Mechanistic and Pharmacological Issues of Aspirin as an Anticancer Agent


Abstract
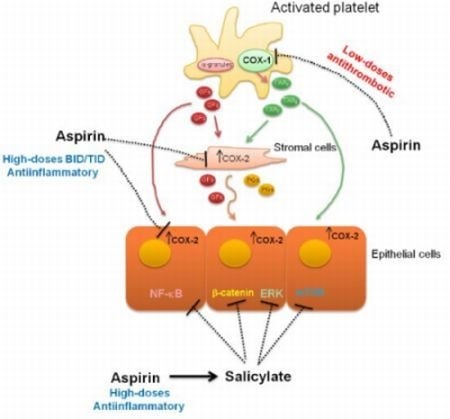
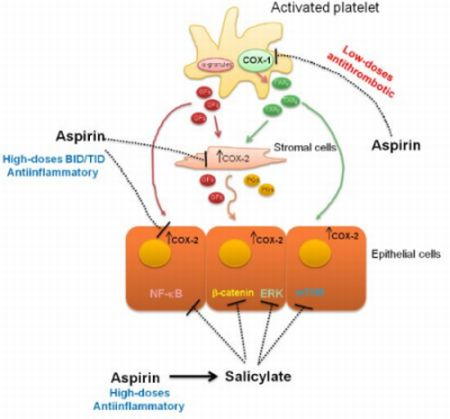
:
1. Introduction
2. Aspirin and Colorectal Cancer Risk: the Results of Clinical Studies

{kind=link}
{kind=link}
{kind=link}
| Study (reference) Patients | Treatment | Primary end-point | Relative risk (RR) (95% Confidence Interval, CI) |
|---|---|---|---|
| AFPPS trial [7,28]: Patients with a recent history of histologically documented (removed) adenomas | Aspirin (81 mg or 325 mg daily) or folic acid (1 mg daily) or placebo for 2.7 years | Proportion of patients in whom one or more colorectal adenomas were detected by colonoscopy | Any adenoma: RR: 0.81 (0.69–0.96), aspirin 81mg versus non aspirin RR: 0.96 (0.81–1.13), aspirin 325 mg versus non aspirin RR: 1.04(0.90–1.20), folic acid versus non folic acid Advanced lesion: RR: 0.59 (0.38–0.92), aspirin 81mg versus non aspirin RR: 0.83 (0.55–1.23), aspirin 325 mg versus non aspirin RR: 1.32 (0.90–1.92), folic acid versus non folic acid |
| CAPS trial [8]: Patients with a histologically documented colon or rectal cancer with a low risk of recurrent disease | Placebo or enteric coated aspirin 325 mg daily for 2.6 years | Detection of adenomas in the large bowel by either colonoscopy or sigmoidoscopy | RR: 0.65(0.46–0.91) |
| APACC trial [9,29]: Patients with a history of colorectal adenomas | Placebo or lysine acetylsalicylate (160 or 300 mg daily) for 1 and 4 years | Proportions of recurrent adenomas and adenomatous polyp burden by colonoscopy | RR: 0.73 (0.52–1.04) for both doses, after 1 year RR: 0.96(0.75–1.22), for both doses, after 4 years |
| ukCAP trial [10]: Patients with an adenoma removed in the 6 months before recruitment | Enteric coated aspirin (300 mg daily) plus placebo or aspirin plus folic acid (0.5 mg/daily) or folic acid plus placebo or double placebo for about 2.6 years | Percentage of patients who developed one or more recurrent colorectal adenomas or cancers by colonoscopy | Any adenoma: RR: 0.79(0.63–0.99), aspirin versus non aspirin RR: 1.07 (0.85–1.34), folic acid versus non folic acid Advanced adenoma: RR: 0.63(0.43–0.91), aspirin versus non aspirin RR: 0.98(0.68–1.40), folic acid versus non folic acid |
| J-CAPP trial [30]: Patients with previous sporadic colorectal tumors | Enteric coated aspirin (100 mg daily) or placebo for 2 years | Presence or absence of new colorectal tumors by colonoscopy | Ongoing |
| Study (reference) Patients | Treatment | Primary end-point | RR or Hazard ratio (HR) (95% CI) |
|---|---|---|---|
| CAPP 2 trial [35]: Lynch syndrome (hereditary non-polyposis colon cancer or HNPCC) | Aspirin (600 mg daily) or aspirin placebo or resistant starch (30g daily) or starch placebo, for up to 4 years | Detection of at least one adenoma or colorectal carcinoma by colonoscopy | HR: 0.63 (0.35–1.13), for the entire post-randomization period (aspirin versus placebo) HR: 0.41(0.19–0.86), for ≥2years of treatment (aspirin versus placebo) |
