Mitochondria-Targeted Antioxidant SS31 Prevents Amyloid Beta-Induced Mitochondrial Abnormalities and Synaptic Degeneration in Alzheimer’s Disease

{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Alzheimer’s Disease Etiology Involves Mitochondrial Dysfunction
3. Mitochondrial Function Is Tightly Coupled to Synaptic Activity
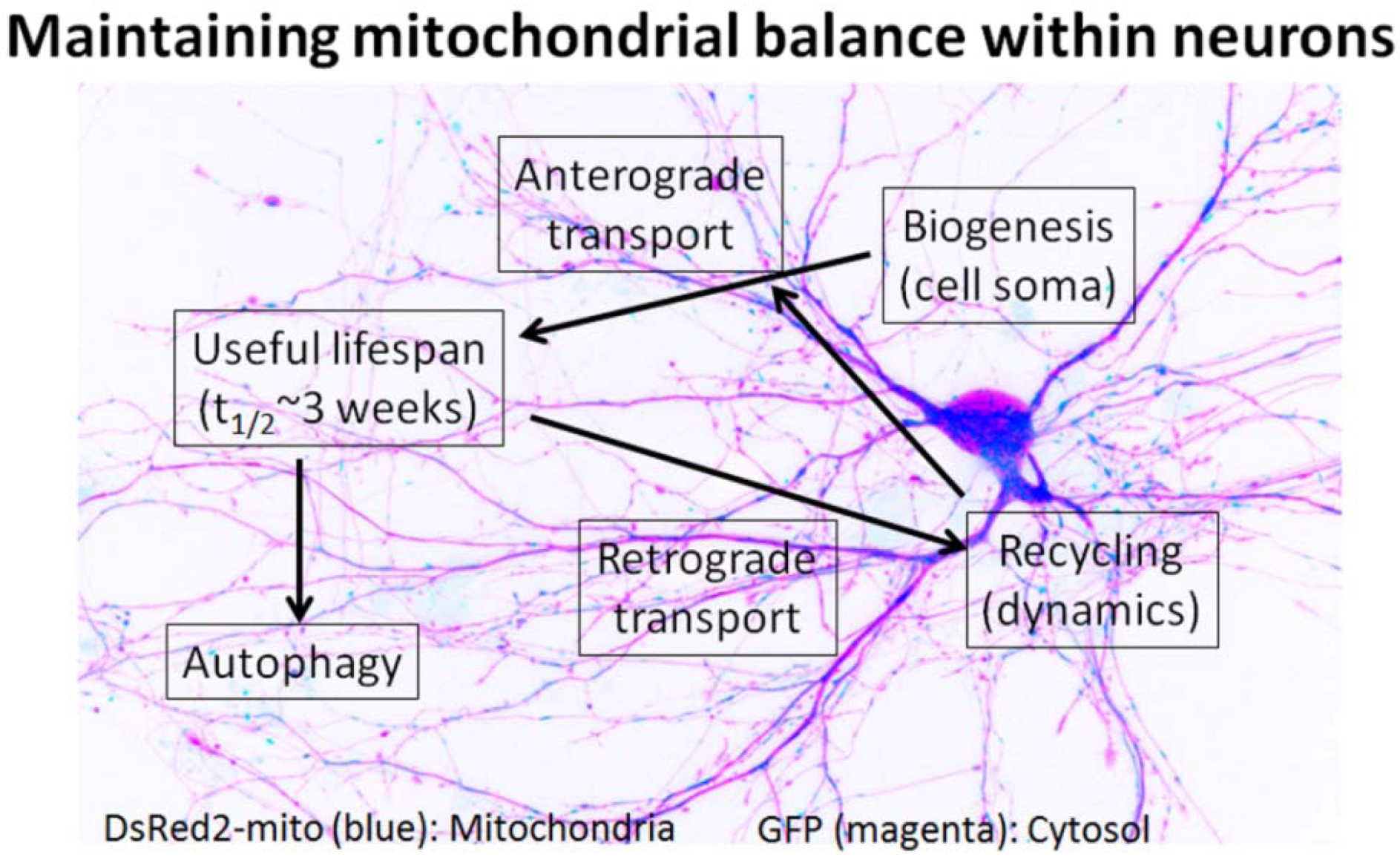
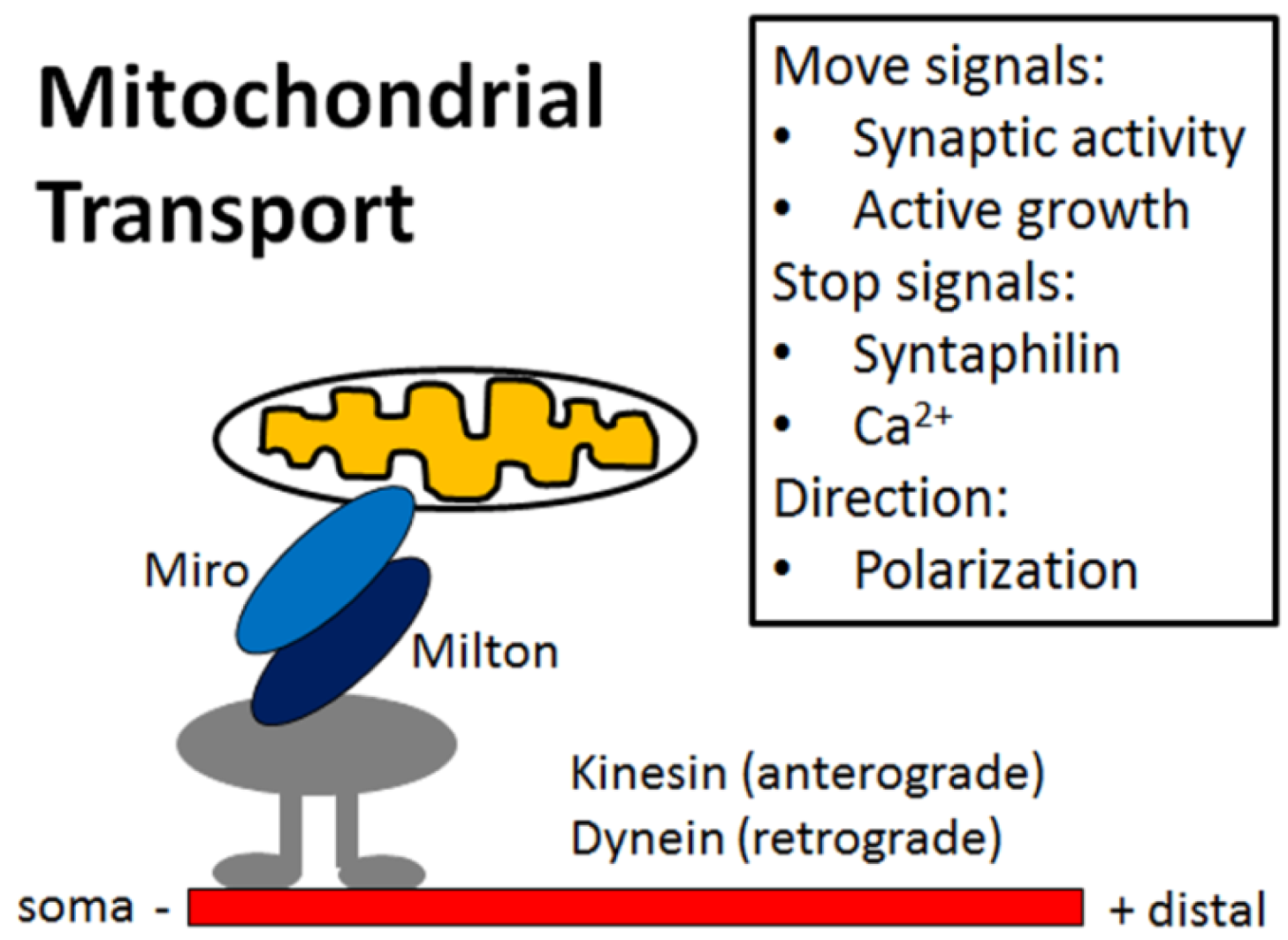
4. Mitochondria Are Highly Regulated in Neurons
