Molecular Dynamics Simulations of Hsp90 with an Eye to Inhibitor Design

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
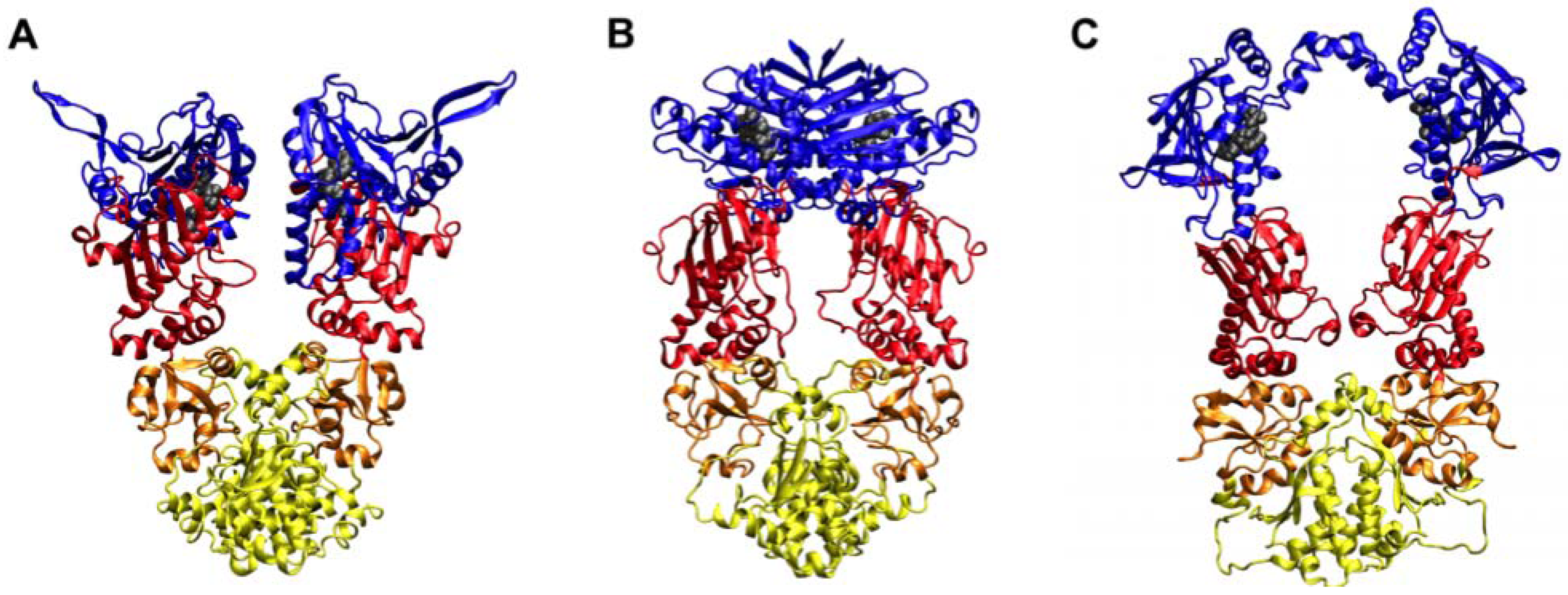
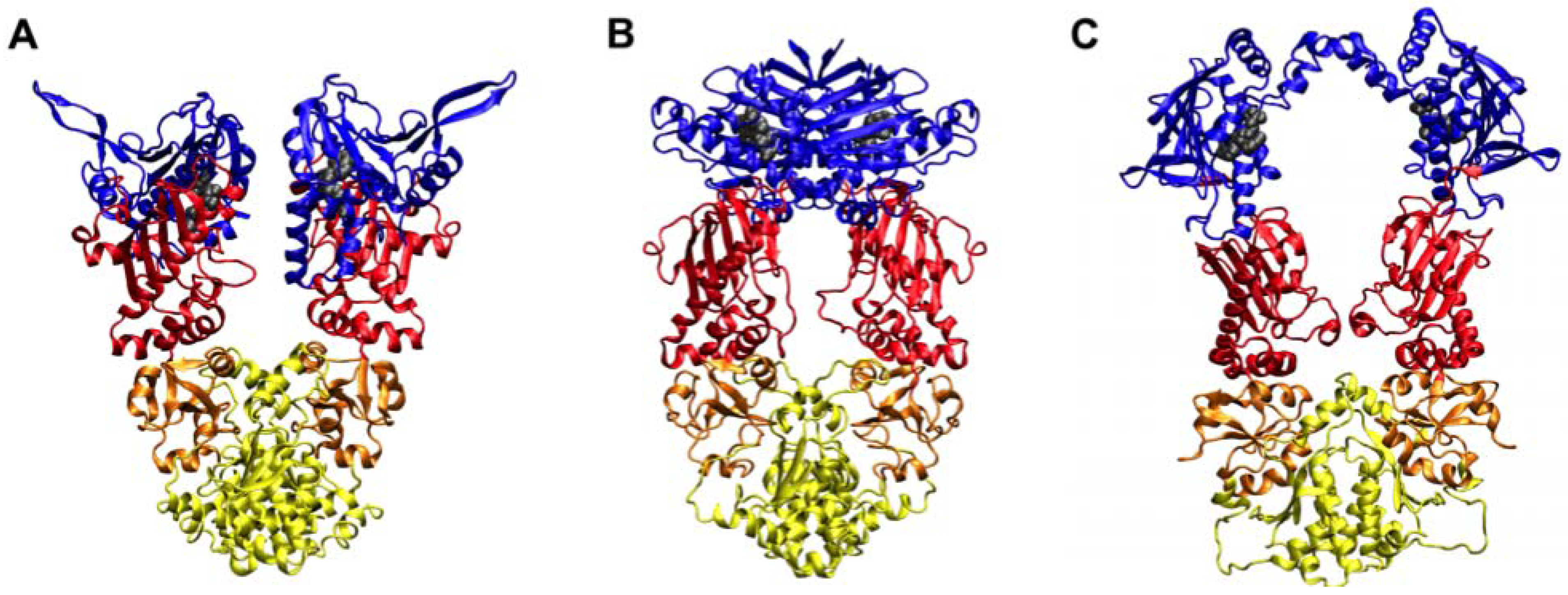
2. Studies of the Isolated Hsp90 N-terminal for Drug Development
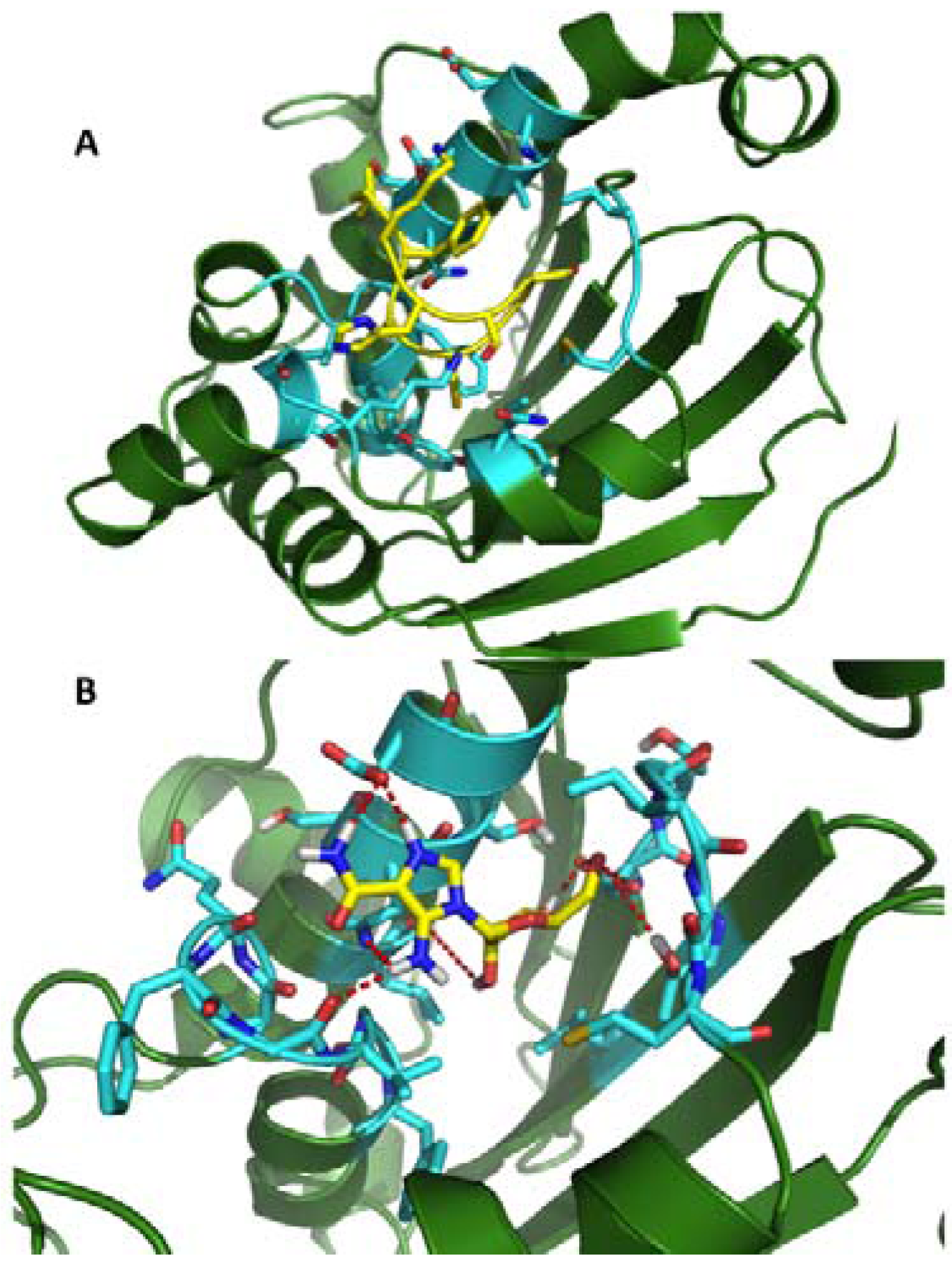
2.1. MD Simulations in the Study of the Recognition between Hsp90-NTD and Ligands
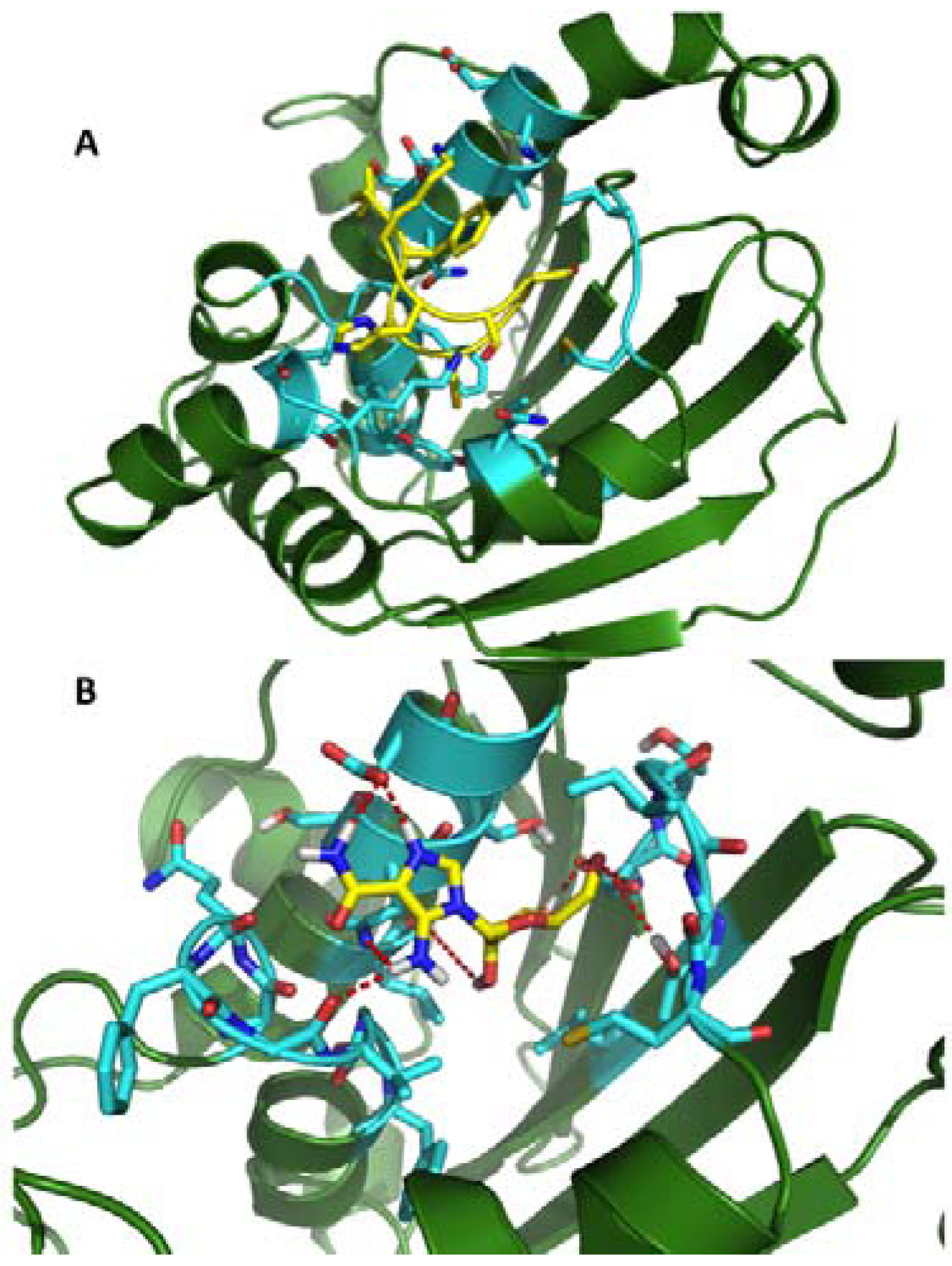
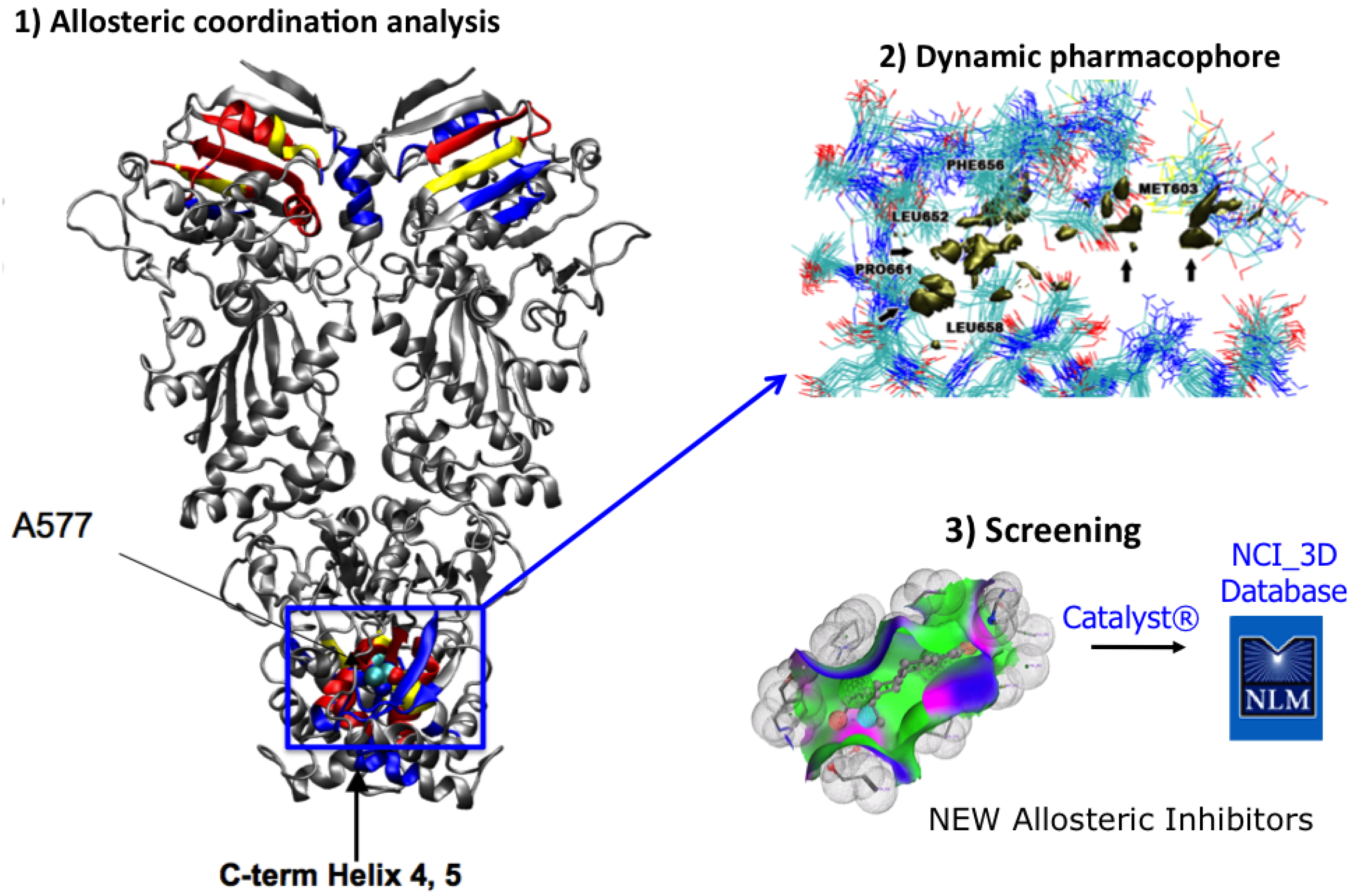
2.2. Dynamic Pharmacophores
3. Studying Ligand-Based Modulation of Hsp90-NTD Dynamics
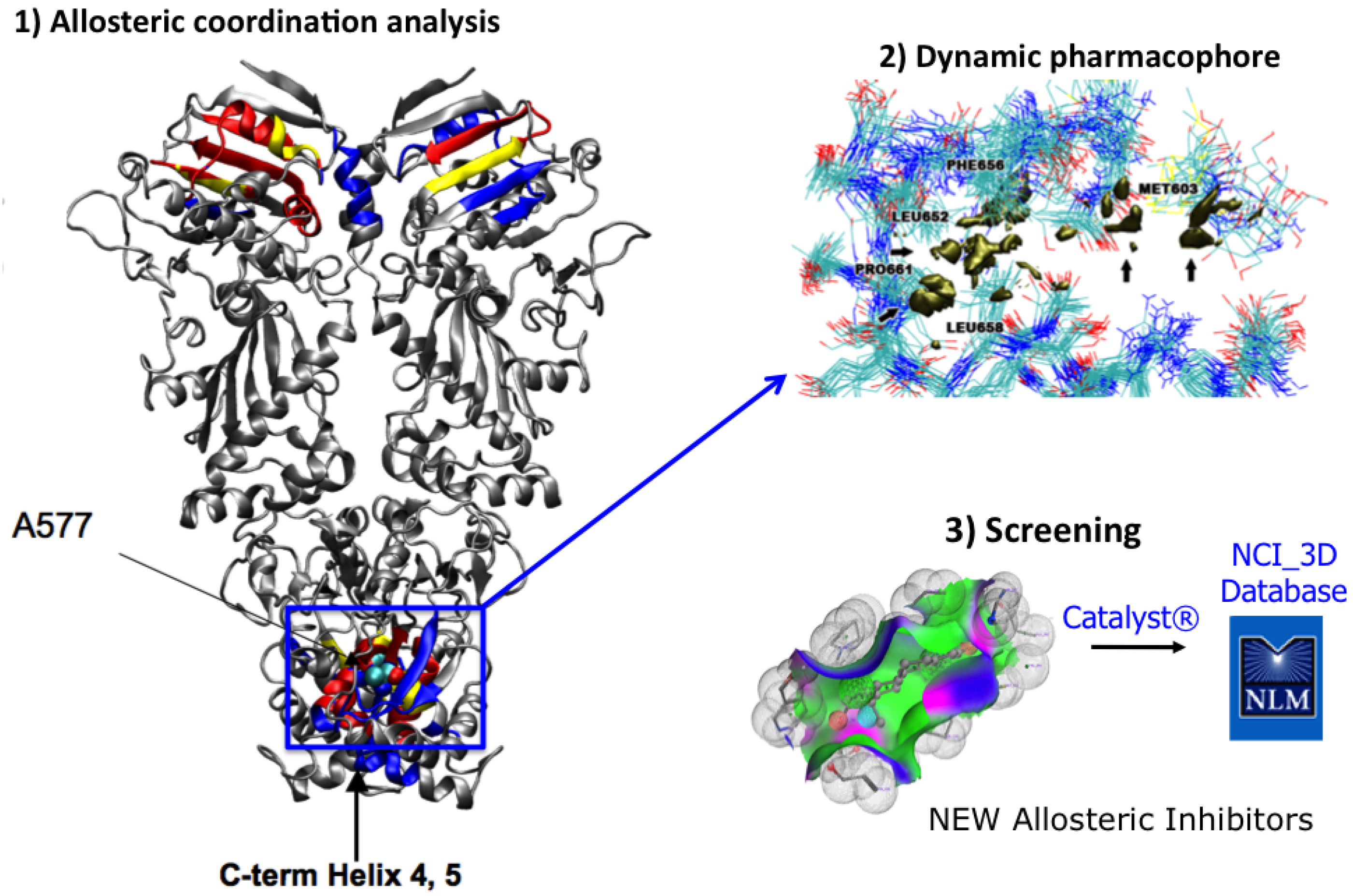
4. MD-Simulations of the Full Length Hsp90: Identification of Possible Allosteric Pockets and Allosteric Drug Design
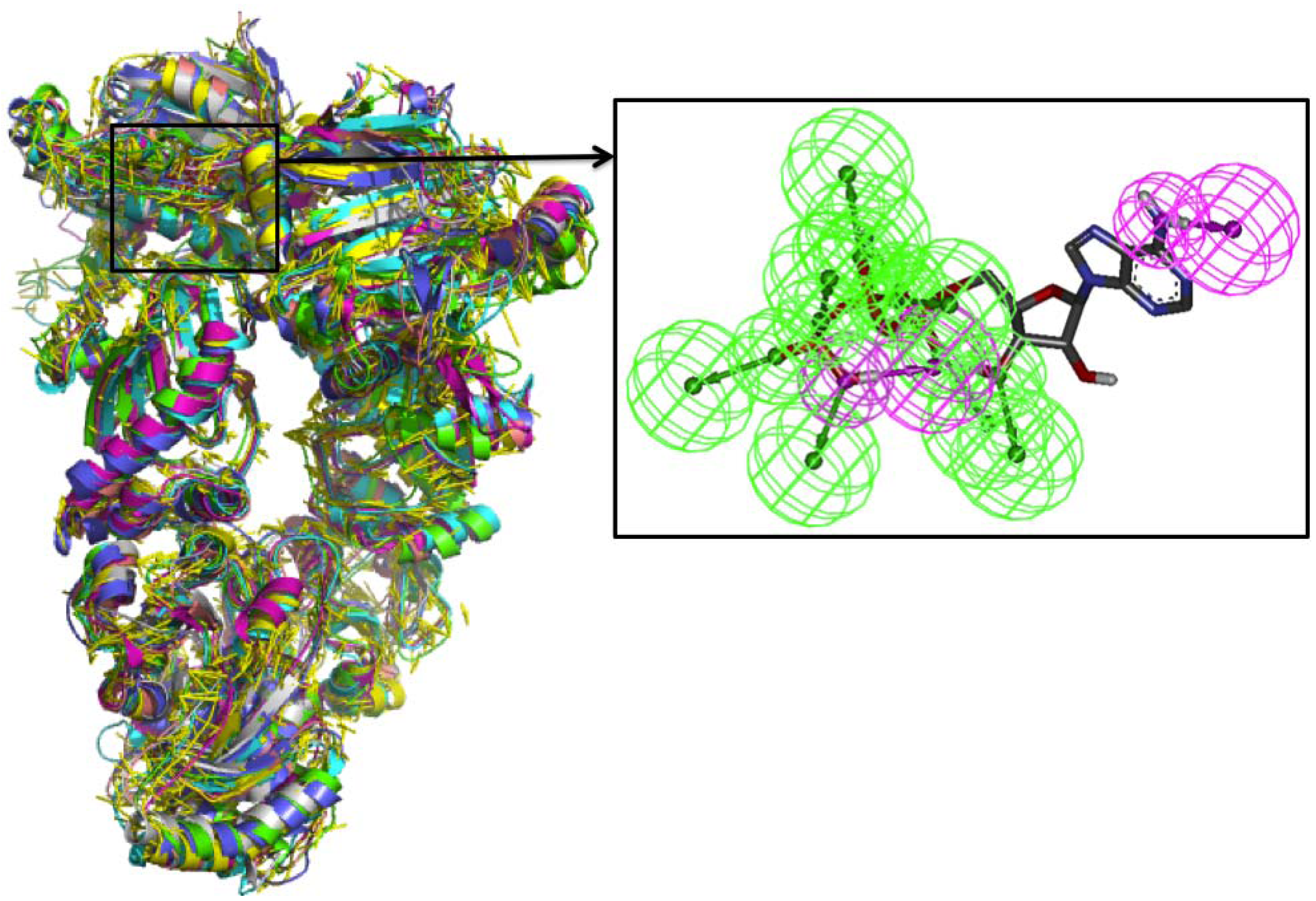
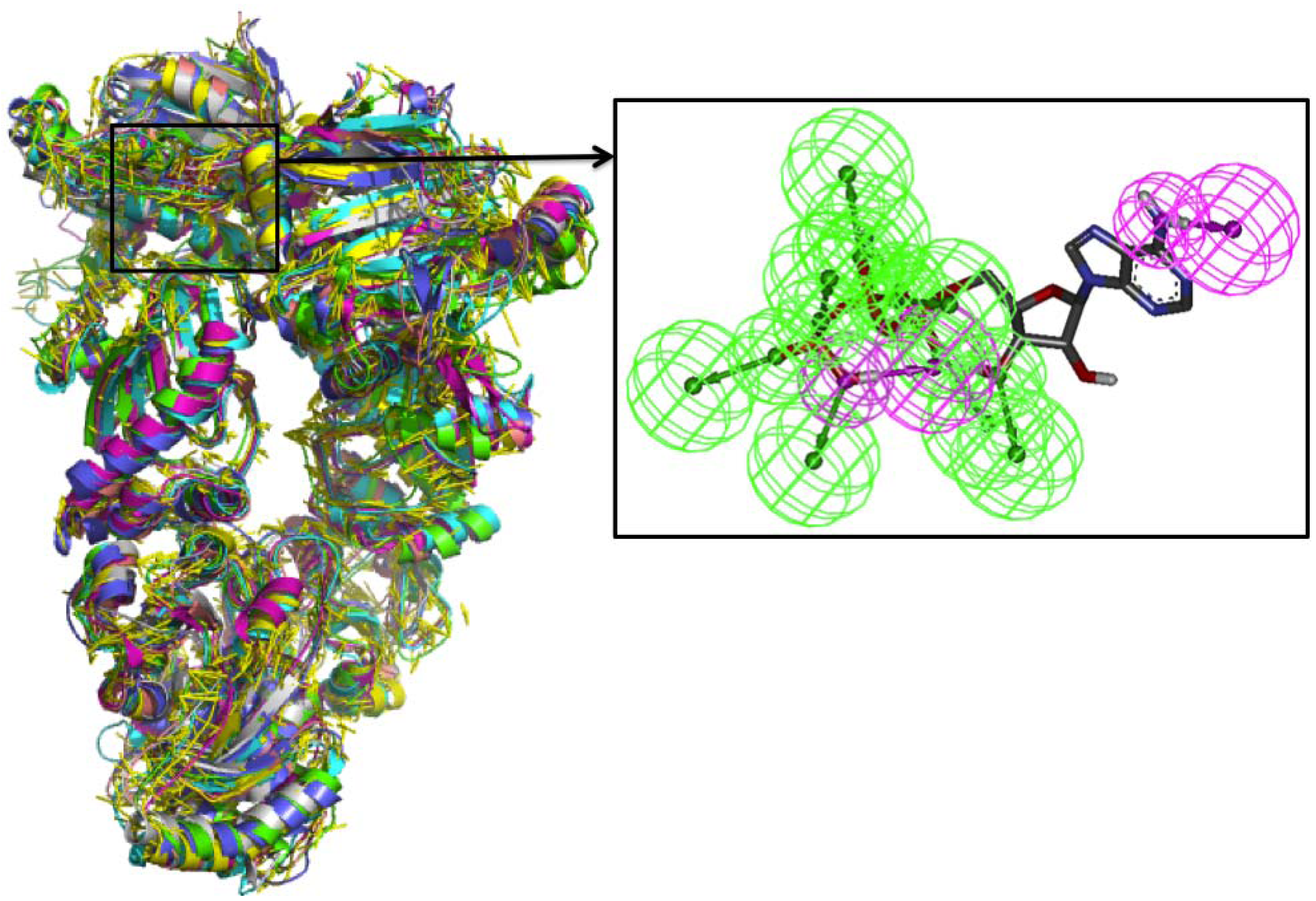
Comparative Dynamics
5. Hsp90 Protein-Protein Interaction Networks and Drug Design
6. Conclusions
Acknowledgments
Conflict of Interest
References
- Aloy, P.; Bottcher, B.; Ceulemans, H.; Leutwein, C.; Mellwig, C.; Fischer, S.; Gavin, A.C.; Bork, P.; Superti-Furga, G.; Serrano, L.; Russell, R.B. Structure-based assembly of protein complexes in yeast. Science 2004, 303, 2026–2029. [Google Scholar] [CrossRef]
- Vogelstein, B.; Kinzler, K.W. Cancer genes and the pathways they control. Nat. Med. 2004, 10, 788–789. [Google Scholar] [CrossRef]
- Taipale, M.; Jarosz, D.F.; Lindquist, S.L. HSP90 at the hub of protein homeostasis: Emerging mechanistic insights. Nat. Rev. Mol. Cell Biol. 2010, 11, 515–528. [Google Scholar] [CrossRef]
- Whitesell, L.; Lindquist, S.L. Hsp90 and the chaperoning of cancer. Nat. Rev. Cancer 2005, 5, 761–772. [Google Scholar] [CrossRef]
- Shames, D.S.; Minna, J.D. IP6K2 is a client for Hsp90 and a target for cancer therapeutics development. Proc. Natl. Acad. Sci. USA 2008, 105, 1389–1390. [Google Scholar] [CrossRef]
- Trepel, J.B.; Mollapour, M.; Giaccone, G.; Neckers, L. Targeting the dynamic Hsp90 complex in cancer. Nat. Rev. Cancer 2010, 10, 537–549. [Google Scholar] [CrossRef]