| CAPP1 trial [31]: FAP young patients (10 to 21 years of age) | Aspirin (600 mg daily) plus placebo or resistant starch (30 g daily) plus placebo or double placebo for 17 years | Polyp number in the rectum and sigmoid colon by colonoscopy | RR: 0.77 (0.54–1.10), aspirin versus non aspirin RR: 1.05(0.73–1.49), resistant starch versus non resistant starch |
| J-FAPP II trial [30]: FAP patients (≥16 years of age) | Placebo versus enteric coated aspirin (100 mg daily for 6-10 months) | Reduction in the number of rectal tumors | Ongoing |
2.1. Chemopreventive Effects of Aspirin in All Cancers
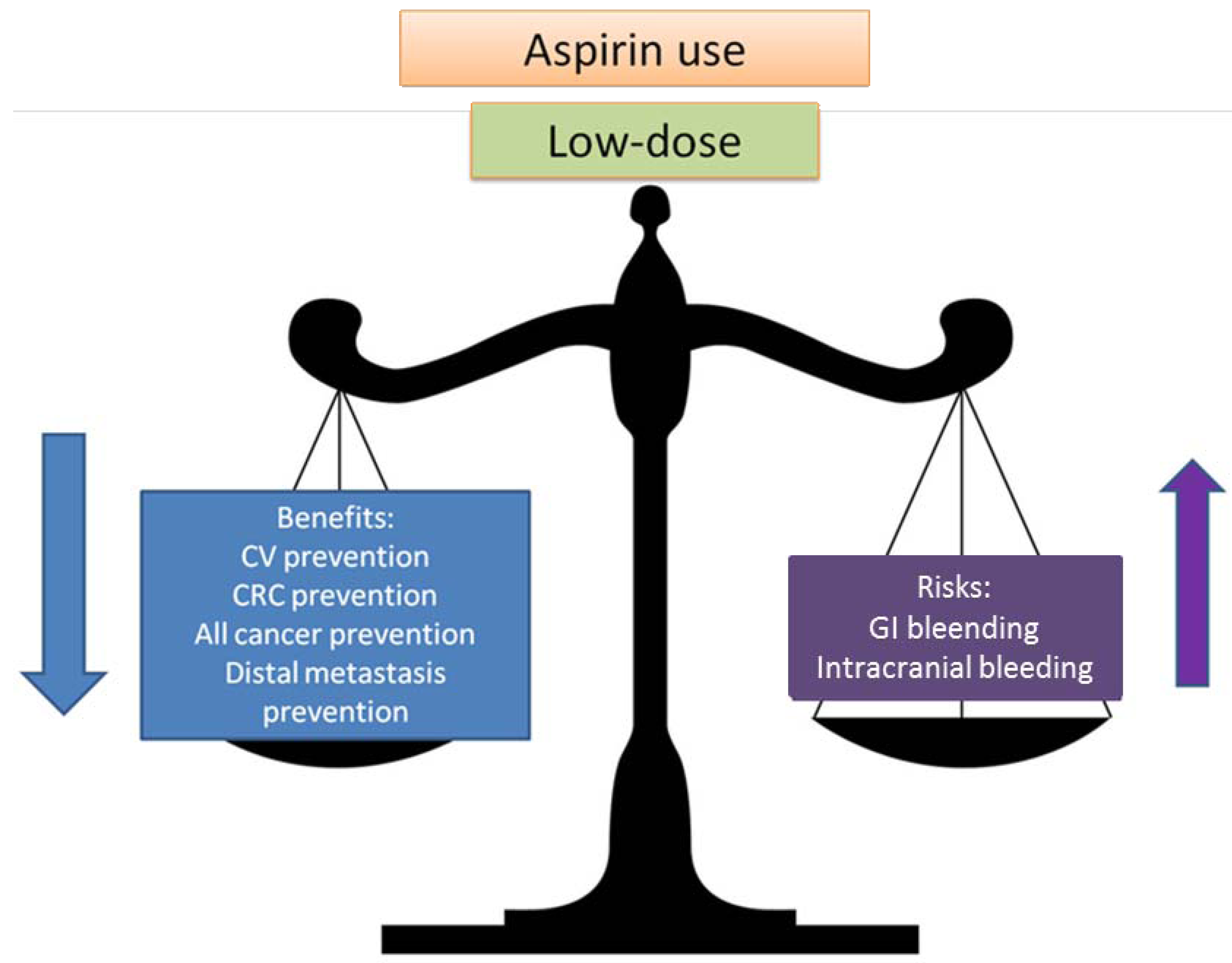
2.2. Balancing Risk and Benefits
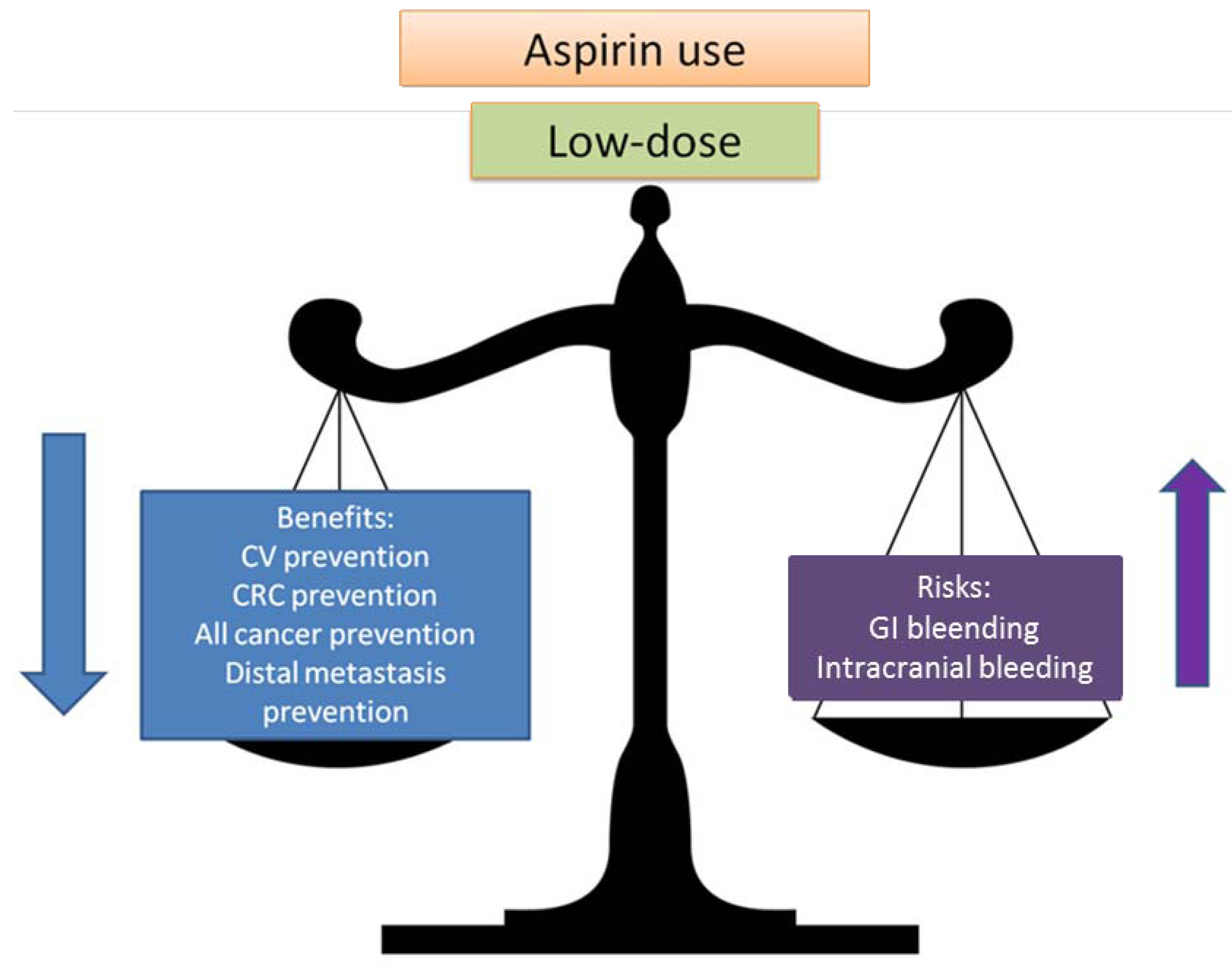
3. Mechanism of Action of Aspirin
| Chemical structure | 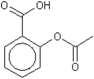 acetylsalicylic acid |
| IC50 (human whole blood assays in vitro ); mean(95% confidence interval, CI) | COX-1:7.9 (4.4–14)μM [65] COX-2: >5000μM [65] |
| Oral bioavailability (%) | 49.2 (40 mg single dose) [68] 48.2 (325 mg single dose) [68] 50.7 (325 mg for 7 days) [68] Approx 30% (enteric coated aspirin) [72,73] |
| Time to maximal plasma concentration | 30–40 minutes [74] 3–4 h (enteric coated aspirin)[72] |
| Maximal plasma concentration (μM; mean±SD) | acetylsalicylic acid: antiplatelet dose(75–100mg/day): 0.52±0.12 μM (controlled release) [5] 7.3±2μM (solution) [5] analgesic dose (325, 600 mg):25±12(68),80±20μM [75] antiinflammatory dose(1.2g): 144±22 μM [75] salicylic acid: antiplatelet dose(75–100mg/day): 4.9±2.5 μM (controlled release) [5] 14.4±5 μM (solution) [5] analgesic dose(600mg/day): 297±96 μM [75] anti-inflammatory dose: (1.2 g/day), 585±164 μM [75] (5.85–6.2 g/day),1800–2300μM [76,77] |
| Half-life (minutes) | 15–20 [74] |
| Volume of distribution (l) | 21.2 (40 mg single dose) [68] 18.4 (325 mg single dose) [68] 19.2 (325 mg daily for 7 days) [68] |
| Bound in plasma (%) | 80–90%[74,78] |
3.1. Pharmacology of Aspirin
3.2. Clinical consequences of COX inhibition by aspirin
4. Evidences for COX-Dependent Mechanisms of the Antitumoral Effects of Aspirin
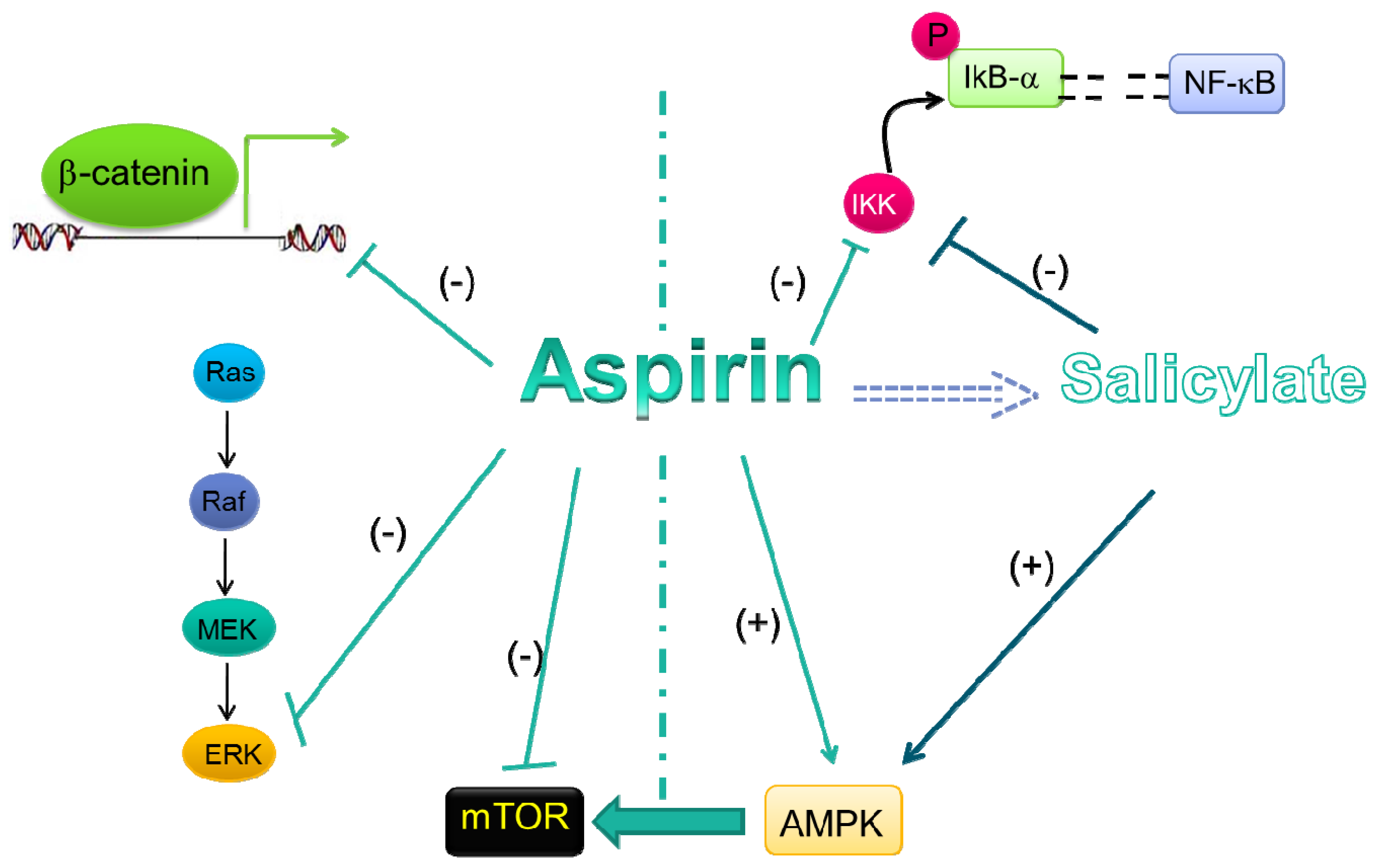
5. Evidences for COX-Independent Mechanisms of the Antitumoral Effects of Aspirin
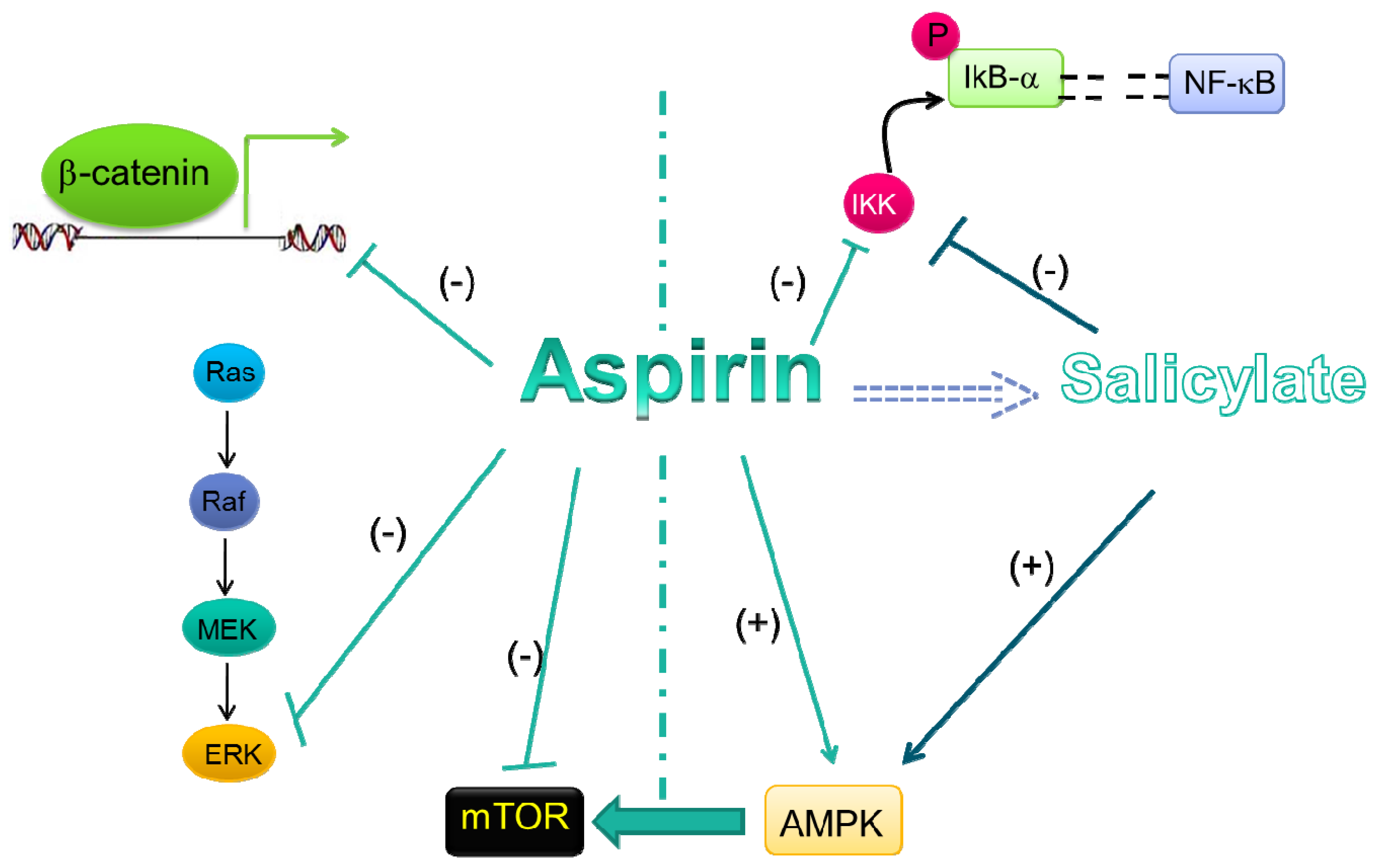
5.1. Wnt/β-Catenin Pathway
5.2. ERK Signaling
5.3. NF-κB Signal Transduction Pathway
5.4. AMPK/mTORsignaling
5.5. Acetylation of Other Proteins by Aspirin
6. Conclusions
Acknowledgements
References
- Thun, M.J.; Jacobs, E.J.; Patrono, C. The role of aspirin in cancer prevention. Nat. Rev. Clin. Oncol. 2012, 9, 259–267. [Google Scholar] [CrossRef]
