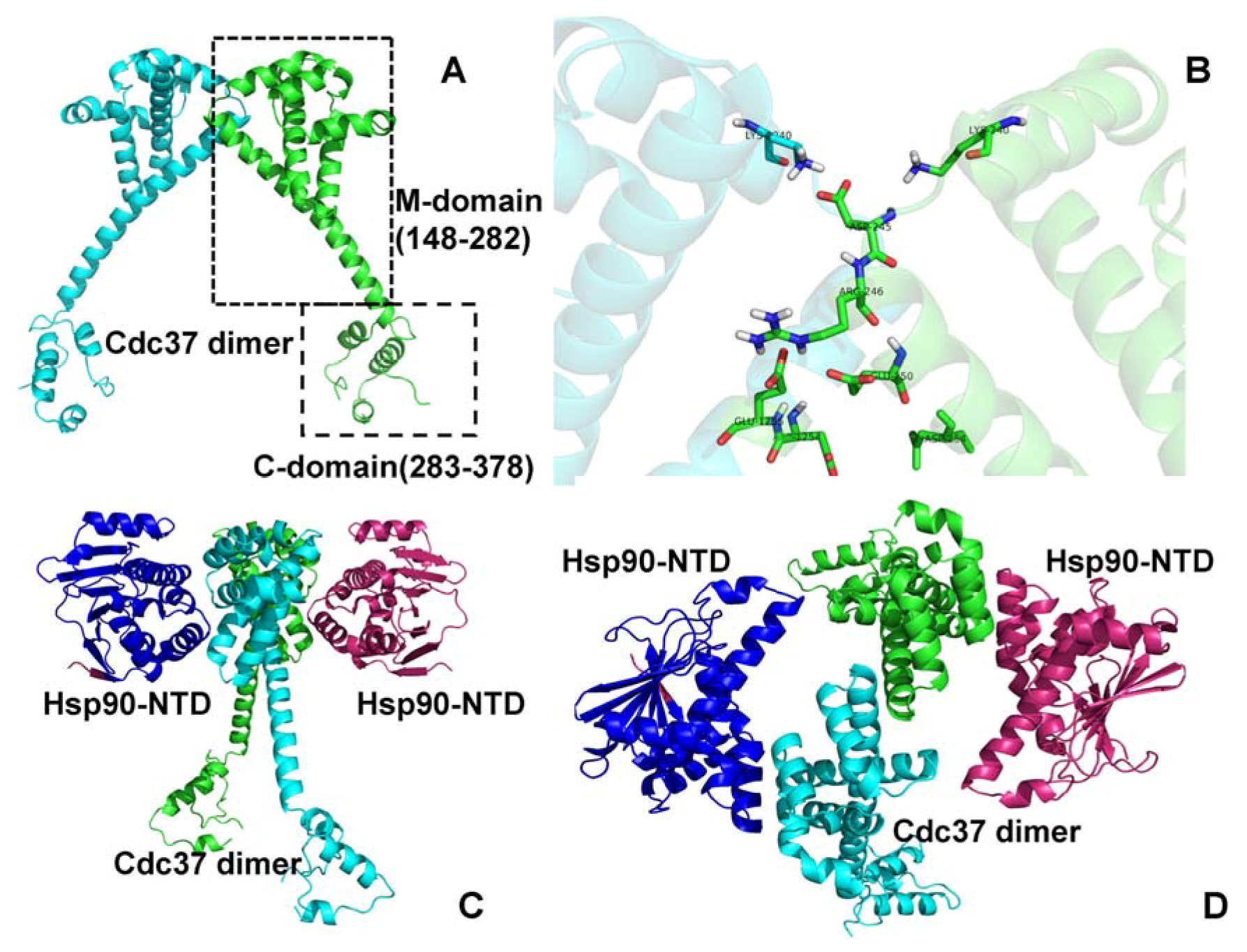
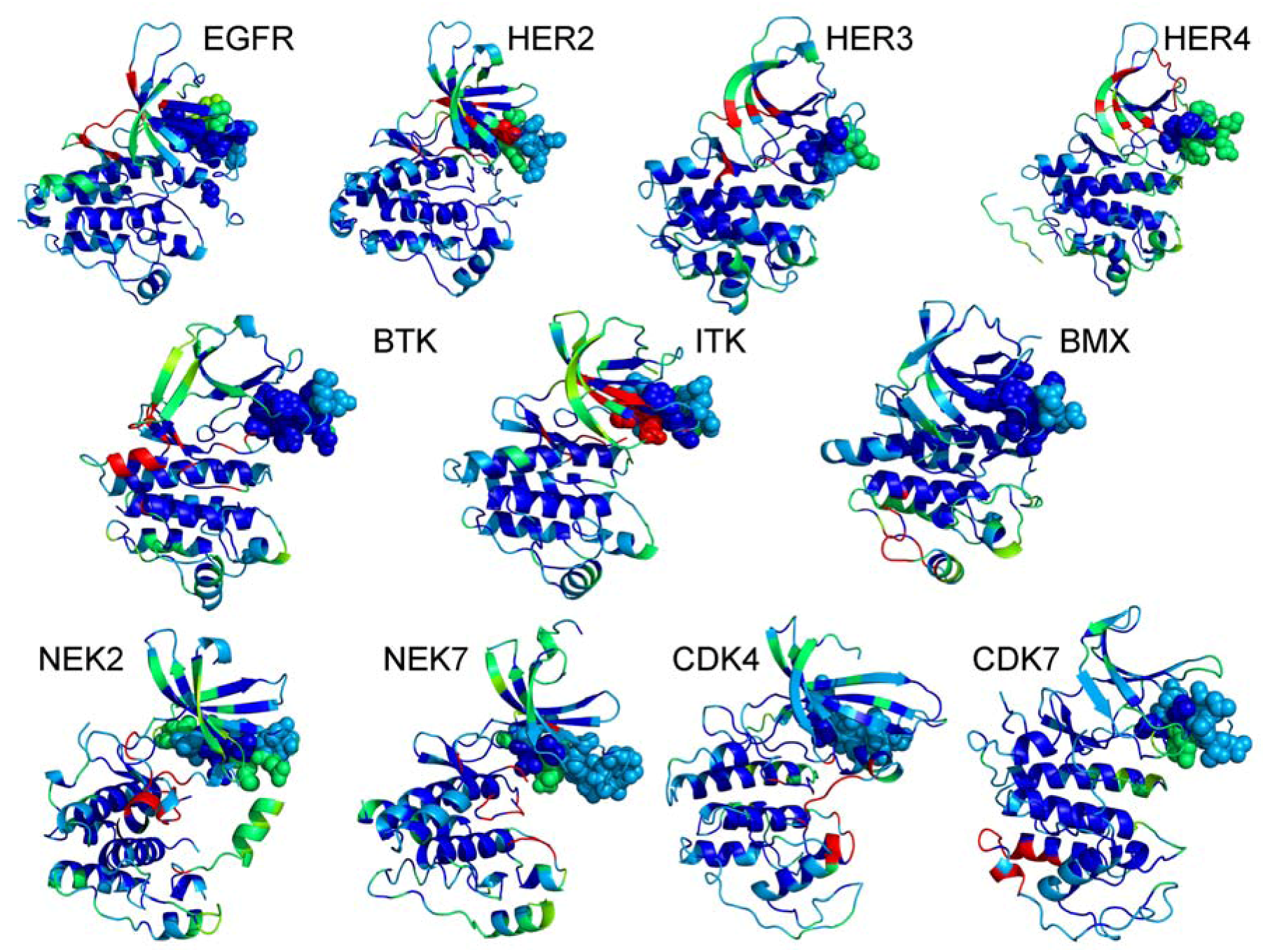
2.1. Bioinformatics Analysis of the Interaction Maps
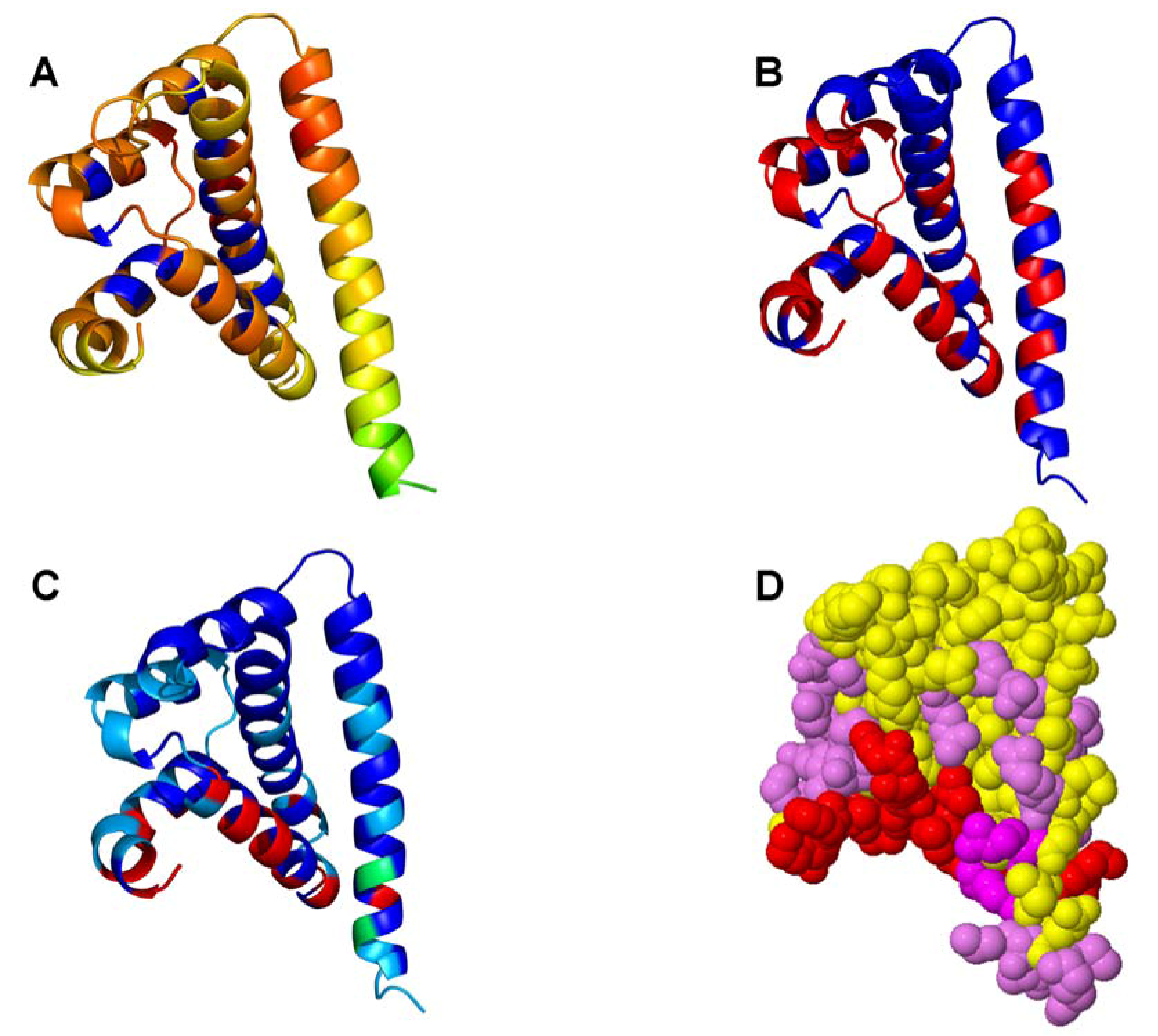
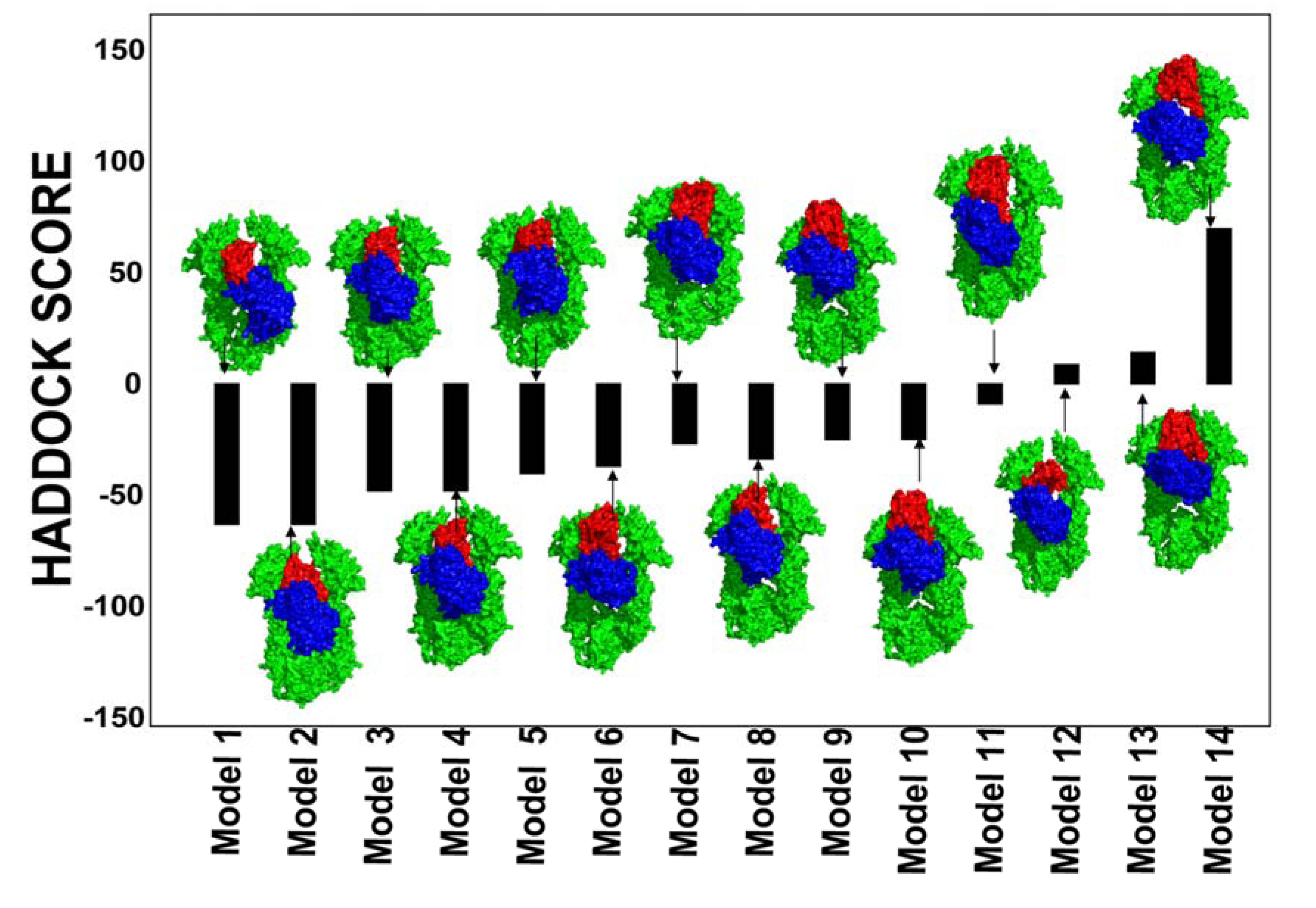
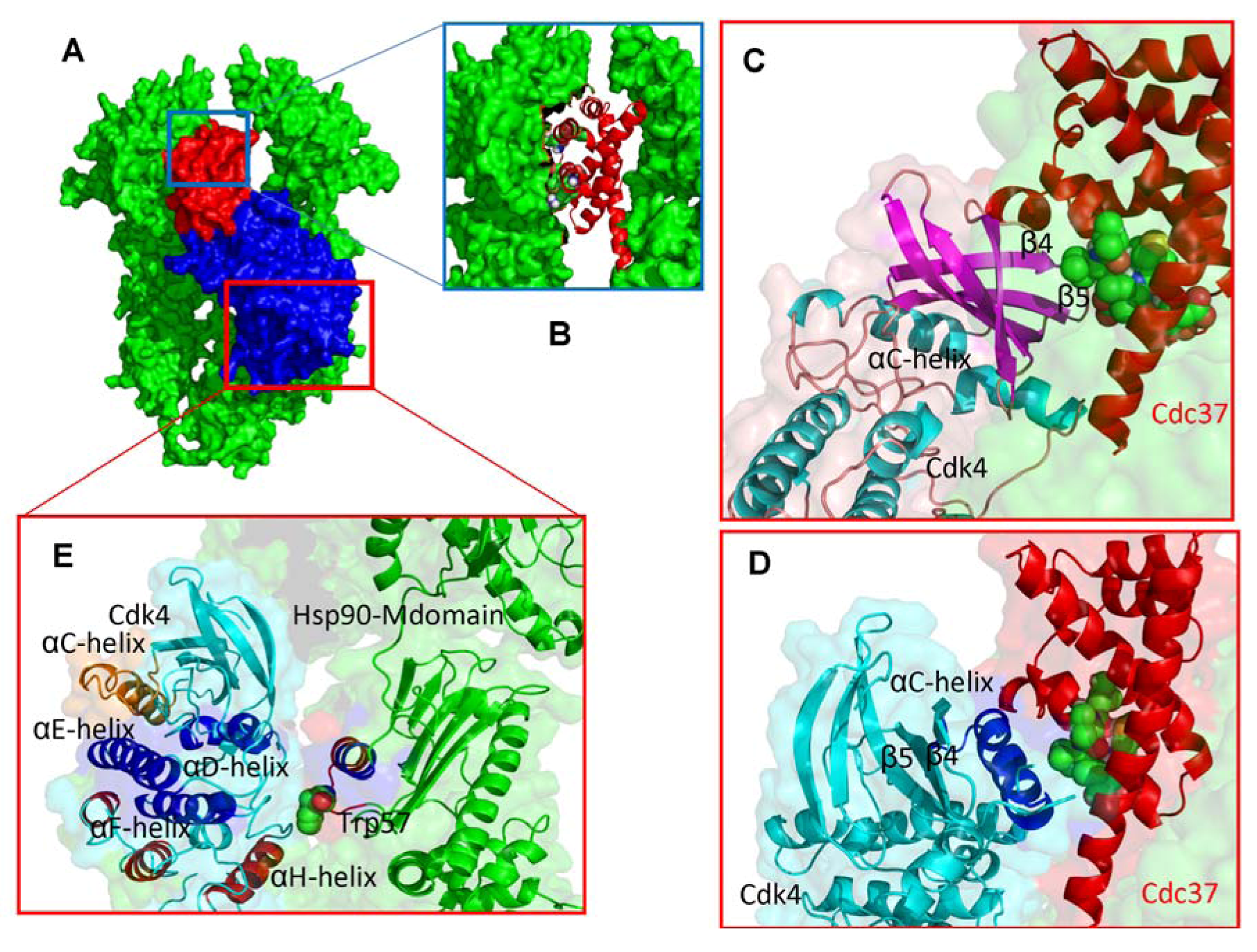
2.2. Experimentally-Guided Docking of the Hsp90-Cdc37-Cdk4 Interactions
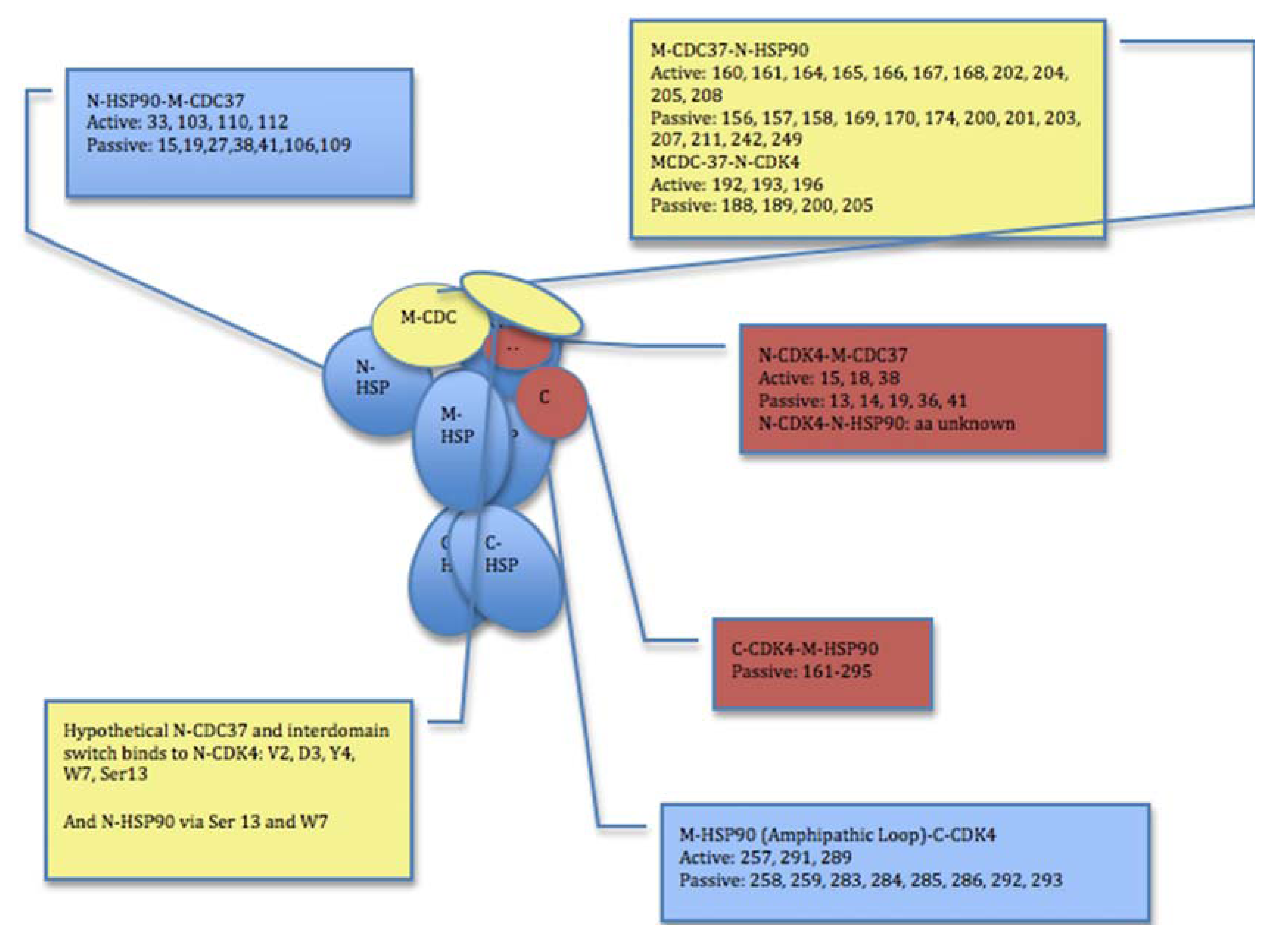
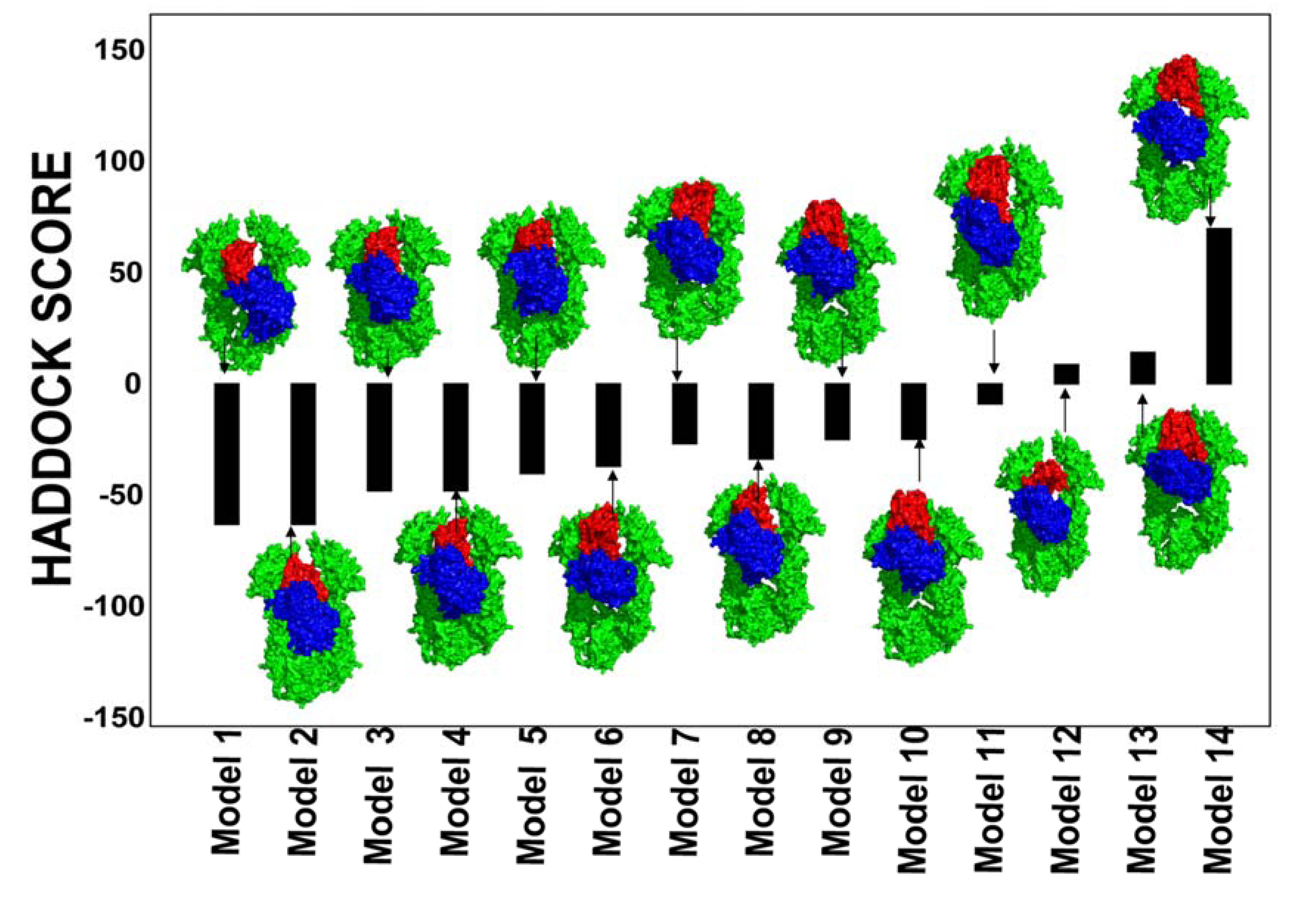
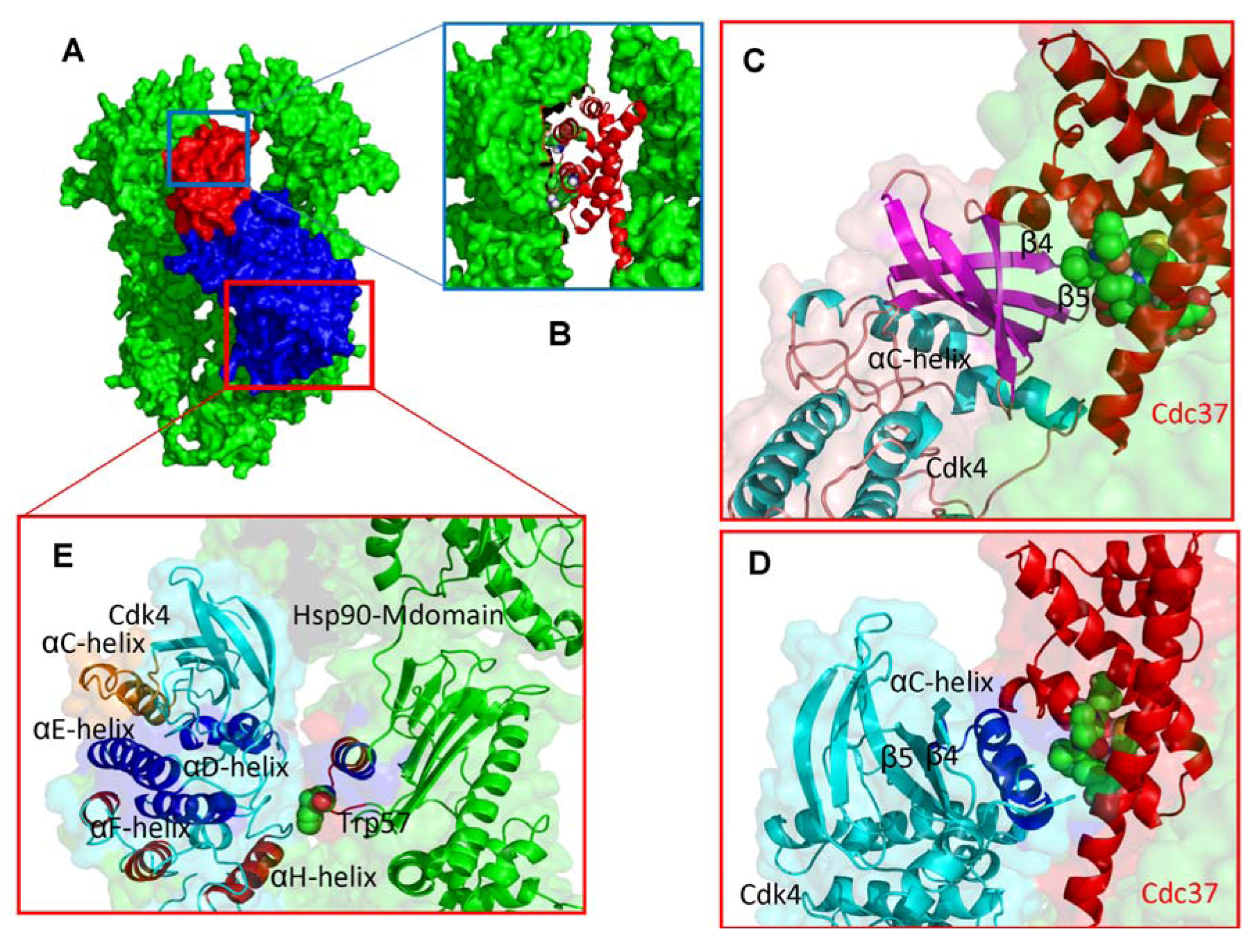
2.3. Towards Allosteric Inhibition of Protein Kinases by Targeting the Hsp90-Cdc37 Chaperone
3. Conclusions
Conflicts of Interest
References
- Pearl, L.H.; Prodromou, C. Structure, function, and mechanism of the Hsp90 molecular chaperone. Adv. Protein Chem. 2001, 59, 157–186. [Google Scholar] [CrossRef]
- Richter, K.; Buchner, J. Hsp90: Chaperoning signal transduction. J. Cell. Physiol. 2001, 188, 281–290. [Google Scholar] [CrossRef]
- Young, J.C.; Moarefi, I.; Hartl, F.U. Hsp90: A specialized but essential protein-folding tool. J. Cell Biol. 2001, 154, 267–273. [Google Scholar] [CrossRef]
- Picard, D. Heat-shock protein 90, a chaperone for folding and regulation. Cell. Mol. Life Sci. 2002, 59, 1640–1648. [Google Scholar] [CrossRef]
- Young, J.C.; Agashe, V.R.; Siegers, K.; Hartl, U.F. Pathways of chaperone-mediated protein folding in the cytosol. Nat. Rev. Mol. Cell. Biol. 2004, 5, 781–791. [Google Scholar] [CrossRef]
- Wayne, N.; Mishra, P.; Bolon, D.N. Hsp90 and client protein maturation. Methods Mol. Biol. 2011, 787, 33–44. [Google Scholar] [CrossRef]
- Zhao, R.; Davey, M.; Hsu, Y.C.; Kaplanek, P.; Tong, A.; Parsons, A.B.; Krogan, N.; Cagney, G.; Mai, D.; Greenblatt, J.; et al. Navigating the chaperone network: an integrative map of physical and genetic interactions mediated by the Hsp90 chaperone. Cell 2005, 120, 715–727. [Google Scholar] [CrossRef]
- McClellan, A.J.; Xia, Y.; Deutschbauer, A.M.; Davis, R.W.; Gerstein, M.; Frydman, J. Diverse cellular functions of the Hsp90 molecular chaperone uncovered using systems approaches. Cell 2007, 131, 121–135. [Google Scholar] [CrossRef]
- Zhao, R.; Houry, W.A. Molecular interaction network of the Hsp90 chaperone system. Adv. Exp. Med. Biol. 2007, 594, 27–36. [Google Scholar] [CrossRef]
- Taipale, M.; Jarosz, D.F.; Lindquist, S. Hsp90 at the hub of protein homeostasis: emerging mechanistic insights. Nat. Rev. Mol. Cell. Biol. 2010, 11, 515–528. [Google Scholar] [CrossRef]
- Manning, G.; Whyte, D.B.; Martinez, R.; Hunter, T.; Sudarsanam, S. The protein kinase complement of the human genome. Science 2002, 298, 1912–1934. [Google Scholar] [CrossRef]
- Manning, G.; Plowman, G.D.; Hunter, T.; Sudarsanam, S. Evolution of protein kinase signaling from yeast to man. Trends Biochem. Sci. 2002, 10, 514–520. [Google Scholar]
- Hunter, T. Signaling—2000 and beyond. Cell 2000, 100, 113–127. [Google Scholar] [CrossRef]
- Lee, P.; Rao, J.; Fliss, A.; Yang, E.; Garrett, S.; Caplan, A.J. The Cdc37 protein kinase-binding domain is sufficient for protein kinase activity and cell viability. J. Cell. Biol. 2002, 159, 1051–1059. [Google Scholar] [CrossRef]
