Cyclooxygenase (COX) Inhibitors and the Newborn Kidney

{kind=link}
{kind=link}
Abstract
:1. Introduction
2. COX Isoforms
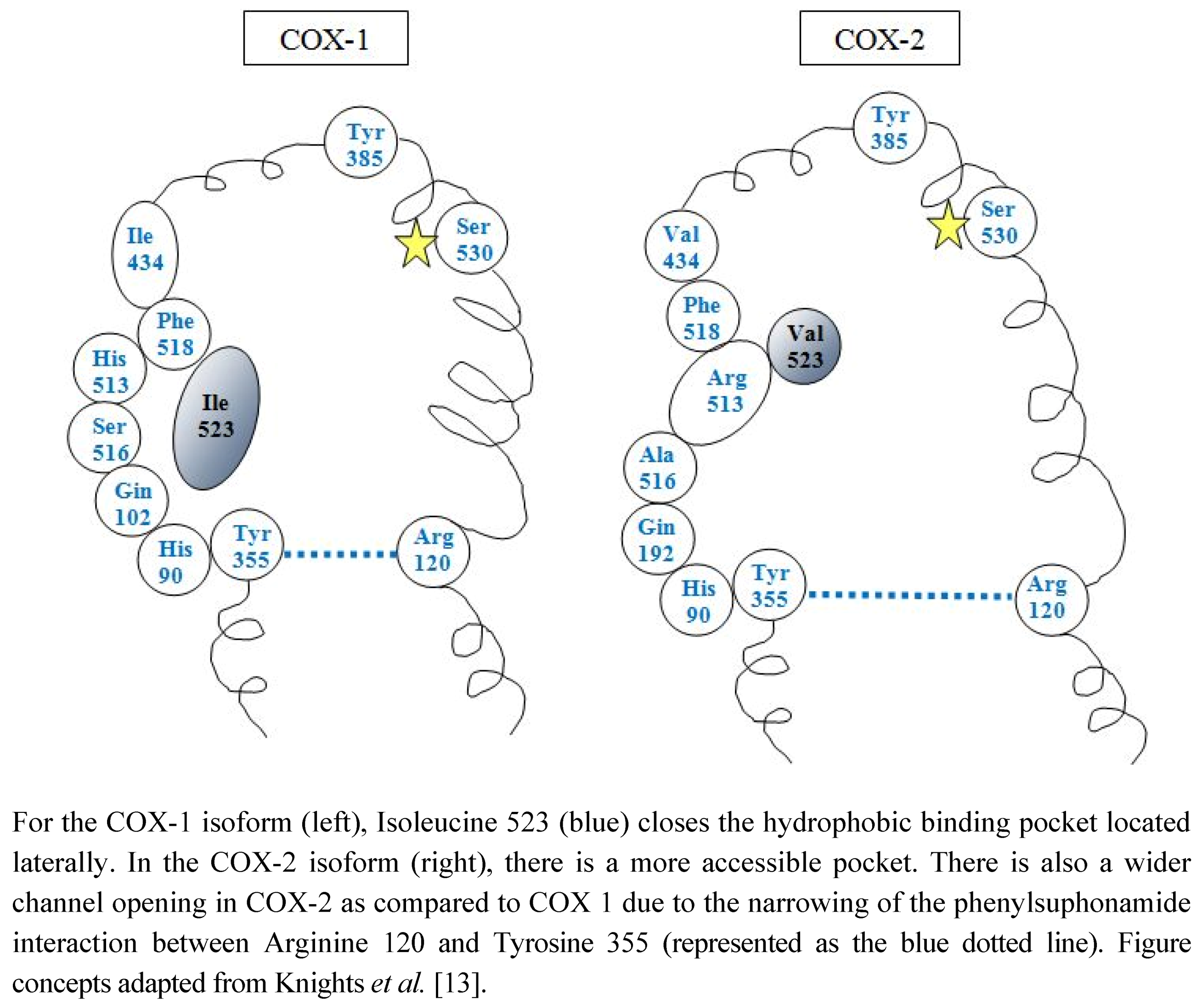
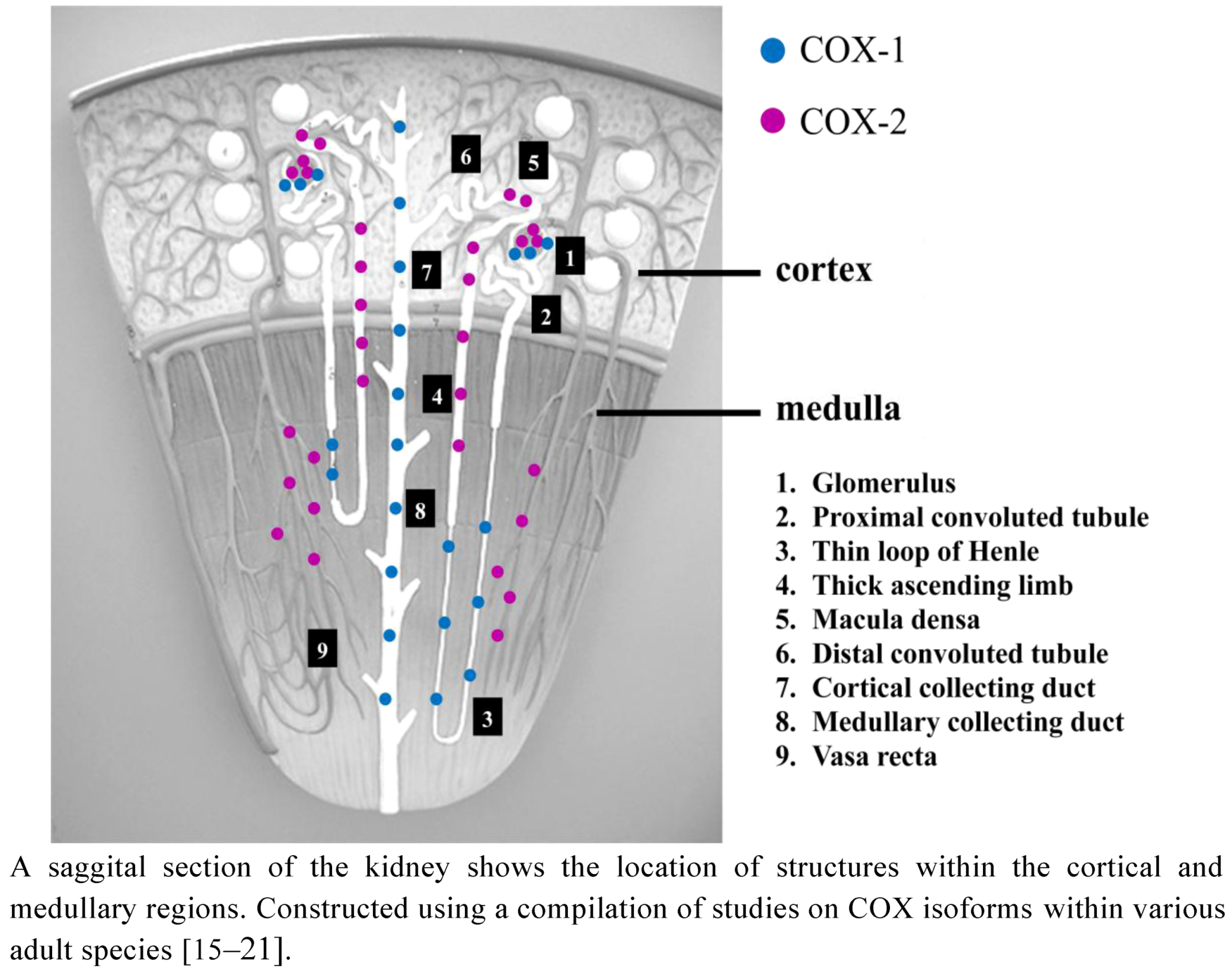
2.1. COX Distribution in the Adult Kidney
2.2. COX Distribution in the Developing Kidney
3. The Role of COX Isoforms in the Developing Kidney
3.1. COXI During Pregnancy: Effects on Fetal and Newborn Renal Function
3.2. COXI in the Newborn Period
3.3. Kidney Effects of Targeted Gene Disruption for COX
3.4. COXI during Postnatal Nephronogenesis
3.5. Physiological Effects of COXI on the Newborn Kidney
4. Summary
5. Conclusions
Acknowledgments
References
- Dubois, R.N.; Abramson, S.B.; Crofford, L.; Gupta, R.A.; Simon, L.S.; van de Putte, L.B.; Lipsky, P.E. Cyclooxygenase in biology and disease. FASEB J. 1998, 12, 1063–1073. [Google Scholar]
- Vane, J.R.; Bakhle, Y.S.; Botting, R.M. Cyclooxygenases 1 and 2. Annu. Rev. Pharmacol. Toxicol. 1998, 38, 97–120. [Google Scholar] [CrossRef]
- Narumiya, S.; Sugimoto, Y.; Ushikubi, F. Prostanoid receptors: Structures, properties, and functions. Physiol. Rev. 1999, 79, 1193–1226. [Google Scholar]
- Breyer, M.D.; Breyer, R.M. Prostaglandin E receptors and the kidney. Am. J. Physiol. Renal Physiol. 2000, 279, F12–F23. [Google Scholar]
- Breyer, M.D.; Breyer, R.M. Prostaglandin receptors: Their role in regulating renal function. Curr. Opin. Nephrol. Hypertens. 2000, 9, 23–29. [Google Scholar] [CrossRef]
- Funk, C.D. Prostaglandins and leukotrienes: Advances in eicosanoid biology. Science 2001, 294, 1871–1875. [Google Scholar] [CrossRef]
- Bonvalet, J.P.; Pradelles, P.; Farman, N. Segmental synthesis and actions of prostaglandins along the nephron. Am. J. Physiol. 1987, 253, F377–F387. [Google Scholar]
- Purdy, K.E.; Arendshorst, W.J. Calcium dependent synthesis of vasodilator renal microvascular prostanoids. Am. J. Nephrol. 1999, 277, F850–F858. [Google Scholar]
- Navar, L.G.; Inscho, E.W.; Majid, D.S.A.; Imag, J.D.; Harrison-Bernard, L.M.; Mitchell, K.D. Paracrine regulation of the renal microcirculation. Physiol. Rev. 1996, 76, 425–536. [Google Scholar]
- Harris, R.C. An update on cyclooxygenase-2 expression and metabolites in the kidney. Curr. Opin. Nephrol. Hypertens. 2008, 17, 64–69. [Google Scholar] [CrossRef]