- Meade, T.W. The Medical Research Council's General Practice Research Framework. Thrombosis prevention trial: randomised trial of low-intensity oral anticoagulation with warfarin and low-dose aspirin in the primary prevention of ischaemic heart disease in men at increased risk. Lancet 1998, 351, 233–241. [Google Scholar] [CrossRef]
- Rothwell, P.M.; Wilson, M.; Elwin, C.E.; Norrving, B.; Algra, A.; Warlow, C.P.; Meade, T.W. Long-term effect of aspirin on colorectal cancer incidence and mortality: 20-year follow-up of five randomised trials. Lancet 2010, 376, 1741–1750. [Google Scholar] [CrossRef]
- Rothwell, P.M.; Fowkes, F.G.; Belch, J.F.; Ogawa, H.; Warlow, C.P.; Meade, T.W. Effect of daily aspirin on long-term risk of death due to cancer: analysis of individual patient data from randomised trials. Lancet 2011, 377, 31–41. [Google Scholar]
- Charman, W.N.; Charman, S.A.; Monkhouse, D.C.; Frisbee, S.E.; Lockhart, E.A.; Weisman, S.; Fitzgerald, G.A. Biopharmaceutical characterisation of a low-dose (75 mg) controlled-release aspirin formulation. Br. J. Clin. Pharmac. 1993, 36, 470–473. [Google Scholar] [CrossRef]
- Clarke, R.J.; Mayo, G.; Price, P.; FitzGerald, G.A. Suppression of thromboxane A2 but not of systemic prostacyclin by controlled-release aspirin. N. Engl. J. Med. 1991, 325, 1137–1141. [Google Scholar] [CrossRef]
- Baron, J.A.; Cole, B.F.; Sandler, R.S.; Haile, R.W.; Ahnen, D.; Bresalier, R.; McKeown-Eyssen, G.; Summers, R.W.; Rothstein, R.; Burke, C.A.; Snover, D.C.; Church, T.R.; Allen, J.I.; Beach, M.; Beck, G.J.; Bond, J.H.; Byers, T.; Greenberg, E.R.; Mandel, J.S.; Marcon, N.; Mott, L.A.; Pearson, L.; Saibil, F.; van Stolk, R.U. A randomized trial of aspirin to prevent colorectal adenomas. N. Engl. J. Med. 2003, 348, 891–899. [Google Scholar]
- Sandler, R.S.; Halabi, S.; Baron, J.A.; Budinger, S.; Paskett, E.; Keresztes, R.; Petrelli, N.; Pipas, J.M.; Karp, D.D.; Loprinzi, C.L.; Steinbach, G.; Schilsky, R. A randomized trial of aspirin to prevent colorectal adenomas in patients with previous colorectal cancer. N. Engl. J. Med. 2003, 348, 883–890. [Google Scholar] [CrossRef]
- Benamouzig, R.; Deyra, J.; Martin, A.; Girard, B.; Jullian, E.; Piednoir, B.; Couturier, D.; Coste, T.; Little, J.; Chaussade, S. Daily soluble aspirin and prevention of colorectal adenoma recurrence: one-year results of the APACC trial. Gastroenterology 2003, 125, 328–336. [Google Scholar] [CrossRef]
- Logan, R.F.; Grainge, M.J.; Shepherd, V.C.; Armitage, N.C.; Muir, K.R. ukCAP Trial Group. Aspirin and folic acid for the prevention of recurrent colorectal adenomas. Gastroenterology 2008, 134, 29–38. [Google Scholar]
- Cuzick, J.; Otto, F.; Baron, J.A.; Brown, P.H.; Burn, J.; Greenwald, P.; Jankowski, J.; La Vecchia, C.; Meyskens, F.; Senn, H.J.; Thun, M. Aspirin and non-steroidal anti-inflammatory drugs for cancer prevention: an international consensus statement. Lancet Oncol. 2009, 10, 501–507. [Google Scholar] [CrossRef]