- MacLean, M.; Picard, D. Cdc37 goes beyond Hsp90 and kinases. Cell. Stress Chaperones 2003, 8, 114–119. [Google Scholar] [CrossRef]
- Pearl, L.H. Hsp90 and Cdc37—A chaperone cancer conspiracy. Curr. Opin. Genet. Dev. 2005, 15, 55–61. [Google Scholar] [CrossRef]
- Gerber, M.R.; Farrell, A.; Deshaies, R.J.; Herskowitz, I.; Morgan, D.O. Cdc37 is required for association of the protein kinase Cdc28 with G1 and mitotic cyclins. Proc. Natl. Acad. Sci. USA 1995, 92, 4651–4655. [Google Scholar]
- Abbas-Terki, T.; Donze, O.; Picard, D. The molecular chaperone Cdc37 is required for Ste11 function and pheromone-induced cell cycle arrest. FEBS Lett. 2000, 467, 111–116. [Google Scholar] [CrossRef]
- Hartson, S.D.; Barrett, D.J.; Burn, P.; Matts, R.L. Hsp90-mediated folding of the lymphoid cell kinase p56lck. Biochemistry 1996, 35, 13451–13559. [Google Scholar] [CrossRef]
- Stepanova, L.; Leng, X.; Parker, S.B.; Harper, J.W. Mammalian p50Cdc37 is a protein kinase-targeting subunit of Hsp90 that binds and stabilizes Cdk4. Genes Dev. 1996, 10, 1491–502. [Google Scholar] [CrossRef]
- Silverstein, A.M.; Grammatikakis, N.; Cochran, B.H.; Chinkers, M.; Pratt, W.B. p50(cdc37) binds directly to the catalytic domain of Raf as well as to a site on hsp90 that is topologically adjacent to the tetratricopeptide repeat binding site. J. Biol. Chem. 1998, 273, 20090–20095. [Google Scholar]
- Grammatikakis, N.; Lin, J.H.; Grammatikakis, A.; Tsichlis, P.N.; Cochran, B.H. p50(cdc37) acting in concert with Hsp90 is required for Raf-1 function. Mol. Cell. Biol. 1999, 19, 1661–1672. [Google Scholar]
- Stepanova, L.; Yang, G.; DeMayo, F.; Wheeler, T.M.; Finegold, M.; Thompson, T.C.; Harper, J.W. Induction of human Cdc37 in prostate cancer correlates with the ability of targeted Cdc37 expression to promote prostatic hyperplasia. Oncogene 2000, 19, 2186–2193. [Google Scholar] [CrossRef]
- Stepanova, L.; Finegold, M.; DeMayo, F.; Schmidt, E.V.; Harper, J.W. The oncoprotein kinase chaperone CDC37 functions as an oncogene in mice and collaborates with both c-myc and cyclin D1 in transformation of multiple tissues. Mol. Cell. Biol. 2000, 20, 4462–4473. [Google Scholar] [CrossRef]
- Hartson, S.D.; Irwin, A.D.; Shao, J.; Scroggins, B.T.; Volk, L.; Huang, W.; Matts, R.L. p50(cdc37) is a nonexclusive Hsp90 cohort which participates intimately in Hsp90-mediated folding of immature kinase molecules. Biochemistry 2000, 39, 7631–7644. [Google Scholar] [CrossRef]
- Shao, J.; Grammatikakis, N.; Scroggins, B.T.; Uma, S.; Huang, W.; Chen, J.J.; Hartson, S.D.; Matts, R.L. Hsp90 regulates p50(cdc37) function during the biogenesis of the active conformation of the heme-regulated eIF2 alpha kinase. J. Biol. Chem. 2001, 276, 206–214. [Google Scholar]
- Mandal, A.K.; Lee, P.; Chen, J.A.; Nillegoda, N.; Heller, A.; DiStasio, S.; Oen, H.; Victor, J.; Nair, D.M.; Brodsky, J.L.; et al. Cdc37 has distinct roles in protein kinase quality control that protect nascent chains from degradation and promote posttranslational maturation. J. Cell. Biol. 2007, 176, 319–328. [Google Scholar] [CrossRef]
