Synthesis, Reactions and Evaluation of the Antimicrobial Activity of Some 4-(p-Halophenyl)-4H-naphthopyran, Pyranopyrimidine and Pyranotriazolopyrimidine Derivatives

Abstract
:1. Introduction
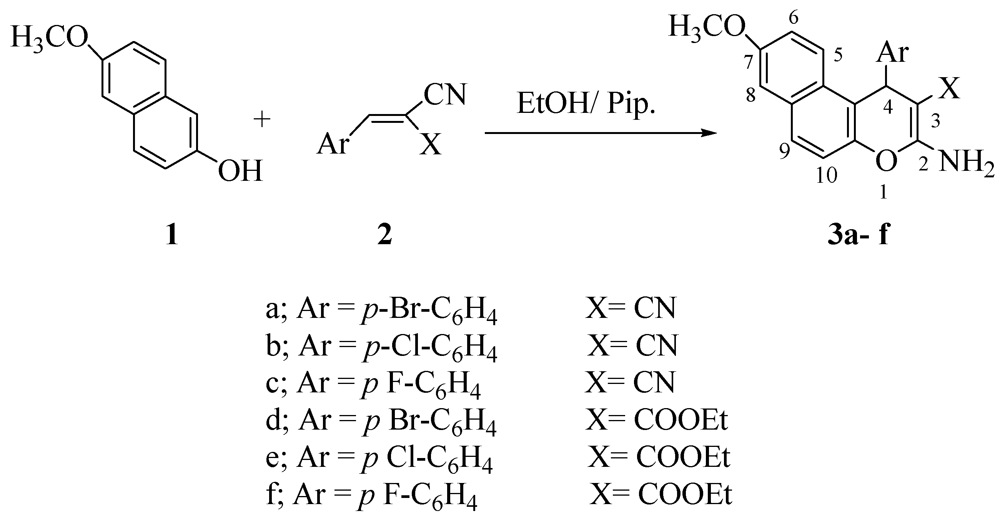
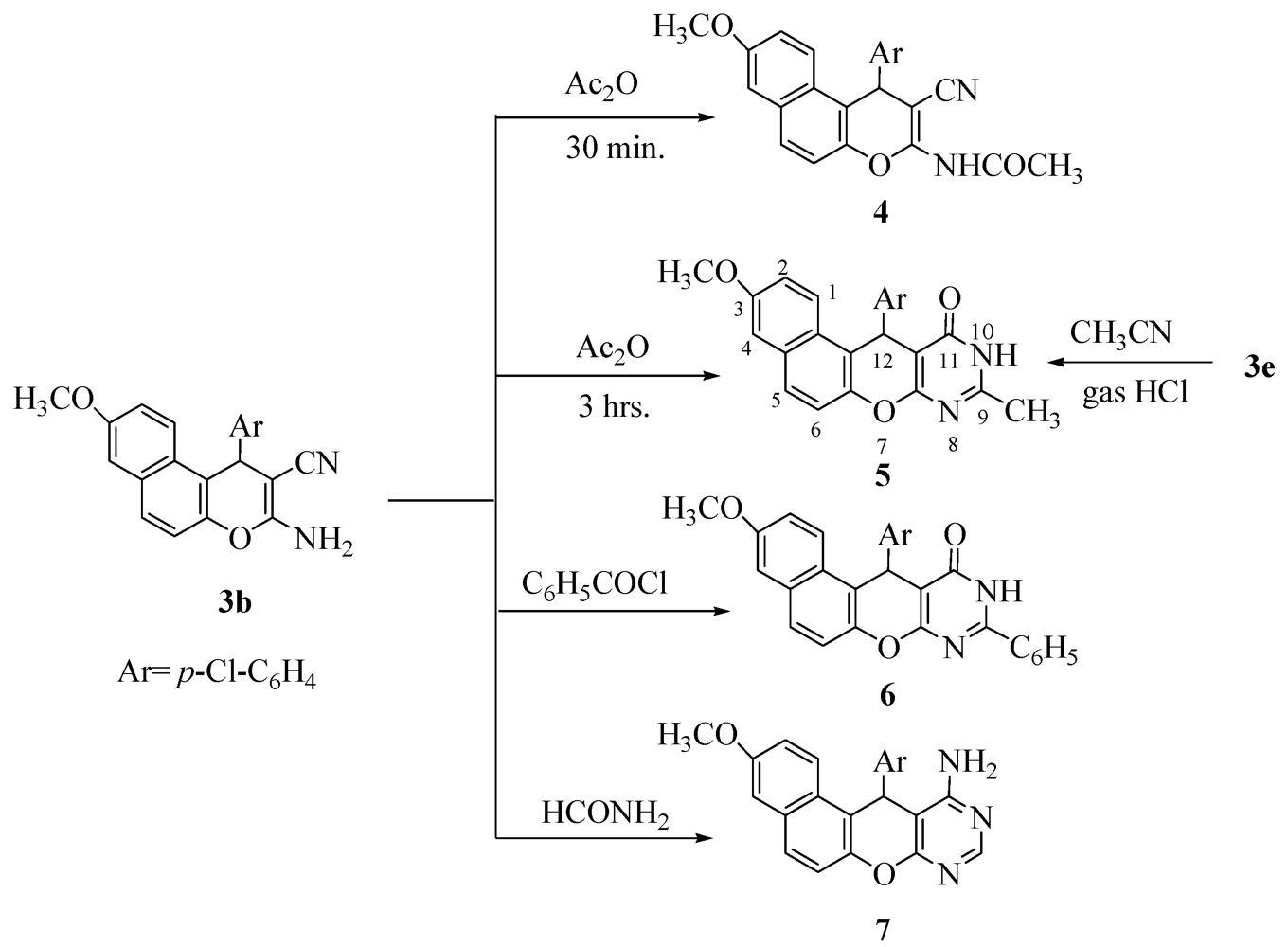
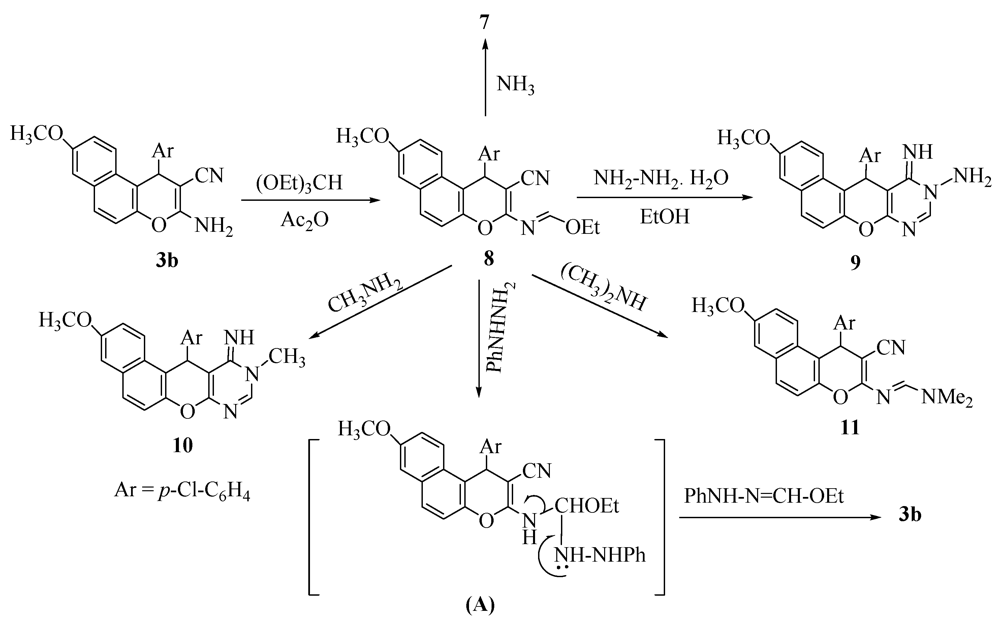
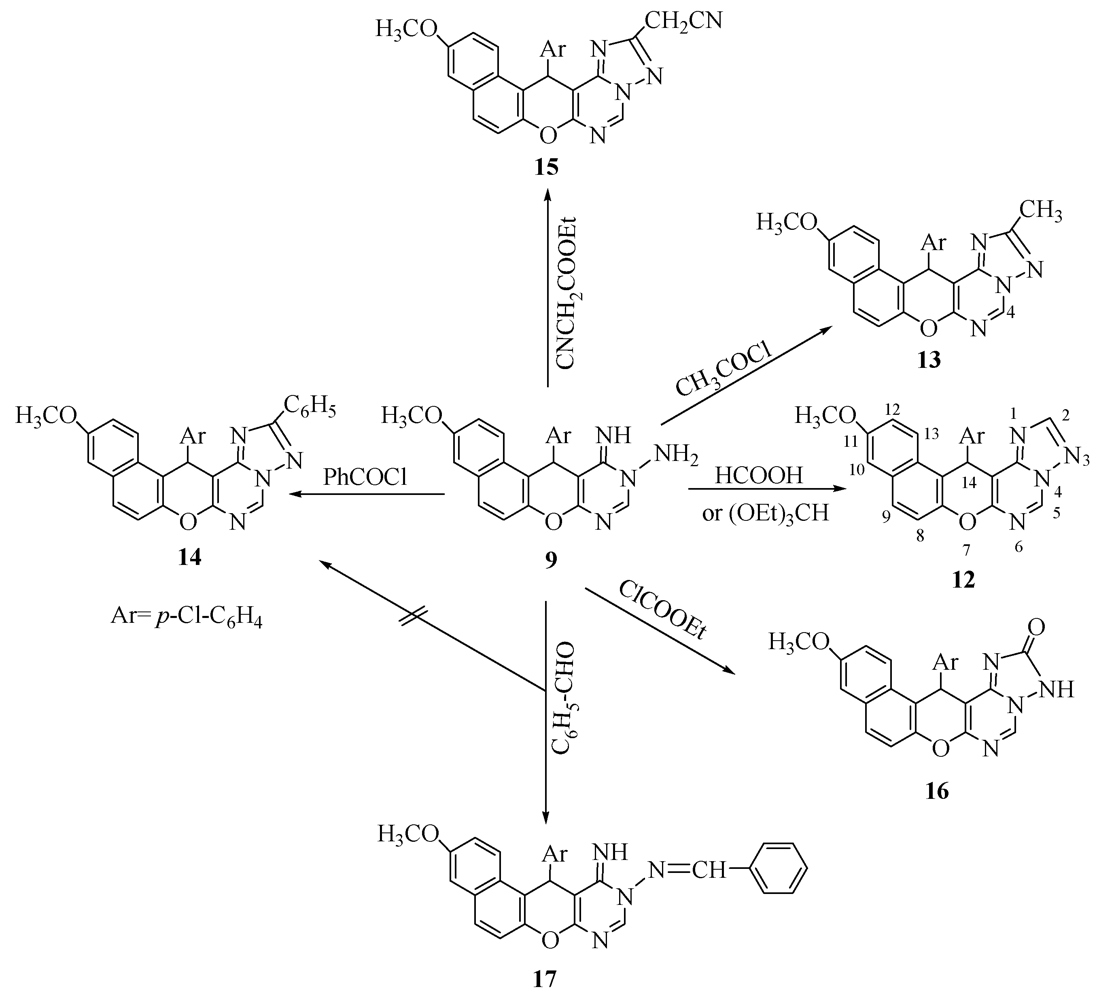
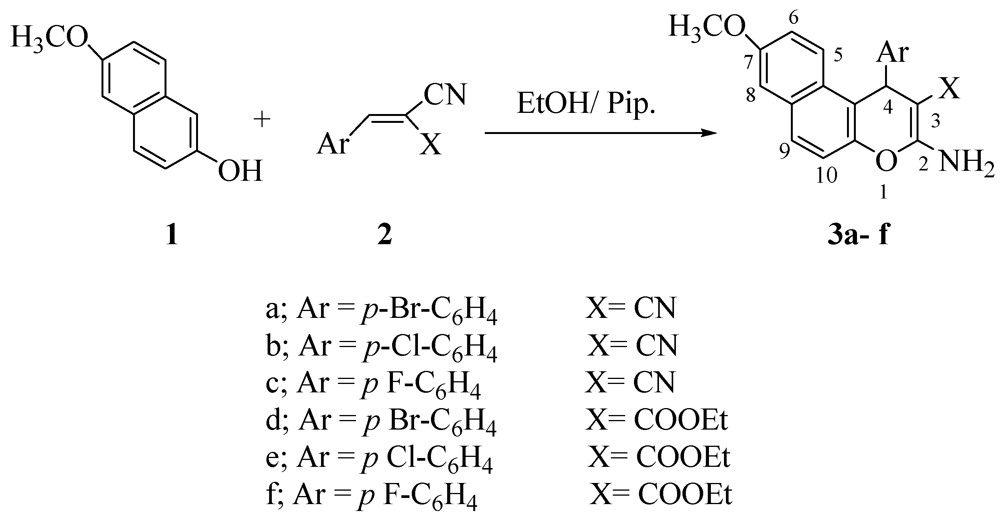
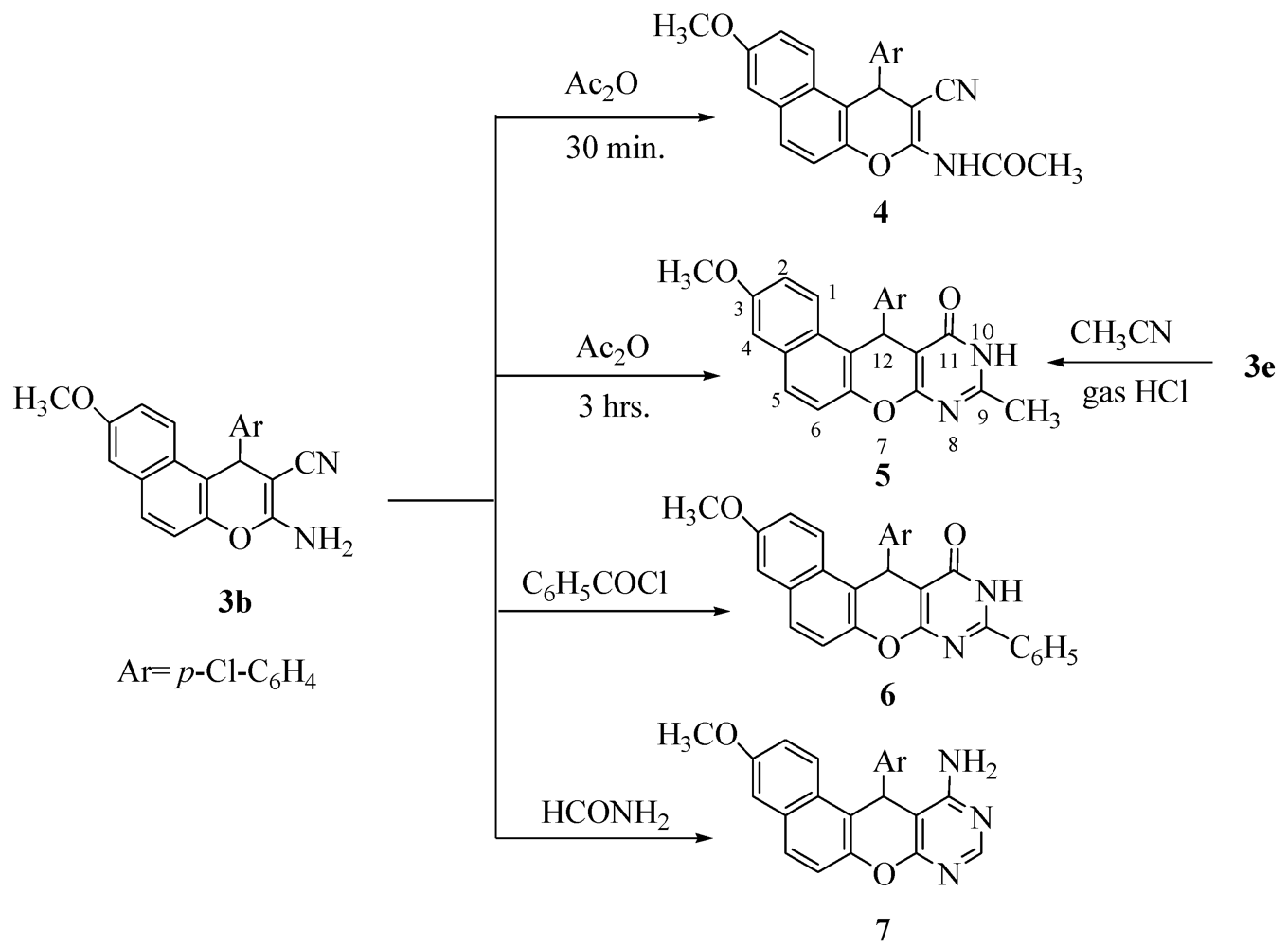
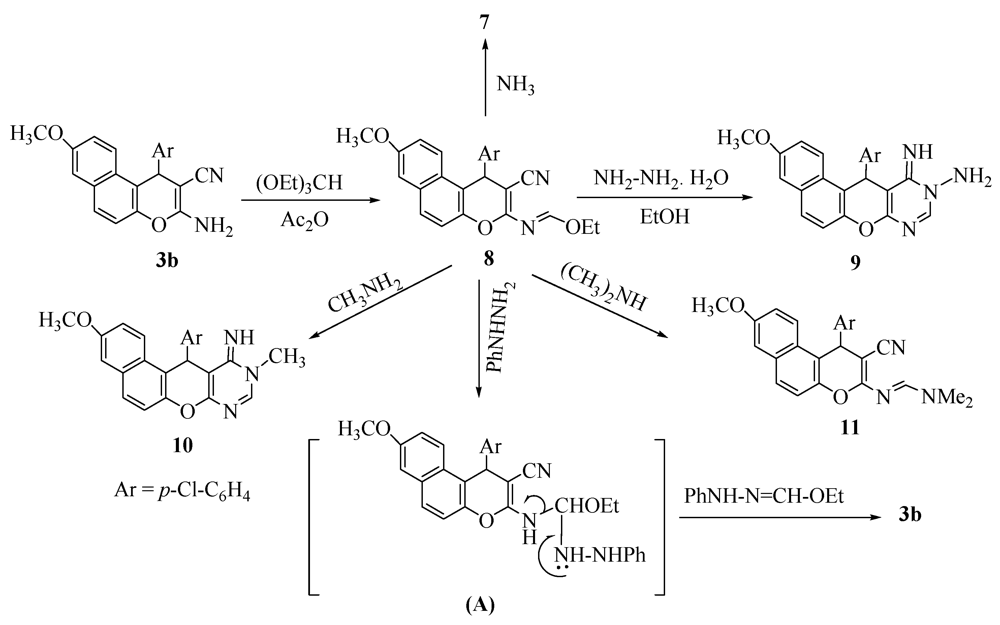
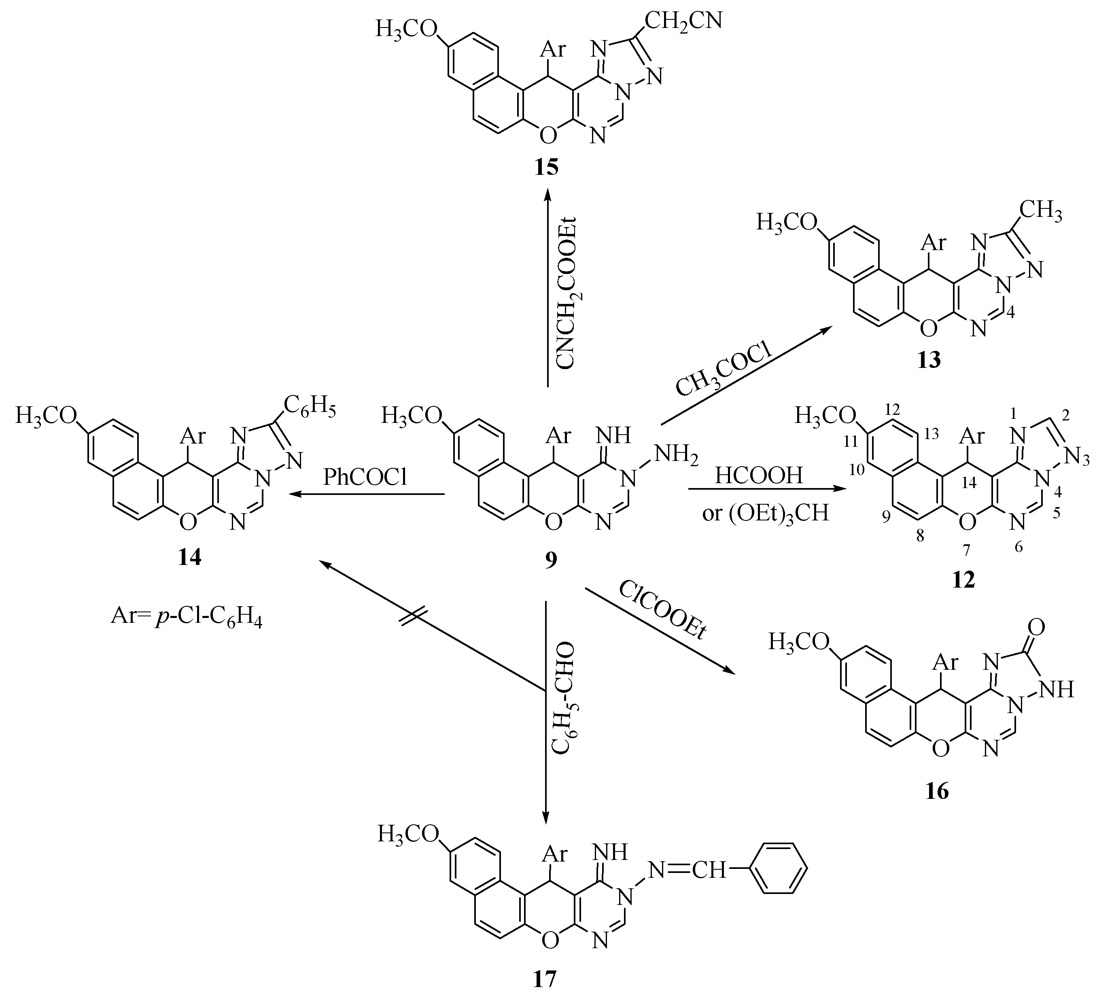
2. Results and Discussion

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compounds | Minimum inhibitory concentration (MIC) in μg/mL | |||||
|---|---|---|---|---|---|---|
| Bacterial strains | Fungal strains | |||||