- Baigent, C. Antithrombotic Trialists' Collaboration. Collaborative meta-analysis of randomised trials of antiplatelet therapy for prevention of death, myocardial infarction, and stroke in high-risk patients. BMJ. 2002, 324, 71–86. [Google Scholar] [CrossRef]
- Sciulli, M.G.; Filabozzi, P.; Tacconelli, S.; Padovano, R.; Ricciotti, E.; Capone, M.L.; Grana, M.; Carnevale, V.; Patrignani, P. Plateletactivation in patients with colorectalcancer. Prostagl. Leukot. Essent. Fatty Acids 2005, 72, 79–83. [Google Scholar] [CrossRef]
- Dovizio, M.; Tacconelli, S.; Ricciotti, E.; Bruno, A.; Maier, T.J.; Anzellotti, P.; Di Francesco, L.; Sala, P.; Signoroni, S.; Bertario, L.; Dixon, D.A.; Lawson, J.A.; Steinhilber, D.; FitzGerald, G.A.; Patrignani, P. Effects of celecoxib on prostanoidbiosynthesis and circulatingangiogenesisproteins in familialadenomatouspolyposis. J. Pharmacol. Exp. Ther. 2012, 341, 242–50. [Google Scholar] [CrossRef]
- Ferlay, J.; Shin, H.R.; Bray, F.; Forman, D.; Mathers, C.; Parkin, D.M. Estimates of worldwide burden of cancer in 2008: GLOBOCAN2008. Int. J. Cancer 2010, 127, 2893–917. [Google Scholar] [CrossRef]
- Shapiro, J.A.; Seeff, L.C.; Thompson, T.D.; Nadel, M.R.; Klabunde, C.N.; Vernon, S.W. Colorectal cancer test use from the 2005 National Health Interview Survey. Cancer Epidemiol. Biomarkers Prev. 2008, 17, 1623–30. [Google Scholar] [CrossRef]
- US Preventive Services Task Force. Screening for colorectal cancer: U.S. Preventive Services Task Force recommendation statement. Ann. Intern. Med. 2008, 149, 627–37.
- Arber, N.; Levin, B. Chemoprevention of colorectal neoplasia: the potential for personalized medicine. Gastroenterology 2008, 134, 1224–1237. [Google Scholar] [CrossRef]
- Bertagnolli, M.M. Chemoprevention of colorectal cancer with cyclooxygenase-2 inhibitors: two steps forward, one step back. Lancet Oncol. 2007, 8, 439–443. [Google Scholar] [CrossRef]
- Giovannucci, E.; Egan, K.M.; Hunter, D.J.; Stampfer, M.J.; Colditz, G.A.; Willett, W.C.; Speizer, F.E. Aspirin and the risk of colorectal cancer in women. N. Engl. J. Med. 1995, 333, 609–614. [Google Scholar] [CrossRef]
- Kune, G.A.; Kune, S.; Watson, L.F. Colorectal cancer risk, chronic illnesses, operations, and medications: case control results from the Melbourne Colorectal Cancer Study. Cancer Res. 1988, 48, 4399–4404. [Google Scholar]
- Flossmann, E.; Rothwell, P.M. Effect of aspirin on long-term risk of colorectal cancer: consistent evidence from randomized and observational studies. Lancet 2007, 369, 1603–1613. [Google Scholar] [CrossRef]
- U.S. Preventive Services Task Force recommendation statement. Routine aspirin or non-steroidal anti-inflammatory drugs for the primary prevention of colorectal cancer. Ann. Intern. Med. 2007, 146, 361–364.