- Mandal, A.K.; Theodoraki, M.A.; Nillegoda, N.B.; Caplan, A.J. Role of molecular chaperones in biogenesis of the protein kinome. Methods Mol. Biol. 2011, 787, 75–81. [Google Scholar] [CrossRef]
- Karnitz, L.M.; Felts, S.J. Cdc37 regulation of the kinome: when to hold ‘em and when to fold’ em. Sci. Signal. Transduct. Knowl. Environ. 2007, 385, e22. [Google Scholar]
- Isaacs, J.S.; Xu, W.; Neckers, L. Heat shock protein 90 as a molecular target for cancer therapeutics. Cancer Cell 2003, 3, 213–217. [Google Scholar] [CrossRef]
- Whitesell, L.; Lindquist, S.L. Hsp90 and the chaperoning of cancer. Nat. Rev. Cancer 2005, 5, 761–772. [Google Scholar] [CrossRef]
- Xu, W.; Neckers, L. Targeting the molecular chaperone heat shock protein 90 provides a multifaceted effect on diverse cell signaling pathways of cancer cells. Clin. Cancer Res. 2007, 13, 1625–1629. [Google Scholar] [CrossRef]
- Meyer, P.; Prodromou, C.; Hu, B.; Vaughan, C.; Roe, M.S.; Panaretou, B.; Piper, P.W.; Pearl, L.H. Structural and functional analysis of the middle segment of hsp90: Implications for ATP hydrolysis and client protein and cochaperone interactions. Mol. Cell 2003, 11, 647–658. [Google Scholar] [CrossRef]
- Ali, M.M.; Roe, S.M.; Vaughan, C.K.; Meyer, P.; Panaretou, B.; Piper, P.W.; Prodromou, C.; Pearl, L.H. Crystal structure of an Hsp90-nucleotide-p23/Sba1 closed chaperone complex. Nature 2006, 440, 1013–1017. [Google Scholar] [CrossRef]
- Shiau, A.K.; Harris, S.F.; Southworth, D.R.; Agard, D.A. Structural analysis of E. coli hsp90 reveals dramatic nucleotide-dependent conformational rearrangements. Cell 2006, 127, 329–340. [Google Scholar] [CrossRef]
- Dollins, D.E.; Warren, J.J.; Immormino, R.M.; Gewirth, D.T. Structures of GRP94-nucleotide complexes reveal mechanistic differences between the hsp90 chaperones. Mol. Cell 2007, 28, 41–56. [Google Scholar] [CrossRef]
- Pearl, L.H.; Prodromou, C. Structure and mechanism of the Hsp90 molecular chaperone machinery. Annu. Rev. Biochem. 2006, 75, 271–294. [Google Scholar] [CrossRef]
- Pearl, L.H.; Prodromou, C.; Workman, P. The Hsp90 molecular chaperone: an open and shut case for treatment. Biochem. J. 2008, 410, 439–453. [Google Scholar] [CrossRef]
- Krukenberg, K.A.; Street, T.O.; Lavery, L.A.; Agard, D.A. Conformational dynamics of the molecular chaperone Hsp90. Q. Rev. Biophys. 2011, 44, 229–255. [Google Scholar] [CrossRef]
- Li, J.; Soroka, J.; Buchner, J. The Hsp90 chaperone machinery: Conformational dynamics and regulation by co-chaperones. Biochim. Biophys. Acta 2012, 1823, 624–635. [Google Scholar] [CrossRef]
- Jackson, S.E. Hsp90: Structure and function. Top. Curr. Chem. 2013, 328, 155–240. [Google Scholar] [CrossRef]
- Graf, C.; Stankiewicz, M.; Kramer, G.; Mayer, M.P. Spatially and kinetically resolved changes in the conformational dynamics of the Hsp90 chaperone machine. EMBO J. 2009, 28, 602–613. [Google Scholar] [CrossRef]