| S. aureus | B. cereus | E. coli | S. marcescens | A. fumigatus | C. albicans | |
| 3a | 100 | 100 | 25 | 50 | 100 | 100 |
| 3b | 125 | 200 | 50 | 50 | 125 | 100 |
| 3c | 100 | 50 | 25 | 50 | 50 | 50 |
| 3d | 250 | 200 | 500 | 500 | 250 | 100 |
| 3e | 25 | 100 | 50 | 100 | 25 | 50 |
| 3f | 500 | 500 | - | 125 | 250 | 500 |
| 4 | 500 | 250 | 250 | 100 | 500 | - |
| 5 | 100 | 125 | - | 500 | - | 250 |
| 6 | 100 | - | 100 | 250 | 500 | 500 |
| 7 | 500 | 200 | - | 200 | - | 250 |
| 8 | 250 | 200 | 500 | 500 | 250 | 100 |
| 9 | - | 500 | - | 100 | 500 | 500 |
| 10 | 500 | 250 | 100 | 250 | 250 | 500 |
| 11 | - | 500 | 250 | - | - | 500 |
| 12 | 25 | 25 | 50 | 25 | 100 | 50 |
| 13 | 25 | 50 | 25 | 50 | 50 | 500 |
| 14 | 50 | 100 | 100 | 25 | 25 | 50 |
| 15 | 100 | 25 | 25 | 125 | 100 | 500 |