- Chandrasekharan, N.V.; Dai, H.; Roos, K.L.; Evanson, N.K.; Tomsik, J.; Elton, T.S.; Simmons, D.L. COX-3, a cyclooxygenase-1 variant inhibited by acetaminophen and other analgesic/antipyretic drugs: cloning, structure, and expression. Proc. Natl. Acad. Sci. USA 2002, 99, 13926–13931. [Google Scholar]
- Hersh, E.V.; Lally, E.T.; Moore, P.A. Update on cyclooxygenase inhibitors: Has a third COX isoform entered the fray? Curr. Med. Res. Opin. 2005, 21, 1217–1226. [Google Scholar] [CrossRef]
- Knights, K.M.; Mangoni, A.A.; Miners, J.O. Defining the COX inhibitor selectivity of NSAIDs: Implications for understanding toxicity. Expert Rev. Clin. Pharmacol. 2010, 3, 769–776. [Google Scholar] [CrossRef]
- Smith, W.L.; Bell, T.G. Immunohistochemical localization of the prostaglandin-forming cyclooxygenase in renal cortex. Am. J. Physiol. 1978, 235, F451–F457. [Google Scholar]
- Nantel, F.; Meadows, E.; Denis, D.; Connolly, B.; Metters, K.M.; Giaid, A. Immunolocalization of cyclooxygenase-2 in the macula densa of human elderly. FEBS Lett. 1999, 457, 475–477. [Google Scholar] [CrossRef]
- Guan, Y.; Chang, M.; Cho, W.; Zhang, Y.; Redha, R.; Davis, L.; Chang, S.; Dubois, R.N.; Hao, C.M.; Breyer, M. Cloning, expression, and regulation of rabbit cyclooxygenase-2 in renal medullary interstitial cells. Am. J. Physiol. 1997, 273, F18–F26. [Google Scholar]
- Komhoff, M.; Grone, H.J.; Klein, T.; Seyberth, H.W.; Nusing, R.M. Localization of cyclooxygenase-1 and -2 in adult and fetal human kidney: implication for renal function. Am. J. Physiol. 1997, 272, F460–F468. [Google Scholar]
- Harris, R.C. Cyclooxygenase-2 in the kidney. J. Am. Soc. Nephrol. 2000, 11, 2387–2394. [Google Scholar]
- Adegboyega, P.A.; Ololade, O. Immunohistochemical expression of cyclooxygenase-2 in normal kidneys. Appl. Immunohistochem. Mol. Morphol. 2004, 12, 71–74. [Google Scholar]
- Therland, K.L.; Stubbe, J.; Thiesson, H.C.; Ottosen, P.D.; Walter, S.; Sorensen, G.L.; Skott, O.; Jensen, B.L. Cycloxygenase-2 is expressed in vasculature of normal and ischemic adult human kidney and is colocalized with vascular prostaglandin E2 EP4 receptors. J. Am. Soc. Nephrol. 2004, 15, 1189–1198. [Google Scholar] [CrossRef]