- Krukenberg, K.A.; Forster, F.; Rice, L.M.; Sali, A.; Agard, D.A. Multiple conformations of E. coli Hsp90 in solution: Insights into the conformational dynamics of Hsp90. Structure 2008, 16, 755–765. [Google Scholar] [CrossRef]
- Ratzke, C.; Mickler, M.; Hellenkamp, B.; Buchner, J.; Hugel, T. Dynamics of heat shock protein 90 C-terminal dimerization is an important part of its conformational cycle. Proc. Natl. Acad. Sci. USA 2010, 107, 16101–16106. [Google Scholar] [CrossRef]
- Shao, J.; Irwin, A.; Hartson, S.D.; Matts, R.L. Functional dissection of Cdc37: Characterization of domain structure and amino acid residues critical for protein kinase binding. Biochemistry 2003, 42, 12577–12588. [Google Scholar] [CrossRef]
- Shao, J.; Prince, T.; Hartson, S.D.; Matts, R.L. Phosphorylation of serine 13 is required for the proper function of the Hsp90 co-chaperone, Cdc37. J. Biol. Chem. 2003, 278, 38117–38120. [Google Scholar]
- Bandhakavi, S.; McCann, R.O.; Hanna, D.E.; Glover, C.V. A positive feedback loop between protein kinase CKII and Cdc37 promotes the activity of multiple protein kinases. J. Biol. Chem. 2003, 278, 2829–2836. [Google Scholar]
- Miyata, Y.; Nishida, E. CK2 controls multiple protein kinases by phosphorylating a kinase targeting molecular chaperone Cdc37. Mol. Cell. Biol. 2004, 24, 4065–4074. [Google Scholar] [CrossRef]
- Zhang, W.; Hirshberg, M.; McLaughlin, S.H.; Lazar, G.A.; Grossmann, J.G.; Nielsen, P.R.; Sobott, F.; Robinson, C.V.; Jackson, S.E.; Laue, E.D. Biochemical and structural studies of the interaction of Cdc37 with Hsp90. J. Mol. Biol. 2004, 340, 891–907. [Google Scholar] [CrossRef]
- Roe, S.M.; Ali, M.M.; Meyer, P.; Vaughan, C.K.; Panaretou, B.; Piper, P.W.; Prodromou, C.; Pearl, L.H. The mechanism of Hsp90 regulation by the protein kinase-specific cochaperone p50(cdc37). Cell 2004, 116, 87–98. [Google Scholar] [CrossRef]
- Siligardi, G.; Panaretou, B.; Meyer, P.; Singh, S.; Woolfson, D.N.; Piper, P.W.; Pearl, L.H.; Prodromou, C. Regulation of Hsp90 ATPase activity by the co-chaperone Cdc37p/p50cdc37. J. Biol. Chem. 2002, 277, 20151–20159. [Google Scholar] [CrossRef]
- Sreeramulu, S.; Jonker, H.R.; Langer, T.; Richter, C.; Lancaster, C.R.; Schwalbe, H. The human Cdc37.Hsp90 complex studied by heteronuclear NMR spectroscopy. J. Biol. Chem. 2009, 284, 3885–3896. [Google Scholar]
- Vaughan, C.K.; Gohlke, U.; Sobott, F.; Good, V.M.; Ali, M.M.; Prodromou, C.; Robinson, C.V.; Saibil, H.R.; Pearl, L.H. Structure of an Hsp90- Cdc37-Cdk4 complex. Mol. Cell 2006, 23, 697–707. [Google Scholar] [CrossRef]
- Millson, S.H.; Truman, A.W.; Wolfram, F.; King, V.; Panaretou, B.; Prodromou, C.; Pearl, L.H.; Piper, P.W. Investigating the protein-protein interactions of the yeast Hsp90 chaperone system by two-hybrid analysis: potential uses and limitations of this approach. Cell. Stress Chaperones 2004, 9, 359–368. [Google Scholar] [CrossRef]