| 16 | 50 | 25 | 50 | 50 | 125 | 100 |
| 17 | 500 | - | 250 | 500 | - | - |
| Ampcillin | 6.25 | 6.25 | 6.25 | 6.25 | 6.25 | 6.25 |
| Ketoconazole | - | - | - | - | 31.25 | 31.25 |
3. Experimental
3.1. General
3.2. General Procedure: Synthesis of 4H-Pyran Derivatives 3a–f
3.3. General Procedure: Synthesis of Pyranopyrimidine Derivatives 9 and 10
3.4. General Procedure: Synthesis of Pyranotriazolopyrimidine Derivatives 12–16
3.5. Antimicrobial Assay
4. Conclusions
References and Notes
- Eid, F.A.; Abd El-Wahab, A.H.F.; El-Hagali, G.A.M.; Khafagy, M.M. Syhthesis and antimicrobial evaluation of naphtho[2,1-b]pyrano[2,3-d]pyrimidine and pyrano[3,2-e][1,2,4]triazolo[1,5-c]pyrimidine derivative. Acta Pharm. 2004, 54, 13–26. [Google Scholar]
- Abd El-Wahab, A.H.F. Synthesis of some new pyrano[2,3-d][1,2,4]triazolo[1,5-c]pyrimidine and pyrimido[1,6-b]triazine derivatives. Acta Pharm. 2003, 58, 701–720. [Google Scholar]
- El-Agrody, A.M.; Abd El-Latif, M.S.; El-Hady, N.A.; Fakery, A.H.; Bedair, A.H. Heteroaromatization with 4-Hyroxycoumarin. Part II: Synthesis of some new pyrano[2,3-d][1,2,4]triazolo[1,5-c]pyrimidine and pyrimido[1,6-b]triazine derivatives. Molecules 2001, 6, 519–527. [Google Scholar] [CrossRef]
- Bedair, A.H.; El-Haddy, N.A.; Abd El-Latif, M.S.; Fakery, A.H.; El-Agrody, A.M. 4-Hyroxycoumarin in heterocycloic synthesis. Part III: Synthesis of some new pyrano[2,3-d]pyrimidine, 2-substituted[1,2,4]triazolo[1,5-c]pyrimidine and pyrimido[1,6-b]triazine derivatives. Farmaco 2000, 55, 708–714. [Google Scholar] [CrossRef]