- Csermely, P.; Schnaider, T.; Soti, C.; Prohaszka, Z.; Nardai, G. The 90-kDa molecular chaperone family: Structure, function, and clinical application. A comprehensive review. Pharmacol. Ther. 1998, 79, 129–168. [Google Scholar] [CrossRef]
- Kamal, A.; Thao, L.; Sensintaffar, J.; Zhang, L.; Boehm, M.F.; Fritz, L.C.; Burrows, F.J. A high-affinity conformation of Hsp90 confers tumor selectivity on Hsp90 inhibitors. Nature 2003, 425, 407–410. [Google Scholar]
- Ali, M.M.U.; Roe, S.M.; Vaughan, C.K.; Meyer, P.; Panaretou, B.; Piper, P.W.; Prodromou, C.; Pearl, L.H. Crystal structure of an Hsp90-nucleotide-p23/Sba1 closed chaperone complex. Nature 2006, 440, 1013–1017. [Google Scholar]
- Eletto, D.; Dersh, D.; Argon, Y. GRP94 in ER quality control and stress responses. Semin. Cell Dev. Biol. 2010, 21, 479–485. [Google Scholar]
- Dollins, D.E.; Warren, J.J.; Immormino, R.M.; Gewirth, D.T. Structures of GRP94-Nucleotide complexes reveal mechanistic differences between the hsp90 chaperones. Mol. Cell 2007, 28, 41–56. [Google Scholar] [CrossRef]
- Shiau, A.K.; Harris, S.F.; Southworth, D.R.; Agard, D.A. Structural analysis of E-coli Hsp90 reveals dramatic nucleotide-dependent conformational rearrangements. Cell 2006, 127, 329–340. [Google Scholar] [CrossRef]
- Krukenberg, K.A.; Forster, F.; Rice, L.M.; Sali, A.; Agard, D.A. Multiple conformations of E-coli Hsp90 in solution: Insights into the conformational dynamics of Hsp90. Structure 2008, 16, 755–765. [Google Scholar] [CrossRef]
- Southworth, D.R.; Agard, D.A. Species-dependent ensembles of conserved conformational states define the Hsp90 chaperone ATPase cycle. Mol. Cell 2008, 32, 631–640. [Google Scholar] [CrossRef]
- Tsutsumi, S.; Mollapour, M.; Graf, C.; Lee, C.T.; Scroggins, B.T.; Xu, W.P.; Haslerova, L.; Hessling, M.; Konstantinova, A.A.; Trepel, J.B.; et al. Hsp90 charged-linker truncation reverses the functional consequences of weakened hydrophobic contacts in the N domain. Nat. Struct. Mol. Biol. 2009, 16, 1141–1147. [Google Scholar]
- Mickler, M.; Hessling, M.; Ratzke, C.; Buchner, J.; Hugel, T. The large conformational changes of Hsp90 are only weakly coupled to ATP hydrolysis. Nat. Struct. Mol. Biol. 2009, 16, 281–286. [Google Scholar]