- Khan, K.N.; Venturini, C.M.; Bunch, R.T.; Brassard, J.A.; Koki, A.T.; Morris, D.L.; Trump, B.F.; Maziasz, T.J.; Alden, C.L. Interspecies differences in renal localization of cyclooxygenase isoforms: implications in nonsteroidal antiinflammatory drug-related nephrotoxicity. Toxicol. Pathol. 1998, 26, 612–620. [Google Scholar]
- Stubbe, J.; Jensen, B.L.; Bachmann, S.; Morsing, P.; Skott, O. Cyclooxygenase-2 contributes to elevated renin in the early postnatal period in rats. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2003, 284, R1179–R1189. [Google Scholar]
- Zhang, M.Z.; Wang, J.L.; Cheng, H.F.; Harris, R.C.; McKanna, J.A. Cyclooxygenase-2 in rat nephron development. Am. J. Physiol. Renal. Physiol. 1997, 273, F994–F1002. [Google Scholar]
- Khan, K.N.M.; Paulson, S.K.; Verburg, K.M.; Lefkowith, J.B.; Maziasz, T.J. Pharmacology of cyclooxygenase-2 inhibition in the kidney. Kidney Int. 2002, 61, 1210–1219. [Google Scholar] [CrossRef]
- Khan, K.N.; Stanfield, K.M.; Dannenberg, A.; Seshan, S.V.; Baergen, R.N.; Baron, D.A.; Soslow, R.A. Cyclooxygenase-2 expression in the developing human kidney. Pediatr. Dev. Pathol. 2001, 4, 461–466. [Google Scholar] [CrossRef]
- Zuckerman, H.; Reiss, M.; Atad, J.; Rubinstein, I. Indomethacin as an inhibitor of premature labor. Harefuah 1975, 89, 201–203. [Google Scholar]
- Moise, K.J., Jr.; Ou, C.N.; Kirshon, B.; Cano, L.E.; Rognerud, C.; Carpenter, R.J., Jr. Placental transfer of indomethacin in the human pregnancy. Am. J. Obstet. Gynecol. 1990, 162, 549–554. [Google Scholar]
- Kirshon, B.; Moise, K.J., Jr.; Wasserstrum, N.; Ou, C.N.; Huhta, J.C. Influence of short-term indomethacin therapy on fetal urine output. Obstet. Gynecol. 1988, 72, 51–53. [Google Scholar]
- Butler-ÓHara, M.; D'Angio, C.T. Risk of persistent renal insufficiency in premature infants following the prenatal use of indomethacin for suppression of preterm labor. J. Perinatol. 2002, 22, 541–546. [Google Scholar] [CrossRef]
- Bernstein, J.; Werner, A.L.; Verani, R. Nonsteroidal anti-inflammatory drug fetal nephrotoxicity. Pediatr. Dev. Pathol. 1998, 1, 153–156. [Google Scholar] [CrossRef]
- Antonucci, R.; Zaffanello, M.; Puxeddu, E.; Porcella, A.; Cuzzolin, L.; Pilloni, M.D.; Fanos, V. Use of non-steroidal anti-inflammatory drugs in pregnancy: Impact on the fetus and newborn. Curr. Drug Metab. 2012, 13, 474–490. [Google Scholar] [CrossRef]