- El-Agrody, A.M.; El-Hakim, M.H.; Abd El-Latif, M.S.; Fakery, A.H.; El-Sayed, E.M.; El-Ghareab, K.A. Synthesis pyrano[2,3-d]pyrimidine and pyrano[3,2-e][1,2,4]triazolo[2,3-c]pyrimidine derivatives with promising antibacterial activities. Acta Pharm. 2000, 50, 111–120. [Google Scholar]
- Taylor, R.N.; Cleasby, A.; Singh, O.; Skarzynski, T. Dihydropyrancarboxamide related to zanamivir, a new series of inhibitors of influrnza virus sialidase. 2-Crystallographic and molecular modeling study of complex 4-amino-4H-pyran-6-carboxamides and sialidase from influenza virus types. J. Med. Chem. 1998, 41, 798–807. [Google Scholar] [CrossRef]
- Hirmoto, K.; Nasuhara, A.; Michiloshi, K.; Kato, T.; Kikugawa, K. DNA strand-breaking activity and mutagenicity of 2,3-dihydro-3,5-dihydroxy-6-methyl-4H-pyran-4-one (DDMP). A Maillard reaction product of glucose and glycine. Mutat. Res. 1997, 395, 47–56. [Google Scholar] [CrossRef]
- Martinez, A.G.; Marco, L.J. Friedlander reaction on 2-amino-3-cyano-4H-pyrans, synthesis of derivatives of 4H-pyran[2,3-b]quinoline, new tacrine analogues. Bioorg. Med. Chem. Lett. 1997, 7, 3165–3170. [Google Scholar] [CrossRef]
- Dell, C.P.; Smith, C.W. Antiproliferative derivatives of 4H-naphtho[1,2-b]pyran and process for their preparation. EP 537949, 21 April 1993. [Google Scholar]
- Bianchi, G.; Tava, A. Synthesis of (2R)(+)-2,3-dihydro-2,6-dimethyl-4H-pyran-one, a homologue of pheromones of a species in the hepialidae family. Agric. Biol. Chem. 1987, 51, 2001–2002. [Google Scholar] [CrossRef]
- Eiden, F.; Denk, F. Synthesis and CNS activity of pyrane derivatives 6,8-dioxabicyclo(3,2,1)octanes. Arch. Pharm. (Weinheim) 1991, 324, 353–354. [Google Scholar] [CrossRef]
- Shishoo, C.J.; Devani, M.B.; Ullas, G.V.; Ananthan, S.; Bahadit, V.S. Studies in the synthesis and interconversion of isomeric triazolopyrimidinees. J. Heterocycl. Chem. 1981, 18, 43–46. [Google Scholar] [CrossRef]
- Noda, K.; Nakagawa, A.; Nakajima, Y.; Ide, H. Pyrido[2,3-d]-s-triazolo[4,3-c]pyrimidine derivatives. Japan Kokai. 1997, 194, 7785, [Chem. Abstr. 1978, 88, P50908q].. [Google Scholar]
- The Chemistry of Heterocyclic Compounds, the Pyrimidines; Brown, D.J.; Evans, R.F.; Cowden, W.B.; Fenn, M.D. (Eds.) John Wiley & Sons: New York, NY, USA, 1994; Volume 23.