- Retzlaff, M.; Hagn, F.; Mitschke, L.; Hessling, M.; Gugel, F.; Kessler, H.; Richter, K.; Buchner, J. Asymmetric activation of the Hsp90 dimer by its cochaperone Aha1. Mol. Cell 2010, 37, 344–354. [Google Scholar] [CrossRef]
- Frey, S.; Leskovar, A.; Reinstein, J.; Buchner, K. The ATPase cycle of the endoplasmic chaperone Grp94. J. Biol. Chem. 2007, 282, 35612–35620. [Google Scholar]
- Grenert, J.P.; Sullivan, W.P.; Fadden, P.; Haystead, T.A.J.; Clark, J.; Mimnaugh, E.; Krutzsch, H.; Ochel, H.-J.; Schulte, T.W.; Sausville, E.; et al. The amino-terminal domain of heat shock protein 90 (Hsp90) that binds geldanamycin is an ATP/ADP switch domain that regulates Hsp90 conformation. J. Biol. Chem. 1997, 272, 23843–23850. [Google Scholar]
- Janin, Y.L. Heat shock protein 90 inhibitors. A text book example of medicinal chemistry? J. Med. Chem. 2005, 48, 7503–7512. [Google Scholar] [CrossRef]
- Roe, S.M.; Prodromou, C.; O’Brien, R.; Ladbury, J.E.; Piper, P.W.; Pearl, L.H. Structural basis for inhibition of the Hsp90 molecular chaperone by the antitumor antibiotics radicicol and geldanamycin. J. Med. Chem. 1999, 42, 260–266. [Google Scholar] [CrossRef]
- Immormino, R.M.; Kang, Y.; Chiosis, G.; Gewirth, D.T. Structural and quantum chemical studies of 8-aryl-sulfanyl adenine class of Hsp90 inhibitors. J. Med. Chem. 2006, 49, 4953–4960. [Google Scholar] [CrossRef]
- Barril, X.; Beswick, M.C.; Collier, A.; Drysdale, M.J.; Dymocka, B.W.; Fink, A.; Grant, K.; Howes, R.; Jordan, A.M.; Massey, A.; et al. 4-Amino derivatives of the Hsp90 inhibitor CCT018159. Bioorg. Med. Chem. Lett. 2006, 16, 2543–2548. [Google Scholar] [CrossRef]
- Wang, M.; Shen, G.; Blagg, B.S.J. Radanamycin, a macrocyclic chimera of radicicol and geldanamycin. Bioorg. Med. Chem. Lett. 2006, 16, 2459–2462. [Google Scholar]
- McCollum, A.K.; Lukasiewitcz, K.B.; TenEyck, C.J.; Lingle, W.L.; Toft, D.O.; Erlichman, C. Cisplatin abogates the geldanamycin-induced heat shock response. Mol. Cancer Ther. 2008, 7, 3256–3264. [Google Scholar]