- Mollapour, M.; Tsutsumi, S.; Truman, A.W.; Xu, W.; Vaughan, C.K.; Beebe, K.; Konstantinova, A.; Vourganti, S.; Panaretou, B.; Piper, P.W.; et al. Threonine 22 phosphorylation attenuates Hsp90 interaction with cochaperones and affects its chaperone activity. Mol. Cell 2011, 41, 672–681. [Google Scholar] [CrossRef]
- Zhang, T.; Li, Y.; Yu, Y.; Zou, P.; Jiang, Y.; Sun, D. Characterization of celastrol to inhibit hsp90 and cdc37 interaction. J. Biol. Chem. 2009, 284, 35381–35389. [Google Scholar]
- Zhang, T.; Hamza, A.; Cao, X.; Wang, B.; Yu, S.; Zhan, C.G.; Sun, D. A novel Hsp90 inhibitor to disrupt Hsp90/Cdc37 complex against pancreatic cancer cells. Mol. Cancer Ther. 2008, 7, 162–170. [Google Scholar] [CrossRef]
- Sreeramulu, S.; Gande, S.L.; Göbel, M.; Schwalbe, H. Molecular mechanism of inhibition of the human protein complex Hsp90-Cdc37, a kinome chaperone-cochaperone, by triterpene celastrol. Angew. Chem. Int. Ed. Engl. 2009, 48, 5853–5855. [Google Scholar] [CrossRef]
- Zhao, Q.; Boschelli, F.; Caplan, A.J.; Arndt, K.T. Identification of a conserved sequence motif that promotes Cdc37 and cyclin D1 binding to Cdk4. J. Biol. Chem. 2004, 279, 12560–12564. [Google Scholar]
- Terasawa, K.; Minami, Y. A client-binding site of Cdc37. FEBS J. 2005, 272, 4684–4690. [Google Scholar] [CrossRef]
- Terasawa, K.; Yoshimatsu, K.; Iemura, S.; Natsume, T.; Tanaka, K.; Minami, Y. Cdc37 Interacts with the Glycine-Rich Loop of Hsp90 Client Kinases. Mol. Cell. Biol. 2006, 26, 3378–3389. [Google Scholar] [CrossRef]
- Scroggins, B.T.; Prince, T.; Shao, J.; Uma, S.; Huang, W.; Guo, Y.; Yun, B.G.; Hedman, K.; Matts, R.L.; Hartson, S.D. High affinity binding of Hsp90 is triggered by multiple discrete segments of its kinase clients. Biochemistry 2003, 42, 12550–12561. [Google Scholar] [CrossRef]
- Prince, T.; Matts, R.L. Definition of proteinkinasesequencemotifs that triggerhighaffinitybinding of Hsp90 and Cdc37. J. Biol. Chem. 2004, 279, 39975–39981. [Google Scholar] [CrossRef]
- Prince, T.; Sun, L.; Matts, R.L. Cdk2: A genuine protein kinase client of Hsp90 and Cdc37. Biochemistry 2005, 44, 15287–15295. [Google Scholar] [CrossRef]
- Citri, A.; Harari, D.; Shohat, G.; Ramakrishnan, P.; Gan, J.; Lavi, S.; Eisenstein, M.; Kimchi, A.; Wallach, D.; Pietrokovski, S.; et al. Hsp90 recognizes a common surface on client kinases. J. Biol. Chem. 2006, 281, 14361–14369. [Google Scholar] [CrossRef]
- Xu, W.; Yuan, X.; Xiang, Z.; Mimnaugh, E.; Marcu, M.; Neckers, L. Surface charge and hydrophobicity determine ErbB2 binding to the Hsp90 chaperone complex. Nat. Struct. Mol. Biol. 2005, 12, 120–126. [Google Scholar] [CrossRef]
- Caplan, A.J.; Mandal, A.K.; Theodoraki, M.A. Molecular chaperones and protein kinase quality control. Trends Cell. Biol. 2007, 17, 87–92. [Google Scholar] [CrossRef]
- Theodoraki, M.A.; Caplan, A.J. Quality control and fate determination of Hsp90 client proteins. Biochim. Biophys. Acta 2012, 1823, 683–688. [Google Scholar] [CrossRef]
- Hartson, S.D.; Matts, R.L. Approaches for defining the Hsp90-dependent proteome. Biochim. Biophys. Acta 2011, 1823, 656–667. [Google Scholar] [CrossRef]