- Zaffanello, M.; Bassareo, P.P.; Cataldi, L.; Antonucci, R.; Biban, P.; Fanos, V. Long-term effects of neonatal drugs on the kidney. J. Matern. Fetal Neonatal. Med. 2010, 23, 87–89. [Google Scholar] [CrossRef]
- Cuzzolin, L.; Fanos, V.; Pinna, B.; di Marzio, M.M.; Perin, M.; Tramontozzi, P.; Tonetto, P.; Cataldi, L. Postnatal renal function in preterm newborns: A role of diseases, drugs and therapeutic interventions. Pediatr. Nephrol. 2006, 21, 931–938. [Google Scholar]
- Groom, K.M.; Shennan, A.H.; Jones, B.A.; Seed, P.; Bennett, P.R. TOCOX-A randomised, double-blind, placebo-controlled trial of rofecoxib a COX-2-specific prostaglandin inhibitor. for the prevention of preterm delivery in women at high risk. Int. J. Obstet. Gynaecol. 2005, 112, 725–730. [Google Scholar] [CrossRef]
- Van Hecken, A.; Schwartz, J.I.; Depre, M.; De Lepeleire, I.; Dallob, A.; Tanaka, W.; Wynants, K.; Buntinx, A.; Arnout, J.; Wong, P.H.; et al. Comparative inhibitory activity of rofecoxib, meloxicam, diclofenac, ibuprofen, and naproxen on COX-2 versus COX-1 in healthy volunteers. J. Clin. Pharmacol. 2000, 40, 1109–1120. [Google Scholar]
- Harris, W.H.; Van Petten, G.R. Placental transfer of indomethacin in the rabbit and sheep. Can. J. Physiol. Pharmacol. 1981, 59, 342–346. [Google Scholar] [CrossRef]
- Chimura, T.; Fujimori, K. The effect of indomethacin on placental permeability and the fetal lung circulation. Nippon Sanka Fujinka Gakkai Zasshi 1983, 35, 425–430. [Google Scholar]
- Stevenson, K.M.; Lumbers, E.R. Effects of indomethacin on fetal renal function, renal and umbilicoplacental blood flow and lung liquid production. J. Dev. Physiol. 1992, 17, 257–264. [Google Scholar]
- Matson, J.R.; Stokesm, J.B.; Robillard, J.E. Effects of inhibition of prostaglandin synthesis on fetal renal function. Kidney Int. 1981, 20, 621–627. [Google Scholar] [CrossRef]
- Rac, V.E.; Small, C.; Scott, C.A.; Adamson, S.L.; Rurak, D.; Challis, J.R.; Lye, S.J. Meloxicam effectively inhibits preterm labor uterine contractions in a chronically catheterized pregnant sheep model: Impact on fetal blood flow and fetal-maternal physiologic parameters. Am. J. Obstet. Gynecol. 2006, 195, 528–534. [Google Scholar] [CrossRef]