- Peto, R.; Gray, R.; Collins, R.; Wheatley, K.; Hennekens, C.; Jamrozik, K.; Warlow, C.; Hafner, B.; Thompson, E.; Norton, S.; et al. Randomised trial of prophylactic daily aspirin in British male doctors. Brit. Med. J. (Clinical Research Ed.) 1988, 296, 313–316. [Google Scholar] [CrossRef]
- Gann, P.H.; Manson, J.E.; Glynn, R.J.; Buring, J.E.; Hennekens, C.H. Low-dose aspirin and incidence of colorectal tumors in a randomized trial. J. Natl. Cancer Inst. 1993, 85, 1220–1224. [Google Scholar] [CrossRef]
- Cook, N.R.; Lee, I.M.; Gaziano, J.M.; Gordon, D.; Ridker, P.M.; Manson, J.E.; Hennekens, C.H.; Buring, J.E. Low-dose aspirin in the primary prevention of cancer: the Women’s Health Study: a randomized controlled trial. JAMA. 2005, 294, 47–55. [Google Scholar]
- Cole, B.F.; Logan, R.F.; Halabi, S.; Benamouzig, R.; Sandler, R.S.; Grainge, M.J.; Chaussade, S.; Baron, J.A. Aspirin for the chemoprevention of colorectal adenomas: meta-analysis of the randomized trials. J. Natl. Cancer Inst. 2009, 101, 256–266. [Google Scholar] [CrossRef]
- Cole, B.F.; Baron, J.A.; Sandler, R.S.; Haile, R.W.; Ahnen, D.J.; Bresalier, R.S.; McKeown-Eyssen, G.; Summers, R.W.; Rothstein, R.I.; Burke, C.A.; Snover, D.C.; Church, T.R.; Allen, J.I.; Robertson, D.J.; Beck, G.J.; Bond, J.H.; Byers, T.; Mandel, J.S.; Mott, L.A.; Pearson, L.H.; Barry, E.L.; Rees, J.R.; Marcon, N.; Saibil, F.; Ueland, P.M.; Greenberg, E.R. Polyp Prevention Study Group. Folic acid for the prevention of colorectal adenomas: a randomized clinical trial. JAMA 2007, 297, 2351–2359. [Google Scholar]
- Benamouzig, R.; Uzzan, B.; Deyra, J.; Martin, A.; Girard, B.; Little, J.; Chaussade, S. Association pour la Prévention par l'Aspirine du Cancer Colorectal Study Group (APACC). Prevention by daily soluble aspirin of colorectal adenoma recurrence: 4-year results of the APACC randomised trial. Gut 2012, 61, 255–261. [Google Scholar] [CrossRef] [Green Version]
- Ishikawa, H.; Nakamura, T.; Kawano, A.; Gondo, N.; Sakai, T. Chemoprevention of colorectal cancer in Japan: a brief introduction to current clinical trials. J. Gastroenterol. 2009, 44, 77–81. [Google Scholar] [CrossRef]
- Burn, J.; Bishop, D.T.; Chapman, P.D.; Elliott, F.; Bertario, L.; Dunlop, M.G.; Eccles, D.; Ellis, A.; Evans, D.G.; Fodde, R.; Maher, E.R.; Möslein, G.; Vasen, H.F.; Coaker, J.; Phillips, R.K.; Bülow, S.; Mathers, J.C. International CAPP consortium. A randomized placebo-controlled prevention trial of aspirin and/or resistant starchin young people with familial adenomatous polyposis. Cancer Prev. Res. 2011, 4, 655–665. [Google Scholar] [CrossRef]
- Chan, A.T. Aspirin and familial adenomatous polyposis: coming full circle. Cancer Prev. Res. 2011, 4, 623–627. [Google Scholar] [CrossRef]
- Burn, J.; Bishop, D.T.; Mecklin, J.P.; Macrae, F.; Möslein, G.; Olschwang, S.; Bisgaard, M.L.; Ramesar, R.; Eccles, D.; Maher, E.R.; et al. CAPP2 Investigators. Effect of aspirin or resistant starch on colorectal neoplasia in the Lynch syndrome. N. Engl. J. Med. 2008, 359, 2567–2578. [Google Scholar] [CrossRef]
- Cooper, K.; Squires, H.; Carroll, C.; Papaioannou, D.; Booth, A.; Logan, R.F.; Maguire, C.; Hind, D.; Tappenden, P. Chemoprevention of colorectal cancer: systematic review and economic evaluation. Health Technol Assess. 2010, 14, 1–206. [Google Scholar]
- Burn, J.; Gerdes, A.M.; Macrae, F.; Mecklin, J.P.; Moeslein, G.; Olschwang, S.; Eccles, D.; Evans, D.G.; Maher, E.R.; Bertario, L.; et al. CAPP2 Investigators. Long-term effect of aspirin on cancer risk in carriers of hereditary colorectal cancer: an analysis from the CAPP2 randomised controlled trial. Lancet 2011, 378, 2081–2087. [Google Scholar]
- Sivak-Sears, N.R.; Schwartzbaum, J.A.; Miike, R.; Moghadassi, M.; Wrensch, M. Case-control study of use of non steroidalantiinflammatory drugs and glioblastomamultiforme. Am. J. Epidemiol. 2004, 159, 1131–1139. [Google Scholar] [CrossRef]
- Amin, R.; Kamitani, H.; Sultana, H.; Taniura, S.; Islam, A.; Sho, A.; Ishibashi, M.; Eling, T.E.; Watanabe, T. Aspirin and indomethacin exhibit anti-proliferative effects and induce apoptosis in T98G human glioblastoma cells. Neurol. Res. 2003, 25, 370–376. [Google Scholar] [CrossRef]
- Rothwell, P.M.; Price, J.F.; Fowkes, F.G.; Zanchetti, A.; Roncaglioni, M.C.; Tognoni, G.; Lee, R.; Belch, J.F.; Wilson, M.; Mehta, Z.; Meade, T.W. Short-term effects of daily aspirin on cancer incidence, mortality, and non-vascular death: analysis of the time course of risks and benefits in 51 randomised controlled trials. Lancet 2012, 379, 1602–1612. [Google Scholar] [CrossRef]
- Rothwell, P.M.; Wilson, M.; Price, J.F.; Belch, J.F.; Meade, T.W.; Mehta, Z. Effect of daily aspirin on risk of cancer metastasis: a study of incident cancers during randomised controlled trials. Lancet 2012, 379, 1591–1601. [Google Scholar]
- Hayden, M.; Pignone, M.; Phillips, C.; Mulrow, C. Aspirin for the primary prevention of cardiovascular events: a summary of the evidence for the US Preventive Services Task Force. Ann. Intern. Med. 2002, 136, 161–172. [Google Scholar]
- McQuaid, K.R.; Laine, L. Systematic review and meta-analysis of adverse events of low-doseaspirin and clopidogrel in randomized controlled trials. Am. J. Med. 2006, 119, 624–638. [Google Scholar] [CrossRef]
- Gorelick, P.B.; Weisman, S.M. Risk of hemorrhagic stroke with aspirin use: an update. Stroke. 2005, 36, 1801–1807. [Google Scholar] [CrossRef]
- Huang, E.S.; Strate, L.L.; Ho, W.W.; Lee, S.S.; Chan, A.T. Long-term use of aspirin and the risk of gastrointestinal bleeding. Am. J. Med. 2011, 124, 426–433. [Google Scholar] [CrossRef]
- Funk, C.D. Prostaglandins and leukotrienes: advances in eicosanoid biology. Science. 2001, 294, 1871–1875. [Google Scholar] [CrossRef]
- Smyth, E.M.; Grosser, T.; Wang, M.; Yu, Y.; FitzGerald, G.A. Prostanoids in health and disease. J. Lipid Res. 2009, 50, S423–428. [Google Scholar]
- Smith, W.L.; DeWitt, D.L.; Garavito, R.M. Cyclooxygenases: structural, cellular, and molecular biology. Annu. Rev. Biochem. 2000, 69, 145–182. [Google Scholar] [CrossRef]