- Tsaytler, P.A.; Krijgsveld, J.; Goerdayal, S.S.; Rüdiger, S.; Egmond, M.R. Novel Hsp90 partners discovered using complementary proteomic approaches. Cell. Stress Chaperones 2009, 14, 629–638. [Google Scholar] [CrossRef]
- Taipale, M.; Krykbaeva, I.; Koeva, M.; Kayatekin, C.; Westover, K.D.; Karras, G.I.; Lindquist, S. Quantitative analysis of HSP90-client interactions reveals principles of substrate recognition. Cell 2012, 150, 987–1001. [Google Scholar] [CrossRef]
- Sharma, K.; Vabulas, R.M.; Macek, B.; Pinkert, S.; Cox, J.; Mann, M.; Hartl, F.U. Quantitative proteomics reveals that Hsp90 inhibition preferentially targets kinases and the DNA damage response. Mol. Cell. Proteomics 2011, 11, M111.014654. [Google Scholar]
- Wu, Z.; Moghaddas Gholami, A.; Kuster, B. Systematic identification of the Hsp90 candidate regulated proteome. Mol. Cell. Proteomics 2012, 11, M111.016675. [Google Scholar]
- Haupt, A.; Joberty, G.; Bantscheff, M.; Frohlich, H.; Stehr, H.; Schweiger, M.R.; Fischer, A.; Kerick, M.; Boerno, S.T.; Dahl, A.; et al. Hsp90 inhibition differentially destabilises MAP kinase and TGF-β signalling components in cancer cells revealed by kinase-targeted chemoproteomics. BMC Cancer 2012, 12, 38. [Google Scholar] [CrossRef]
- Colombo, G.; Morra, G.; Meli, M.; Verkhivker, G. Understanding ligand-based modulation of the Hsp90 molecular chaperone dynamics at atomic resolution. Proc. Natl. Acad. Sci. USA 2008, 105, 7976–7981. [Google Scholar] [CrossRef]
- Morra, G.; Verkhivker, G.; Colombo, G. Modeling signal propagation mechanisms and ligand-based conformational dynamics of the Hsp90 molecular chaperone full length dimer. PLoS Comput. Biol. 2009, 5, e1000323. [Google Scholar] [CrossRef]
- Verkhivker, G.M.; Dixit, A.; Morra, G.; Colombo, G. Structural and computational biology of the molecular chaperone Hsp90: From understanding molecular mechanisms to computer-based inhibitor design. Curr. Top. Med. Chem. 2009, 9, 1369–1385. [Google Scholar] [CrossRef]
- Dixit, A.; Verkhivker, G.M. Probing molecular mechanisms of the Hsp90 chaperone: Biophysical modeling identifies key regulators of functional dynamics. PLoS One 2012, 7, e37605. [Google Scholar] [CrossRef]
- Dixit, A.; Verkhivker, G. Hierarchical modeling of activation mechanisms in the ABL and EGFR kinase domains: Thermodynamic and mechanistic catalysts of kinase activation by cancer mutations. PLoS Comput. Biol. 2009, 5, e1000487. [Google Scholar] [CrossRef]
- Dixit, A.; Verkhivker, G. Computational modeling of allosteric communication reveals organizing principles of mutation-induced signaling in ABL and EGFR kinases. PLoS Comput. Biol. 2011, 7, e1002179. [Google Scholar] [CrossRef]
- Neuvirth, H.; Raz, R.; Schreiber, G. ProMate: A structure based prediction program to identify the location of protein-protein binding sites. J. Mol. Biol. 2010, 33, 181–199. [Google Scholar]
- De Vries, S.J.; van Dijk, A.D.; Bonvin, A.M. WHISCY: What information does surface conservation yield? Application to data-driven docking. Proteins 2006, 63, 479–489. [Google Scholar] [CrossRef]