- Kaur, J.; Ralhan, R. Induction of apoptosis by abrogation of Hsp70 expresion in human oral cancer cells. Int. J. Cancer 2000, 85, 1–5. [Google Scholar] [CrossRef]
- Marcu, M.G.; Chadli, A.; Bouhouche, I.; Catelli, M.; Neckers, L.M. The heat shock protein 90 antagonist novobiocin interacts with a previously unrecognized ATP-binding domain in the carboxyl terminus of the chaperone. J. Biol. Chem. 2000, 275, 37181–37186. [Google Scholar]
- Donnelly, A.; Blagg, B.S.J. Novobiocin and additional inhibitors of the Hsp90 C-terminal nucleotide-binding Pocket. Curr. Med. Chem. 2008, 15, 2702–2717. [Google Scholar] [CrossRef]
- Cox, M.B.; Miller, C.A., III. Cooperation of heat shock protein 90 and p23 in aryl hydrocarbon receptor signaling. Cell Stress Chaperones 2004, 9, 4–20. [Google Scholar]
- Zhao, H.P.; Yan, B.; Peterson, L.B.; Blagg, B.S.J. 3-Arylcoumarin derivatives manifest anti-proliferative activity through Hsp90 inhibition. ACS Med. Chem. Lett. 2012, 3, 327–331. [Google Scholar] [CrossRef]
- Kusuma, B.R.; Peterson, L.B.; Zhao, H.P.; Vielhauer, G.; Holzbeierlein, J.; Blagg, B.S.J. Targeting the heat shock protein 90 dimer with dimeric inhibitors. J. Med. Chem. 2011, 54, 6234–6253. [Google Scholar] [CrossRef]
- Zhao, H.P.; Donnelly, A.C.; Kusuma, B.R.; Brandt, G.E.L.; Brown, D.; Rajewski, R.A.; Vielhauer, G.; Holzbeierlein, J.; Cohen, M.S.; Blagg, B.S.J. Engineering an antibiotic to fight cancer: Optimization of the novobiocin scaffold to produce anti-proliferative agents. J. Med. Chem. 2011, 54, 3839–3853. [Google Scholar] [CrossRef]
- Dixit, A.; Verkhivker, G. Probing molecular mechanisms of the Hsp90 chaperone: Biophysical modeling identifies key regulators of functional dynamics. PLoS One 2012, 7, e37605. [Google Scholar]
- Pandini, A.; Fornili, A.; Fraternali, F.; Kleinjung, J. Detection of allosteric signal transmission by information-theoretic analysis of protein dynamics. FASEB J. 2012, 26, 868–881. [Google Scholar] [CrossRef]