- Walker, M.P.R.; Moore, T.R.; Cheung, C.Y.; Brace, R.A. Indomethacin-induced urinary flow rate reduction in the ovine fetus is associated with reduced free water clearance and elevated plasma arginine vasopressin levels. Am. J. Obstet. Gynecol. 1992, 167, 1723–1731. [Google Scholar]
- Walker, M.P.R.; Moore, T.R.; Brace, R.A. Indomethacin and arginine vasopressin interaction in the fetal kidney: A mechanism of oliguria. Am. J. Obstet. Gynecol. 1994, 171, 1234–1241. [Google Scholar]
- Saez, F.; Reverte, V.; Salazar, F.; Castells, M.T.; Llinas, M.T.; Salazar, F.J. Hypertension and sex differences in the age-related renal changes when cyclooxygenase-2 activity is reduced during nephrogenesis. Hypertension 2009, 53, 331–337. [Google Scholar]
- Komhoff, M.; Wang, J.L.; Cheng, H.F.; Langenbach, R.; McKanna, J.A.; Harris, R.C.; Breyer, M.D. Cyclooxygenase-2-selective inhibitors impair glomerulogenesis and renal cortical development. Kidney Int. 2000, 57, 414–422. [Google Scholar]
- Langenbach, R.; Loftin, C.; Lee, C.; Tiano, H. Cyclooxygenase knockout mice: Models for elucidating isoform-specific functions. Biochem. Pharmacol. 1999, 58, 1237–1246. [Google Scholar] [CrossRef]
- Loftin, C.D.; Trivedi, D.B.; Langenbach, R. Cyclooxygenase-1-selective inhibition prolongs gestation in mice without adverse effects on the ductus arteriosus. J. Clin. Invest. 2002, 110, 549–557. [Google Scholar]
- Athirakul, K.; Kim, H.S.; Audoly, L.P.; Smithies, O.; Coffman, T.M. Deficiency of COX-1 causes natriuresis and enhanced sensitivity to ACE inhibition. Kidney Int. 2001, 60, 2324–2329. [Google Scholar] [CrossRef]
- Kawada, N.; Solis, G.; Ivey, N.; Connors, S.; Dennehy, K.; Modlinger, P.; Hamel, R.; Kawada, J.T.; Imai, E.; Langenbach, R.; et al. Cyclooxygenase-1-deficient mice have high sleep-to-wake blood pressure ratios and renal vasoconstriction. Hypertension 2005, 45, 1131–1138. [Google Scholar] [CrossRef]
- Morham, S.G.; Langenbach, R.; Loftin, C.D.; Tiano, H.F.; Vouloumanos, N.; Jennette, J.C.; Mahler, J.F.; Kluckman, K.D.; Ledford, A.; Lee, C.A.; et al. Prostaglandin synthase 2 gene disruption causes severe renal pathology in the mouse. Cell 1995, 83, 473–482. [Google Scholar] [CrossRef]
- Norwood, V.F.; Morham, S.G.; Smithies, O. Postnatal development and progression of renal dysplasia in cyclooxygenase-2 null mice. Kidney Int. 2000, 58, 2291–2300. [Google Scholar] [CrossRef]