- Armon, A.; Graur, D.; Ben-Tal, N. ConSurf: an algorithmic tool for the identification of functional regions in proteins by surface mapping of phylogenetic information. J. Mol. Biol. 2001, 307, 447–463. [Google Scholar] [CrossRef]
- Negi, S.S.; Schein, C.H.; Oezguen, N.; Power, T.D.; Braun, W. InterProSurf: a web server for predicting interacting sites on protein surfaces. Bioinformatics 2007, 23, 3397–3399. [Google Scholar] [CrossRef]
- Guo, Y.; Li, M.; Pu, X.; Li, G.; Guang, X.; Xiong, W.; Li, J. PRED_PPI: A server for predicting protein-protein interactions based on sequence data with probability assignment. BMC Res. Notes 2010, 3, 145. [Google Scholar] [CrossRef]
- Porollo, A.; Meller, J. Prediction-based fingerprints of protein-protein interactions. Proteins 2007, 66, 630–645. [Google Scholar] [CrossRef]
- Huang, B.; Schroeder, M. Using protein binding site prediction to improve protein docking. Gene 2008, 422, 14–21. [Google Scholar] [CrossRef]
- Ota, A.; Zhang, J.; Ping, P.; Han, J.; Wang, Y. Specific regulation of noncanonical p38alpha activation by Hsp90-Cdc37 chaperone complex in cardiomyocyte. Circ. Res. 2010, 106, 1404–1412. [Google Scholar] [CrossRef]
- Morelli, X.J.; Palma, P.N.; Guerlesquin, F.; Rigby, A.C. A novel approach for assessing macromolecular complexes combining soft-docking calculations with NMR data. Protein Sci. 2001, 10, 2131–2137. [Google Scholar] [CrossRef]
- Van Dijk, A.D.; Boelens, R.; Bonvin, A.M. Data-driven docking for the study of biomolecular complexes. FEBS J. 2005, 272, 293–312. [Google Scholar] [CrossRef]
- Dominguez, C.; Boelens, R.; Bonvin, A.M. HADDOCK: A protein-protein docking approach based on biochemical or biophysical information. J. Am. Chem. Soc. 2003, 125, 1731–1737. [Google Scholar] [CrossRef]
- Van Dijk, A.D.; Bonvin, A.M. Solvated docking: introducing water into the modeling of biomolecular complexes. Bioinformatics 2006, 22, 2340–2347. [Google Scholar] [CrossRef]
- Day, P.J.; Cleasby, A.; Tickle, I.J.; O'Reilly, M.; Coyle, J.E.; Holding, F.P.; McMenamin, R.L.; Yon, J.; Chopra, R.; Lengauer, C.; et al. Crystal structure of human CDK4 in complex with a D-type cyclin. Proc. Natl. Acad. Sci. USA 2009, 106, 4166–4170. [Google Scholar] [CrossRef]
- Zheng, W.; Brooks, B.R. Modeling protein conformational changes by iterative fitting of distance constraints using reoriented normal modes. Biophys. J. 2006, 90, 4327–4336. [Google Scholar] [CrossRef]
- Chaudhury, S.; Lyskov, S.; Gray, J.J. PyRosetta: a script-based interface for implementing molecular modeling algorithms using Rosetta. Bioinformatics 2010, 26, 689–691. [Google Scholar] [CrossRef]
- Baugh, E.H.; Lyskov, S.; Weitzner, B.D.; Gray, J.J. Real-time PyMOL visualization for Rosetta and PyRosetta. PLoS One 2011, 6, e21931. [Google Scholar]
- Taylor, S.S.; Kornev, A.P. Protein kinases: Evolution of dynamic regulatory proteins. Trends Biochem. Sci. 2011, 36, 65–77. [Google Scholar] [CrossRef]