- Fanelli, F.; Seeber, M. Structural insights into retinitis pigmentosa from unfolding simulations of rhodopsin mutants. FASEB J. 2010, 24, 3196–3209. [Google Scholar] [CrossRef]
- Colombo, G.; Ottolina, G.; Carrea, G.; Bernardi, A.; Scolastico, C. Application of structure-based thermodynamic calculations to the rationalization of the enantioselectivity of subtilisin in organic solvents. Tetrahedron Asymmetry 1998, 9, 1205–1214. [Google Scholar]
- Rothlisberger, D.; Khersonsky, O.; Wollacott, A.M.; Jiang, L.; DeChancie, J.; Betker, J.; Gallaher, J.L.; Althoff, E.A.; Zanghellini, A.; Dym, O.; et al. Kemp elimination catalysts by computational enzyme design. Nature 2008, 453, 190–195. [Google Scholar]
- Siegel, J.B.; Zanghellini, A.; Lovick, H.M.; Kiss, G.; Lambert, A.R.; Clair, J.L.S.; Gallaher, J.L.; Hilvert, D.; Gelb, M.H.; Stoddard, B.L.; et al. Computational design of an enzyme catalyst for a stereoselective bimolecular Diels-Alder reaction. Science 2010, 329, 309–313. [Google Scholar] [CrossRef]
- Colombo, G.; Ottolina, G.; Carrea, G.; Merz, K.M., Jr. Modelling the enantioselectivity of subtilisin in water and organic solvents: Insights from molecular dynamics and quantum mechanical/molecular mechanical studies. Chem. Commun. 2000, 559–560. [Google Scholar]
- Colombo, G.; Toba, S.; Merz, K.M., Jr. Rationalization of the enantioselectivity of subtilisin in DMF. J. Am. Chem. Soc. 1999, 121, 3486–3493. [Google Scholar] [CrossRef]
- Agarwal, P.K.; Billeter, S.R.; Rajagopalan, P.T.R.; Benkovic, S.J.; Hammes-Schiffer, S. Network of coupled promoting motions in enzyme catalysis. Proc. Natl. Acad. Sci. USA 2002, 99, 2794–2799. [Google Scholar]
- Morra, G.; Colombo, G. Relationship between energy distribution and fold stability: Insights from molecular dynamics simulations of native and mutant proteins. Proteins 2008, 72, 660–672. [Google Scholar] [CrossRef]
- Boukharta, L.; Keranen, H.; Stary-Weinzinger, A.; Wallin, G.; de Groot, B.L.; Aqvist, J. Computer simulations of structure-activity relationships for hERG channel blockers. Biochemistry 2011, 50, 6146–6156. [Google Scholar] [CrossRef]