- Dinchuk, J.E.; Car, B.D.; Focht, R.J.; Johnston, J.J.; Jaffee, B.D.; Covington, M.B.; Contel, N.R.; Eng, V.M.; Collins, R.J.; Czerniak, P.M. Renal abnormalities and an altered inflammatory response in mice lacking cyclooxygenase II. Nature 1995, 378, 406–409. [Google Scholar] [CrossRef]
- Yang, T.; Huang, Y.G.; Ye, W.; Hansen, P.; Schnermann, J.B.; Briggs, J.P. Influence of genetic background and gender on hypertension and renal failure in COX-2-deficient mice. Am. J. Physiol. Renal Physiol. 2005, 288, F1125–F1132. [Google Scholar] [CrossRef]
- Seta, F.; Chung, A.D.; Turner, P.V.; Mewburn, J.D.; Yu, Y.; Funk, C.D. Renal and cardiovascular characterization of COX-2 knockdown mice. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2009, 296, R1751–R1760. [Google Scholar] [CrossRef]
- Hasan, J.; Beharry, K.D.; Gharraee, Z.; Stavitsky, Y.; bad-Santos, P.; bad-Santos, M.; Aranda, J.V.; Modanlou, H.D. Early postnatal ibuprofen and indomethacin effects in suckling and weanling rat kidneys. Prostaglandins Other Lipid Mediat. 2008, 85, 81–88. [Google Scholar] [CrossRef]
- Mitchell, J.A.; Akarasereenont, P.; Thiemermann, C.; Flower, R.J.; Vane, J.R. Selectivity of nonsteroidal antiinflammatory drugs as inhibitors of constitutive and inducible cyclooxygenase. Proc. Natl. Acad. Sci. USA 1993, 90, 11693–11697. [Google Scholar] [CrossRef]
- Chandrasekharan, N.V.; Simmons, D.L. The cyclooxygenases. Genome Biol. 2004, 5, 241. [Google Scholar] [CrossRef]
- Olliges, A.; Wimmer, S.; Nusing, R.M. Defects in mouse nephrogenesis induced by selective and non-selective cyclooxygenase-2 inhibitors. Br. J. Pharmacol. 2011, 163, 927–936. [Google Scholar] [CrossRef]
- Kent, A.L.; Douglas-Denton, R.; Shadbolt, B.; Dahlstrom, J.E.; Maxwell, L.E.; Koina, M.E.; Falk, M.C.; Willenborg, D.; Bertram, J.F. Indomethacin, ibuprofen and gentamicin administered during late stages of glomerulogenesis do not reduce glomerular number at 14 days of age in the neonatal rat. Pediatr. Nephrol. 2009, 24, 1143–1149. [Google Scholar] [CrossRef]
- Heymann, M.A.; Rudolph, A.M.; Silverman, N.H. Closure of the ductus arteriosus in premature infants by inhibition of prostaglandin synthesis. N. Engl. J. Med. 1976, 295, 530–533. [Google Scholar] [CrossRef]
- Antonucci, R.; Fanos, V. NSAIDs, prostaglandins and the neonatal kidney. J. Matern. Fetal. Neonatal. Med. 2009, 22, 23–26. [Google Scholar] [CrossRef]