- Kulmacz, R.J.; Lands, W.E. Stoichiometry and kinetics of the interaction of prostaglandin H synthase with anti-inflammatory agents. J Biol. Chem. 1985, 260, 12572–12578. [Google Scholar]
- Yuan, C.; Sidhu, R.S.; Kuklev, D.V.; Kado, Y.; Wada, M.; Song, I.; Smith, W.L. Cyclooxygenase Allosterism, Fatty Acid-mediated Cross-talk between Monomers of Cyclooxygenase Homodimers. J. Biol. Chem. 2009, 284, 10046–10055. [Google Scholar]
- Yuan, C.; Rieke, C.J.; Rimon, G.; Wingerd, B.A.; Smith, W.L. Partnering between monomers of cyclooxygenase-2 homodimers. Proc. Natl. Acad. Sci. USA 2006, 103, 6142–6147. [Google Scholar]
- Patrono, C.; Patrignani, P.; García Rodríguez, L.A. Cyclooxygenase-selective inhibition of prostanoid formation: transducing biochemical selectivity into clinical read-outs. J. Clin. Invest. 2001, 108, 7–13. [Google Scholar]
- Kang, Y.J.; Mbonye, U.R.; DeLong, C.J.; Wada, M.; Smith, W.L. Regulation of intracellular cyclooxygenase levels by gene transcription and protein degradation. Prog. Lipid Res. 2007, 46, 108–125. [Google Scholar] [CrossRef]
- Harper, K.A.; Tyson-Capper, A.J. Complexity of COX-2 gene regulation. Biochem. Soc. Trans. 2008, 36, 543–545. [Google Scholar] [CrossRef]
- Dixon, D.A.; Tolley, N.D.; King, P.H.; Nabors, L.B.; McIntyre, T.M.; Zimmerman, G.A.; Prescott, S.M. Altered expression of the mRNA stability factor HuR promotes cyclooxygenase-2 expression in colon cancer cells. J. Clin. Invest. 2001, 2108, 1657–1665. [Google Scholar]
- Wang, D.; Dubois, R.N. Eicosanoids and cancer. Nat. Rev. Cancer. 2000, 10, 181–193. [Google Scholar]
- Patrono, C.; Baigent, C.; Hirsh, J.; Roth, G. American College of Chest Physicians. Antiplatelet drugs: American College of Chest Physicians Evidence-Based Clinical Practice Guidelines (8th Edition). Chest. 2008, 133, 199–233. [Google Scholar] [CrossRef]
- Sidhu, R.S.; Lee, J.Y.; Yuan, C.; Smith, W.L. Comparison of cyclooxygenase-1 crystal structures: cross-talk between monomers comprising cyclooxygenase-1 homodimers. Biochemistry 2010, 49, 7069–7079. [Google Scholar]
- Rimon, G.; Sidhu, R.S.; Lauver, D.A.; Lee, J.Y.; Sharma, N.P.; Yuan, C.; Frieler, R.A.; Trievel, R.C.; Lucchesi, B.R.; Smith, W.L. Coxibs interfere with the action of aspirin by binding tightly to one monomer of cyclooxygenase-1. Proc. Natl. Acad. Sci. USA 2010, 107, 28–33. [Google Scholar]
- Sharma, N.P.; Dong, L.; Yuan, C.; Noon, K.R.; Smith, W.L. Asymmetric acetylation of the cyclooxygenase-2 homodimer by aspirin and its effects on the oxygenation of arachidonic, eicosapentaenoic, and docosahexaenoic acids. Mol. Pharmacol. 2010, 77, 979–986. [Google Scholar] [CrossRef]
- Serhan, C.N. Lipoxins and aspirin-triggered 15-epi-lipoxins are the first lipid mediators of endogenous anti-inflammation and resolution. Prostagl. Leukot. Essent. Fatty Acids 2005, 73, 141–162. [Google Scholar] [CrossRef]
- Romano, M. Lipid mediators: lipoxin and aspirin-triggered 15-epi-lipoxins. Inflamm. Allergy Drug Targets. 2006, 5, 81–90. [Google Scholar] [CrossRef]
- Janakiram, N.B.; Mohammed, A.; Rao, C.V. Role of lipoxins, resolvins, and other bioactive lipids in colon and pancreatic cancer. Cancer Metastasis Rev. 2011, 30, 507–23. [Google Scholar] [CrossRef]
- Dovizio, M.; Bruno, A.; Tacconelli, S.; Patrignani, P. Mode of action of aspirin as a chemopreventive agent. Recent Results Cancer Res. 2013, 191, 39–65. [Google Scholar] [CrossRef]
- Patrignani, P. Non-steroidal anti-inflammatory drugs, COX-2 and colorectal cancer. Toxicol. Lett. 2000, 112/113, 493–498. [Google Scholar] [CrossRef]
- Warner, T.D.; Giuliano, F.; Vojnovic, I.; Bukasa, A.; Mitchell, J.A.; Vane, J.R. Nonsteroid drug selectivities for cyclo-oxygenase-1 rather than cyclo-oxygenase-2 are associated with human gastrointestinal toxicity: a full in vitro analysis. Proc. Natl. Acad. Sci. USA 1999, 96, 7563–7568. [Google Scholar]
- Ricciotti, E.; Dovizio, M.; Di Francesco, L.; Anzellotti, P.; Salvatore, T.; Di Francesco, A.; Sciulli, M.G.; Pistritto, G.; Monopoli, A.; Patrignani, P. NCX 4040, a nitric oxide-donating aspirin, exerts anti-inflammatory effects through inhibition of I kappa B-alpha degradation in human monocytes. J. Immunol. 2010, 184, 2140–2147. [Google Scholar] [CrossRef]
- Patrignani, P.; Filabozzi, P.; Patrono, C. Selective cumulative inhibition of platelet thromboxane production by low-dose aspirin in healthy subjects. J. Clin. Invest. 1982, 69, 1366–1372. [Google Scholar] [CrossRef]
- Evangelista, V.; Manarini, S.; Di Santo, A.; Capone, M.L.; Ricciotti, E.; Di Francesco, L.; Tacconelli, S.; Sacchetti, A.; D'Angelo, S.; Scilimati, A.; Sciulli, M.G.; Patrignani, P. De novo synthesis of cyclooxygenase-1 counteracts the suppression of platelet thromboxane biosynthesis by aspirin. Circ. Res. 2006, 98, 593–595. [Google Scholar] [CrossRef]
- Pedersen, A.K.; FitzGerald, G.A. Dose-related kinetics of aspirin: presystemic acetylation of platelet cyclo-oxygenase. N. Engl. J. Med. 1984, 311, 1206–1211. [Google Scholar] [CrossRef]
- Capone, M.L.; Tacconelli, S.; Sciulli, M.G.; Grana, M.; Ricciotti, E.; Minuz, P.; Di Gregorio, P.; Merciaro, G.; Patrono, C.; Patrignani, P. Clinical pharmacology of platelet, monocyte, and vascular cyclooxygenase inhibition by naproxen and low-dose aspirin in healthy subjects. Circulation. 2004, 109, 1468–1471. [Google Scholar] [CrossRef]
- Patrono, C.; García Rodríguez, L.A.; Landolfi, R.; Baigent, C. Low-dose aspirin for the prevention of atherothrombosis. N. Engl. J. Med. 2005, 353, 2373–2383. [Google Scholar] [CrossRef]
- FitzGerald, G.A.; Oates, J.A.; Hawiger, J.; Maas, R.L.; Roberts, L.J., 2nd; Lawson, J.A.; Brash, A.R. Endogenous biosynthesis of prostacyclin and thromboxane and platelet function during chronic administration of aspirin in man. J. Clin. Invest. 1983, 71, 676–688. [Google Scholar] [CrossRef]
- Sagar, K.A.; Smyth, M.R. A comparative bioavailability study of different aspirin formulations using on-line multidimensional chromatography. J. Pharm. Biomed. Anal. 1999, 21, 383–392. [Google Scholar] [CrossRef]