- Damm, K.L.; Carlson, H.A. Exploring experimental sources of multiple protein conformations in structure-based drug design. J. Am. Chem. Soc. 2007, 129, 8225–8235. [Google Scholar]
- Sukuru, S.K.; Arora, S.; Zavodszky, M.I.; Kron, M.A.; Kuhn, L.A. Flexible protein structure-based screening and docking: Applications to anti-filarial drug design. Protein Sci. 2004, 13, 279. [Google Scholar]
- Teodoro, M.L.; Kavraki, L.E. Conformational flexibility models for the receptor in structure based drug design. Curr. Pharm. Des. 2003, 9, 1635–1648. [Google Scholar] [CrossRef]
- Carlson, H.A. Protein flexibility and drug design: How to hit a moving target. Curr. Opin. Chem. Biol. 2002, 6, 447–452. [Google Scholar] [CrossRef]
- Van Gunsteren, W.F.; Burgi, R.; Peter, C.; Daura, X. The key to solving the protein folding problem lies in the accurate description of the denatured state. Angew. Chem. Int. Ed. Engl. 2001, 40, 352–355. [Google Scholar]
- Van Gunsteren, W.F.; Bakowies, D.; Baron, R.; Chandrasekhar, I.; Christen, M.; Daura, X.; Gee, P.; Geerke, D.P.; Glättli, A.; Hünenberger, P.H.; et al. Biomolecular modeling: Goals, problems, perspectives. Angew. Chem. Int. Ed. Engl. 2006, 45, 4064–4092. [Google Scholar]
- Plescia, J.; Salz, W.; Xia, F.; Pennati, M.; Zaffaroni, N.; Daidone, M.G.; Meli, M.; Dohi, T.; Fortugno, P.; Nefedova, Y.; et al. Rational design of Shepherdin, a novel anticancer agent. Cancer Cell 2005, 7, 457–467. [Google Scholar] [CrossRef]
- Fortugno, P.; Beltrami, E.; Plescia, J.; Fontana, J.; Pradhan, D.; Marchisio, P.C.; Sessa, W.C.; Altieri, D.C. Regulation of survivin function by Hsp90. Proc. Natl. Acad. Sci. USA 2003, 100, 13791–13796. [Google Scholar]
- Morris, G.M.; Goodsell, D.S.; Halliday, R.S.; Huey, R.; Hart, W.E.; Belew, R.K.; Olson, A.J. Automated docking using a lamarckian genetic algorithm and and empirical binding free energy function. J. Comput. Chem. 1998, 19, 1639–1662. [Google Scholar] [CrossRef]
- Stebbins, C.E.; Russo, A.A.; Schneider, C.; Rosen, N.; Hartl, F.U.; Pavletich, N.P. Crystal structure of Hsp90-geldanamycin complex: Targeting of a protein chaperone by an antitumor agent. Cell 1997, 89, 239–250. [Google Scholar]