- Guignard, J.-P. The adverse renal effects of prostaglandin-synthesis inhibitors in the newborn rabbit. Semin. Perinatol. 2002, 26, 398–405. [Google Scholar] [CrossRef]
- Pezzati, M.; Vangi, V.; Biagiotti, R.; Bertini, G.; Cianciulli, D.; Rubaltelli, F.F. Effects of indomethacin and ibuprofen on mesenteric and renal blood flow in preterm infants with patent ductus arteriosus. J. Pediatr. 1999, 135, 733–738. [Google Scholar] [CrossRef]
- Rodriguez, M.M.; Gomez, A.H.; Abitbol, C.L.; Chandar, J.J.; Duara, S.; Zilleruelo, G.E. Histomorphometric analysis of postnatal glomerulogenesis in extremely preterm infants. Pediatr. Dev. Pathol. 2004, 7, 17–25. [Google Scholar]
- Vieux, R.; Desandes, R.; Boubred, F.; Semama, D.; Guillemin, F.; Buchweiller, M.C. Ibuprofen in very preterm infants impairs renal function for the first month of life. Pediatr. Nephrol. 2010, 25, 267–274. [Google Scholar] [CrossRef]
- Allegaert, K.; Vanhole, C.; de Hoon, J.; Guignard, J.P.; Tibboel, D.; Devlieger, H.; Van Overmeire, B. Nonselective cyclo-oxygenase inhibitors and glomerular filtration rate in preterm neonates. Pediatr. Nephrol. 2005, 20, 1557–1561. [Google Scholar] [CrossRef]
- Friedman, Z.; Demers, L.M. Prostaglandin synthetase in the human neonatal kidney. Pediatr. Res. 1979, 14, 190–193. [Google Scholar] [CrossRef]
- Herin, P.; Aperia, A. The effect of prostaglandin inhibition on renal function in the developing anesthetized lamb. Acta Physiol. Scand. 1982, 114, 75–79. [Google Scholar] [CrossRef]
- Winther, J.B.; Hoskins, E.; Printz, M.P.; Mendoza, S.A.; Kirkpatrick, S.E.; Friedman, W.F. Influence of indomethacin on renal function in conscious newborn lambs. Biol. Neonate 1980, 38, 76–84. [Google Scholar] [CrossRef]
- Osborn, J.L.; Hook, J.B.; Bailie, M.D. Effect of saralasin and indomethacin on renal function in developing piglets. Am. J. Physiol. Regul. Integr. Comp. Physiol. 1980, 238, R438–R442. [Google Scholar]
- Chamaa, N.S.; Mosig, D.; Drukker, A.; Guignard, J.P. The renal hemodynamic effects of ibuprofen in the newborn rabbit. Pediatr. Res. 2000, 48, 600–605. [Google Scholar] [CrossRef]
- Drukker, A.; Mosig, D.; Guignard, J.P. The renal hemodynamic effects of aspirin in newborn and young adult rabbits. Pediatr. Nephrol. 2001, 16, 713–718. [Google Scholar] [CrossRef]