- Taylor, S.S.; Keshwani, M.M.; Steichen, J.M.; Kornev, A.P. Evolution of the eukaryotic protein kinases as dynamic molecular switches. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2012, 367, 2517–2528. [Google Scholar] [CrossRef]
- Sato, S.; Fujita, N.; Tsuruo, T. Modulation of Akt kinase activity by binding to Hsp90. Proc. Natl. Acad. Sci. USA 2000, 97, 10832–10837. [Google Scholar] [CrossRef]
- Jura, N.; Zhang, X.; Endres, N.F.; Seeliger, M.A.; Schindler, T.; Kuriyan, J. Catalytic control in the EGF receptor and its connection to general kinase regulatory mechanisms. Mol. Cell 2011, 42, 9–22. [Google Scholar] [CrossRef]
- Southworth, D.R.; Agard, D.A. Client-loading conformation of the Hsp90 molecular chaperone revealed in the cryo-EM structure of the human Hsp90:Hop complex. Mol. Cell 2011, 42, 771–781. [Google Scholar] [CrossRef]
- Street, T.O.; Lavery, L.A.; Agard, D.A. Substrate binding drives large-scale conformational changes in the Hsp90 molecular chaperone. Mol. Cell 2011, 42, 96–105. [Google Scholar] [CrossRef]
- Kim, Y.S.; Alarcon, S.V.; Lee, S.; Lee, M.J.; Giaccone, G.; Neckers, L.; Trepel, J.B. Update on Hsp90 inhibitors in clinical trial. Curr. Top. Med. Chem. 2009, 9, 1479–1492. [Google Scholar] [CrossRef]
- Hieronymus, H.; Lamb, J.; Ross, K.N.; Peng, X.P.; Clement, C.; Rodina, A.; Nieto, M.; Du, J.; Stegmaier, K.; Raj, S.M. Gene expression signature-based chemical genomic prediction identifies a novel class of HSP90 pathway modulators. Cancer Cell 2006, 10, 321–330. [Google Scholar] [CrossRef]
- Rice, J.W.; Veal, J.M.; Barabasz, A.; Foley, B.; Fadden, P.; Scott, A.; Huang, K.; Steed, P.; Hall, S. Targeting of multiple signaling pathways by the Hsp90 inhibitor SNX-2112 in EGFR resistance models as a single agent or in combination with erlotinib. Oncol. Res. 2009, 18, 229–242. [Google Scholar] [CrossRef]
- Kang, B.H.; Altieri, D.C. Compartmentalized cancer drug discovery targeting mitochondrial Hsp90 chaperones. Oncogene 2009, 28, 3681–3688. [Google Scholar] [CrossRef]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Lawless, N.; Blacklock, K.; Berrigan, E.; Verkhivker, G. Structural Bioinformatics and Protein Docking Analysis of the Molecular Chaperone-Kinase Interactions: Towards Allosteric Inhibition of Protein Kinases by Targeting the Hsp90-Cdc37 Chaperone Machinery. Pharmaceuticals 2013, 6, 1407-1428. https://doi.org/10.3390/ph6111407
Lawless N, Blacklock K, Berrigan E, Verkhivker G. Structural Bioinformatics and Protein Docking Analysis of the Molecular Chaperone-Kinase Interactions: Towards Allosteric Inhibition of Protein Kinases by Targeting the Hsp90-Cdc37 Chaperone Machinery. Pharmaceuticals. 2013; 6(11):1407-1428. https://doi.org/10.3390/ph6111407
Chicago/Turabian StyleLawless, Nathan, Kristin Blacklock, Elizabeth Berrigan, and Gennady Verkhivker. 2013. "Structural Bioinformatics and Protein Docking Analysis of the Molecular Chaperone-Kinase Interactions: Towards Allosteric Inhibition of Protein Kinases by Targeting the Hsp90-Cdc37 Chaperone Machinery" Pharmaceuticals 6, no. 11: 1407-1428. https://doi.org/10.3390/ph6111407