- Meli, M.; Pennati, M.; Curto, M.; Daidone, M.G.; Plescia, J.; Toba, S.; Altieri, D.C.; Zaffaroni, N.; Colombo, G. Small-molecule targeting of heat shock protein 90 chaperone function: Rational identification of a new anticancer lead. J. Med. Chem. 2006, 49, 7721–7730. [Google Scholar]
- Tomaselli, S.; Meli, M.; Plescia, J.; Zetta, L.; Altieri, D.C.; Colombo, G.; Ragona, L. Combined in silico and experimental approach for drug design: The binding mode of peptidic and non-peptidic inhibitors to Hsp90 N-terminal domain. Chem. Biol. Drug Des. 2010, 76, 382–391. [Google Scholar] [CrossRef]
- Colombo, G.; Morra, G.; Meli, M.; Verkhivker, G.M. Understanding ligand-based modulation of the Hsp90 molecular chaperone dynamics at atomic resolution. Proc. Natl. Acad. Sci. USA 2008, 105, 7676–7681. [Google Scholar] [CrossRef]
- Morra, G.; Verkhivker, G.M.; Colombo, G. Modeling signal propagation mechanisms and ligand-based conformational dynamics of the Hsp90 molecular chaperone full length dimer. PLoS Comput. Biol. 2009, 5, e1000323. [Google Scholar] [CrossRef]
- Morra, G.; Neves, M.A.C.; Plescia, C.J.; Tsutsumi, S.; Neckers, L.; Verkhivker, G.; Altieri, D.C.; Colombo, G. Dynamics-based discovery of allosteric inhibitors: Selection of new ligands for the C-terminal domain of Hsp90. J. Chem. Theory Comput. 2010, 6, 2978–2989. [Google Scholar]
- Genoni, A.; Pennati, M.; Morra, G.; Zaffaroni, N.; Colombo, G. Ligand selection from the analysis of protein conformational substates: New leads targeting the N-terminal domain of Hsp90. RSC Adv. 2012, 2, 4268–4283. [Google Scholar]
- Morra, G.; Potestio, R.; Micheletti, C.; Colombo, G. Corresponding functional dynamics across the Hsp90 chaperone family: Insights from a multiscale analysis of MD simulations. PLoS Comput. Biol. 2012, 8, e1002433. [Google Scholar] [CrossRef]
- Nathan, D.F.; Vos, M.H.; Lindquist, S. In vivo functions of the Saccharomyces cerevisiae Hsp90 chaperone. Proc. Natl. Acad. Sci. USA 1997, 94, 12949–12956. [Google Scholar] [CrossRef]