- Prevot, A.; Mosig, D.; Martini, S.; Guignard, J.P. Nimesulide, a cyclooxygenase-2 preferential inhibitor, impairs renal function in the newborn rabbit. Pediatr. Res. 2004, 55, 254–260. [Google Scholar] [CrossRef]
- Ebenezar, K.K.; Ghane, F.S.; Smith, F.G. Effects of indomethacin on systemic and renal haemodynamics in conscious lambs. Exp. Physiol. 2007, 92, 575–581. [Google Scholar] [CrossRef]
- Sener, A.; Smith, F.G. Renal hemodynamic effects of L-NAME during postnatal maturation in conscious lambs. Pediatr. Nephrol. 2001, 16, 868–873. [Google Scholar] [CrossRef]
- Sener, A.; Smith, F.G. Dose dependent effects of nitric oxide synthase inhibition on systemic and renal haemodynamics in conscious lambs. Can. J. Physiol. Pharmacol. 1999, 77, 1–7. [Google Scholar] [CrossRef]
- Sener, A.; Smith, F.G. Glomerular and tubular responses to NG-nitro-L-arginine methyl ester are age dependent in conscious lambs. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2002, 282, R1512–R1520. [Google Scholar]
- Ebenezar, K.K.; Ghane, F.S.; Qi, W.; Smith, F.G. Do prostaglandins modulate renal haemodynamic effects of endothelin-1in conscious lambs? Can. J. Physiol. Pharmacol. 2010, 88, 161–167. [Google Scholar] [CrossRef]
- Ebenezar, K.K.; Wong, A.K.O.; Smith, F.G. Haemodynamic responses to angiotensin II in conscious lambs: Role of nitric oxide and prostaglandins. Pflügers Arch. Eur. J. Physiol. 2012, 463, 399–404. [Google Scholar]
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Smith, F.G.; Wade, A.W.; Lewis, M.L.; Qi, W. Cyclooxygenase (COX) Inhibitors and the Newborn Kidney. Pharmaceuticals 2012, 5, 1160-1176. https://doi.org/10.3390/ph5111160
Smith FG, Wade AW, Lewis ML, Qi W. Cyclooxygenase (COX) Inhibitors and the Newborn Kidney. Pharmaceuticals. 2012; 5(11):1160-1176. https://doi.org/10.3390/ph5111160
Chicago/Turabian StyleSmith, Francine G., Andrew W. Wade, Megan L. Lewis, and Wei Qi. 2012. "Cyclooxygenase (COX) Inhibitors and the Newborn Kidney" Pharmaceuticals 5, no. 11: 1160-1176. https://doi.org/10.3390/ph5111160
APA StyleSmith, F. G., Wade, A. W., Lewis, M. L., & Qi, W. (2012). Cyclooxygenase (COX) Inhibitors and the Newborn Kidney. Pharmaceuticals, 5(11), 1160-1176. https://doi.org/10.3390/ph5111160