- Cox, D.; Maree, A.O.; Dooley, M.; Conroy, R.; Byrne, M.F.; Fitzgerald, D.J. Effect of enteric coating on antiplatelet activity of low-dose aspirin in healthy volunteers. Stroke 2006, 7, 2153–2158. [Google Scholar]
- Burke, A.; Smyth, E.; FitzGerald, G.A. Analgesic-antipyretic agents: pharmacotherapy of gout. In Goodman & Gilman’s the Pharmacological Basis of Therapeutics, 11th; Brunton, L.L., Lazo, J.S., Parker, K.L., Eds.; McGraw-Hill: New York, NY, USA, 2006; pp. 671–715. [Google Scholar]
- Seymour, R.A.; Rawlins, M.D. Efficacy and pharmacokinetics of aspirin in post-operative dental pain. J. Clin. Pharmac. 1982, 13, 807–810. [Google Scholar] [CrossRef]
- Hundal, R.S.; Petersen, K.F.; Mayerson, A.B.; Randhawa, P.S.; Inzucchi, S.; Shoelson, S.E.; Shulman, G.I. Mechanism by which high-dose aspirin improves glucose metabolism in type 2 diabetes. J. Clin. Invest. 2002, 109, 1321–1326. [Google Scholar]
- Day, R.O.; Graham, G.G.; Bieri, D.; Brown, M.; Cairns, D.; Harris, G.; Hounsell, J.; Platt-Hepworth, S.; Reeve, R.; Sambrook, P,N.; Smith, J. Concentration-response relationships for salicylate-induced ototoxicity in normal volunteers. Br. J. Clin. Pharmacol. 1989, 28, 695–702. [Google Scholar] [CrossRef]
- Roberts, M.S.; Rumble, R.H.; Wanwimolruk, S.; Thomas, D.; Brooks, P.M. Pharmacokinetics of aspirin and salicylate in elderly subjects and in patients with alcoholic liver disease. Eur. J. Clin. Pharmacol. 1983, 25, 253–261. [Google Scholar] [CrossRef]
- Capone, M.L.; Tacconelli, S.; Sciulli, M.G.; Anzellotti, P.; Di Francesco, L.; Merciaro, G.; Di Gregorio, P.; Patrignani, P. Human pharmacology of naproxensodium. J. Pharmacol. Exp. Ther. 2007, 322, 453–460. [Google Scholar] [CrossRef]
- García-Rodríguez, L.A.; Tacconelli, S.; Patrignani, P. Role of dose potency in the prediction of risk of myocardial infarction associated with nonsteroidal anti-inflammatory drugs in the general population. J. Am. Coll. Cardiol. 2008, 52, 1628–36. [Google Scholar] [CrossRef]
- Grosser, T.; Fries, S.; FitzGerald, G.A. Biological basis for the cardiovascular consequences of COX-2 inhibition: therapeutic challenges and opportunities. J. Clin. Invest. 2006, 116, 4–15. [Google Scholar]
- FitzGerald, G.A.; Patrono, C. The coxibs, selective inhibitors of cyclooxygenase-2. N. Engl. J. Med. 2001, 345, 433–442. [Google Scholar] [CrossRef]
- Breder, C.D.; Dewitt, D.; Kraig, R.P. Characterization of inducible cyclooxygenase in rat brain. J. Comp. Neurol. 1995, 355, 296–315. [Google Scholar] [CrossRef]
- García Rodríguez, L.A.; Hernández-Díaz, S.; de Abajo, F.J. Association between aspirin and upper gastrointestinal complications: systematic review of epidemiologic studies. Br. J. Clin, Pharmacol. 2001, 52, 563–571. [Google Scholar] [CrossRef]
- García Rodríguez, L.A.; Lin, K.J.; Hernández-Díaz, S.; Johansson, S. Risk of upper gastrointestinal bleeding with low-dose acetylsalicylic acid alone and in combination with clopidogrel and other medications. Circulation 2011, 123, 1108–1115. [Google Scholar] [CrossRef]
- Gay, L.J.; Felding-Habermann, B. Contribution of platelets to tumour metastasis. Nat. Rev. Cancer 2011, 11, 123–134. [Google Scholar] [CrossRef]
- Ulrych, T.; Böhm, A.; Polzin, A.; Daum, G.; Nüsing, R.M.; Geisslinger, G.; Hohlfeld, T.; Schrör, K.; Rauch, B.H. Release of sphingosine-1-phosphate from human platelets is dependent on thromboxane formation. J. Thromb. Haemost. 2011, 9, 790–798. [Google Scholar] [CrossRef]
- Kawamori, T.; Kaneshiro, T.; Okumura, M.; Maalouf, S.; Uflacker, A.; Bielawski, J.; Hannun, Y.A.; Obeid, L.M. Role for sphingosine kinase 1 in colon carcinogenesis. FASEB J. 2009, 23, 405–414. [Google Scholar]
- Ogretmen, B.; Hannun, Y.A. Biologically active sphingolipids in cancer pathogenesis and treatment. Nat. Rev. Cancer. 2004, 4, 604–616. [Google Scholar] [CrossRef]
- Tani, M.; Sano, T.; Ito, M.; Igarashi, Y. Mechanisms of sphingosine and sphingosine 1-phosphate generation in human platelets. J. Lipid Res. 2005, 46, 2458–2467. [Google Scholar] [CrossRef]
- Yatomi, Y. Sphingosine 1-phosphate in vascular biology: possible therapeutic strategies to control vascular diseases. Curr. Pharm. Des. 2006, 12, 575–587. [Google Scholar] [CrossRef]
- Sample, D.; Wargovich, M.; Fischer, S.M.; Inamdar, N.; Schwartz, P.; Wang, X.; Do, K.A.; Sinicrope, F.A. A dose-finding study of aspirin for chemoprevention utilizing rectal mucosal prostaglandin E(2) levels as a biomarker. Cancer Epidemiol. Biomarkers. Prev. 2002, 11, 275–279. [Google Scholar]
- Barnes, C.J.; Hamby-Mason, R.L.; Hardman, W.E.; Cameron, I.L.; Speeg, K.V.; Lee, M. Effect of aspirin on prostaglandin E2 formation and transforming growth factor alpha expression in human rectal mucosa from individuals with a history of adenomatous polyps of the colon. Cancer Epidemiol. Biomarkers Prev. 1999, 8, 311–315. [Google Scholar]
- Hanif, R.; Pittas, A.; Feng, Y.; Koutsos, M.I.; Qiao, L.; Staiano-Coico, L.; Shiff, S.I.; Rigas, B. Effects of nonsteroidal anti-inflammatory drugs on proliferation and on induction of apoptosis in colon cancer cells by a prostaglandin-independent pathway. Biochem. Pharmacol. 1996, 52, 237–425. [Google Scholar]
- Miller, J.R.; Hocking, A.M.; Brown, J.D.; Moon, R.T. Mechanism and function of signal transduction by the Wnt/beta-catenin and Wnt/Ca2+ pathways. Oncogene 1999, 18, 7860–7872. [Google Scholar]
- Polakis, P. The adenomatous polyposis coli (APC) tumor suppressor. Biochim. Biophys. Acta 1997, 1332, F127–147. [Google Scholar]
- Mann, D.J.; Child, E.S.; Swanton, C.; Laman, H.; Jones, N. Modulation of p27(Kip1) levels by the cyclin encoded by Kaposi's sarcoma-associated herpesvirus. EMBO J. 1999, 18, 654–663. [Google Scholar] [CrossRef]
- Bos, C.L.; Kodach, L.L.; van den Brink, G.R.; Diks, S.H.; van Santen, M.M.; Richel, D.J.; Peppelenbosch, M.P.; Hardwick, J.C. Effect of aspirin on the Wnt/beta-catenin pathway is mediated via protein phosphatase 2A. Oncogene 2006, 25, 6447–6456. [Google Scholar]