- Matloobi, M.; Kappe, C.O. Microwave assisted solution- and solid-phase synthesis of amino-4-arylpyrimidine derivatives. J. Comb. Chem. 2007, 9, 275–284. [Google Scholar] [CrossRef]
- Gadhachanda, V.R.; Wu, B.; Wang, Z.; Kuhen, K.L.; Caldwell, J.; Zondler, H.; Walter, H.; Havenhand, M.; He, Y. 4-Amino-pyrimidines as novel HIV-1 inhibitors. Bioorg. Med. Chem. Lett. 2007, 17, 260–265. [Google Scholar]
- Agarwal, A.; Srivastava, K.; Puri, S.K.; Chauhan, P.M.S. Synthesis of 4-pyrido-6-aryl-2-substituted amino pyrimidines as a new class of antimalarial agents. Bioorg. Med. Chem. 2005, 13, 6226–6232. [Google Scholar] [CrossRef]
- Chang, L.C.W.; Spanjersberg, R.F.; Frijtag, D.K.; Mulder-K, T.; Hout, G.V.; Beukers, M.W.; Brussee, J.; Jzerman, A.P. 2,4,6-Trisubstituted pyrimidines as a new class of selective adenosine A1 receptor antagonists. J. Med. Chem. 2004, 47, 6529–6540. [Google Scholar]
- Capdeville, R.; Buchdunger, E.; Zimmermann, J.; Matter, A. Glivec (STI571, imatinib), a rationally developed, targeted anticancer drug. Nat. Rev. Drug Discov. 2002, 1, 493–502. [Google Scholar] [CrossRef]
- Sondhi, S.M.; Dinodia, M.; Rani, R.S.; Shukla, R.; Raghubir, R. Synthesis, anti-inflammatory and analgesic activity evaluation of some pyrimidine derivatives. Indian J. Chem. 2009, 48, 273–281. [Google Scholar]
- Shanker, K.; Gupta, G.P.; Singh, S. Synthesis of novel pyrimidinediones and thiazolidinones as Cardiovascular agents. Indian J. Chem. 1985, 24, 1094–1097. [Google Scholar]
- Ozeki, K.; Ichikawa, T.; Takehara, H.; Tanimura, K.; Sato, M.; Yaginuma, H. Studies on antiallergy agents. III. Synthesis of 2-anilino-1, 6-dihydro-6-oxo-5-pyrimidine carboxylic acids and related compounds. Chem. Pharm. Bull. 1989, 37, 1780–1787. [Google Scholar]
- Elagamey, A.G.A.; El-Taweel, F.M.A.; Sowellim, S.Z.A.; Sofan, M.A.; Elnagdi, M.H. Nitriles in heterocyclic synthesis, a novel route for the synthesis of naphthodipyrans, pyridines, 2H- and 4H-pyrans. Collect. Czech. Chem. Commun. 1990, 55, 524–534. [Google Scholar] [CrossRef]
- Elngdi, M.H.; Elghandour, A.H.H.; Ibrahim, M.K.A.; Hafez, I.S.A. Studies with polyfunctionally substituted heterocyclic, synthesis of new pyrimidines, naphtha[1,2-b]pyrans, pyrazolo[3,4-b]pyrimidines and pyrazolo[1,5-a]pyrimidines. Z. Naturforsch. 1992, 47, 572–578. [Google Scholar]
- Dave, K.G.; Shishoo, C.J.; Devani, M.B.; Kalyanaraman, R.; Ananthan, S.; Ullas, G.V.; Bahadit, V.S. Reaction of nitriles under acidic conditions. Part I: A general method of synthesis of condensed pyridines. J. Heterocycl. Chem. 1980, 17, 1497–1501. [Google Scholar]
- European Committee for Antimicrobial Susceptibility Testing (EUCAST) of the European Society of Clinical Microbiology and Infectious Diseases (ESCMID). Determination of minimum inhibitory concentrations (MICs) of antibacterial agents by agar dilution. Clin. Microbiol. Infect. 2000, 6, 509. [CrossRef]
- National Committee for Clinical Laboratory Standards, Methods for Dilution AntimicrobialSusceptibility Tests for Bacteria That Grow Aerobicallyl; Approved Standard M7-A5, 5th ed; NCCLS: Wayne, PA, USA, 2000.
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Abd El-Wahab, A.H.F. Synthesis, Reactions and Evaluation of the Antimicrobial Activity of Some 4-(p-Halophenyl)-4H-naphthopyran, Pyranopyrimidine and Pyranotriazolopyrimidine Derivatives. Pharmaceuticals 2012, 5, 745-757. https://doi.org/10.3390/ph5070745
Abd El-Wahab AHF. Synthesis, Reactions and Evaluation of the Antimicrobial Activity of Some 4-(p-Halophenyl)-4H-naphthopyran, Pyranopyrimidine and Pyranotriazolopyrimidine Derivatives. Pharmaceuticals. 2012; 5(7):745-757. https://doi.org/10.3390/ph5070745
Chicago/Turabian StyleAbd El-Wahab, Ashraf H. F. 2012. "Synthesis, Reactions and Evaluation of the Antimicrobial Activity of Some 4-(p-Halophenyl)-4H-naphthopyran, Pyranopyrimidine and Pyranotriazolopyrimidine Derivatives" Pharmaceuticals 5, no. 7: 745-757. https://doi.org/10.3390/ph5070745