- Zhao, R.; Davey, M.; Hsu, Y.C.; Kaplanek, P.; Tong, A.; Parsons, A.B.; Krogan, N.; Cagney, G.; Mai, D.; Greenblatt, J.; et al. Navigating the chaperone network: An integrative map of physical and genetic interactions mediated by the hsp90 chaperone. Cell 2005, 120, 715–727. [Google Scholar] [CrossRef]
- Echeverria, P.C.; Bernthaler, A.; Dupuis, P.; Mayer, B.; Picard, D. An interaction network predicted from public data as a discovery tool: Application to the Hsp90 molecular chaperone machine. PLoS One 2012, 6, e26044. [Google Scholar]
- Bandyopadhyay, S.; Chiang, C.Y.; Srivastava, J.; Gersten, M.; White, S.; Bell, R.; Kurschner, C.; Martin, C.H.; Smoot, M.; Sahasrabudhe, S.; et al. A human MAP kinase interactome. Nat. Methods 2010, 7, 801–805. [Google Scholar] [CrossRef]
- Bergman, A.; Siegal, M.L. Evolutionary capacitance as a general feature of complex gene networks. Nature 2003, 424, 549–552. [Google Scholar] [CrossRef]
- Hagn, F.; Lagleder, S.; Retzlaff, M.; Rohrberg, J.; Demmer, O.; Richter, K.; Buchner, J.; Kessler, H. Structural analysis of the interaction between Hsp90 and the tumor suppressor protein p53. Nat. Struct. Mol. Biol. 2011, 18, 1086–1093. [Google Scholar]
- Scarabelli, G.; Morra, G.; Colombo, G. Predicting interaction sited from the energetics of isolated proteins: A new approach to epitope mapping. Biophys. J. 2010, 98, 1966–1975. [Google Scholar] [CrossRef]
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Moroni, E.; Morra, G.; Colombo, G. Molecular Dynamics Simulations of Hsp90 with an Eye to Inhibitor Design. Pharmaceuticals 2012, 5, 944-962. https://doi.org/10.3390/ph5090944
Moroni E, Morra G, Colombo G. Molecular Dynamics Simulations of Hsp90 with an Eye to Inhibitor Design. Pharmaceuticals. 2012; 5(9):944-962. https://doi.org/10.3390/ph5090944
Chicago/Turabian StyleMoroni, Elisabetta, Giulia Morra, and Giorgio Colombo. 2012. "Molecular Dynamics Simulations of Hsp90 with an Eye to Inhibitor Design" Pharmaceuticals 5, no. 9: 944-962. https://doi.org/10.3390/ph5090944