- Seger, R.; Krebs, E.G. The MAPK signaling cascade. FASEB J. 1995, 9, 726–735. [Google Scholar]
- Pan, M.R.; Chang, H.C. Non-steroidal anti-inflammatory drugs suppress the ERK signaling pathway via block of Ras/c-Raf interaction and activation of MAP kinase phosphatases. Cell. Signal. 2008, 20, 1134–1141. [Google Scholar] [CrossRef]
- Shao, J.; Fujiwara, T.; Kadowaki, Y.; Fukazawa, T.; Waku, T.; Itoshima, T.; Yamatsuji, T.; Nishizaki, M.; Roth, J.A.; Tanaka, N. Overexpression of the wild-type p53 gene inhibits NF-kappaB activity and synergizes with aspirin to induce apoptosis in human colon cancer cells. Oncogene 2000, 19, 726–736. [Google Scholar] [CrossRef]
- Takada, Y.; Singh, S.; Aggarwal, B.B. Identification of a p65 peptide that selectively inhibits NF-kB activation induced by various inflammatory stimuli and its role in down-regulation of NF-κB-mediated gene expression and upregulation of apoptosis. J. Biol. Chem. 2004, 279, 15096–15104. [Google Scholar]
- Chen, Z.; Hagler, J.; Palombella, V.J.; Melandri, F.; Scherer, D.; Ballard, D.; Maniatis, T. Signal-induced site-specific phosphorylation targets I kappa B alpha to the ubiquitin-proteasome pathway. Genes Dev. 1995, 9, 1586–1597. [Google Scholar] [CrossRef]
- Yin, M.J.; Yamamoto, Y.; Gaynor, R.B. The anti-inflammatory agents aspirin and salicylate inhibit the activity of I(kappa)B kinase-beta. Nature 1998, 396, 77–80. [Google Scholar] [CrossRef]
- Kopp, E.; Ghosh, S. Inhibition of NF-kappa B by sodium salicylate and aspirin. Science 1994, 265, 956–959. [Google Scholar]
- Grilli, M.; Pizzi, M.; Memo, M.; Spano, P. Neuroprotection by aspirin and sodium salicylate through blockade of NF-kB activation. Science 1996, 274, 1383–1385. [Google Scholar] [CrossRef]
- Hardie, D.G. AMP-activated protein kinase: an energy sensor that regulates all aspects of cellfunction. Genes Dev. 2011, 25, 1895–908. [Google Scholar] [CrossRef]
- Luo, Z.; Zang, M.; Guo, W. AMPK as a metabolic tumor suppressor: control of metabolism and cell growth. Future Oncol. 2010, 6, 457–470. [Google Scholar] [CrossRef]
- Luo, Z.; Saha, A.K.; Xiang, X.; Ruderman, N.B. AMPK, the metabolic syndrome and cancer. Trends Pharmacol. Sci. 2005, 26, 69–76. [Google Scholar] [CrossRef]
- Wullschleger, S.; Loewith, R.; Hall, M.N. TOR signaling in growth and metabolism. Cell. 2006, 124, 471–484. [Google Scholar] [CrossRef]
- Din, F.V.; Valanciute, A.; Houde, V.P.; Zibrova, D.; Green, K.A.; Sakamoto, K.; Alessi, D.R.; Dunlop, M.G. Aspirin inhibits mTOR signaling, activates AMP-activated protein kinase, and induces autophagy in colorectal cancer cells. Gastroenterology 2012, 142, 1504–15.e3. [Google Scholar] [CrossRef]
- Hawley, S.A.; Fullerton, M.D.; Ross, F.A.; Schertzer, J.D.; Chevtzoff, C.; Walker, K.J.; Peggie, M.W.; Zibrova, D.; Green, K.A.; Mustard, K.J.; Kemp, B.E.; Sakamoto, K.; Steinberg, G.R.; Hardie, D.G. The ancient drug salicylate directly activates AMP-activated protein kinase. Science. 2012, 336, 918–922. [Google Scholar]
- Pincard, R.N.; Hawkins, D.; Farr, R.S. in vitro acetylation of plasma proteins, enzymes and DNA by aspirin. Nature 1968, 219, 68–69. [Google Scholar] [CrossRef]
- Alfonso, L.F.; Srivenugopal, K.S.; Bhat, G.J. Does aspirin acetylate multiple cellular proteins? Mol. Med. Report 2009, 2, 533–537. [Google Scholar]
- Alfonso, L.F.; Srivenugopal, K.S.; Arumugam, T.V.; Abbruscato, T.J.; Weidanz, J.A.; Bhat, G.J. Aspirin inhibits camptothecin-induced p21CI P1 levels and potentiates apoptosis in human breast cancer cells. Int. J. Oncol. 2009, 34, 597–608. [Google Scholar]
- Harris, S.L.; Levine, A.J. The p53 pathway: positive and negative feedback loops. Oncogene 2005, 24, 2899–2908. [Google Scholar] [CrossRef]
- Marimuthu, S.; Chivukula, R.S.; Alfonso, L.F.; Moridani, M.; Hagen, F.K.; Bhat, G.J. Aspirin acetylates multiple cellular proteins in HCT-116 colon cancer cells: Identification of novel targets. Int. J. Oncol. 2011, 39, 1273–1283. [Google Scholar]
- Wang, D.; DuBois, R.N. Pro-inflammatory prostaglandins and progression of colorectal cancer. Cancer Lett. 2008, 267, 197–203. [Google Scholar] [CrossRef]
- Dixon, D.A.; Blanco, F.F.; Bruno, A.; Patrignani, P. Mechanistic aspects of COX-2 expression in colorectal neoplasia. Recent Results Cancer Res. 2013, 191, 7–37. [Google Scholar] [CrossRef]
- Chulada, P.C.; Thompson, M.B.; Mahler, J.F.; Doyle, C.M.; Gaul, B.W.; Lee, C.; Tiano, H.F.; Morham, S.G.; Smithies, O.; Langenbach, R. Genetic disruption of Ptgs-1, as well as Ptgs-2, reduces intestinal tumorigenesis in Min mice. Cancer Res. 2000, 60, 4705–4758. [Google Scholar]
- Oshima, M.; Dinchuk, J.E.; Kargman, S.L.; Oshima, H.; Hancock, B.; Kwong, E.; Trzaskos, J.M.; Evans, J.F.; Taketo, M.M. Suppression of intestinal polyposis in Apc delta716 knockout mice by inhibition of cyclooxygenase 2 (COX-2). Cell 1996, 87, 803–809. [Google Scholar] [CrossRef]
- Liao, X.; Lochhead, P.; Nishihara, R.; Morikawa, T.; Kuchiba, A.; Yamauchi, M.; Imamura, Y.; Qian, Z.R.; Baba, Y.; Shima, K.; Sun, R.; Nosho, K.; Meyerhardt, J.A.; Giovannucci, E.; Fuchs, C.S.; Chan, A.; Ogino, S. Aspirin use, tumor PIK3CA mutation, and colorectal-cancer survival. N Engl. J. Med. 2012, 367, 1596–1606. [Google Scholar] [CrossRef]
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Dovizio, M.; Tacconelli, S.; Sostres, C.; Ricciotti, E.; Patrignani, P. Mechanistic and Pharmacological Issues of Aspirin as an Anticancer Agent. Pharmaceuticals 2012, 5, 1346-1371. https://doi.org/10.3390/ph5121346
Dovizio M, Tacconelli S, Sostres C, Ricciotti E, Patrignani P. Mechanistic and Pharmacological Issues of Aspirin as an Anticancer Agent. Pharmaceuticals. 2012; 5(12):1346-1371. https://doi.org/10.3390/ph5121346
Chicago/Turabian StyleDovizio, Melania, Stefania Tacconelli, Carlos Sostres, Emanuela Ricciotti, and Paola Patrignani. 2012. "Mechanistic and Pharmacological Issues of Aspirin as an Anticancer Agent" Pharmaceuticals 5, no. 12: 1346-1371. https://doi.org/10.3390/ph5121346