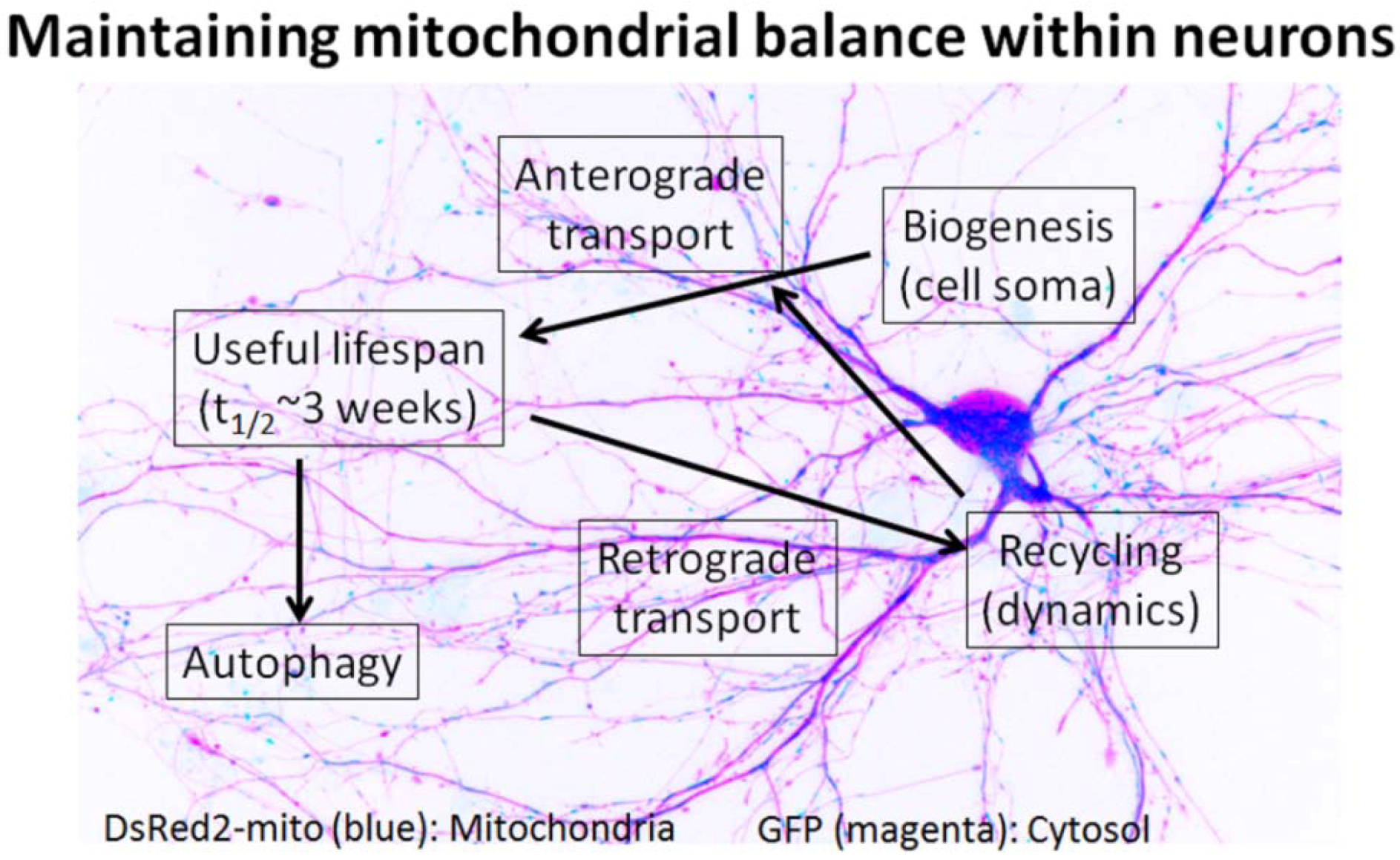
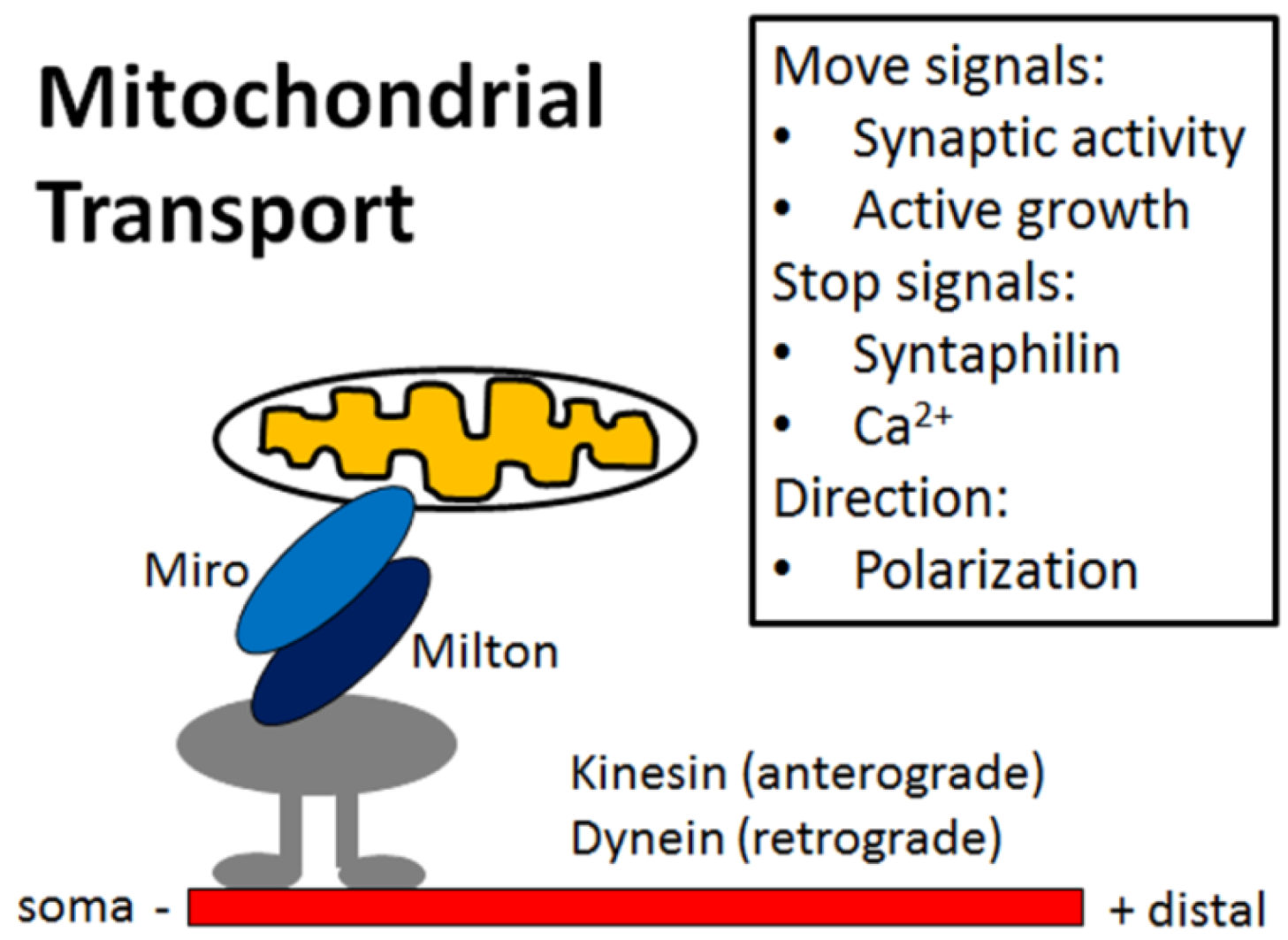
5. Impaired Mitochondrial Dynamics in Alzheimer’s Disease Neurons
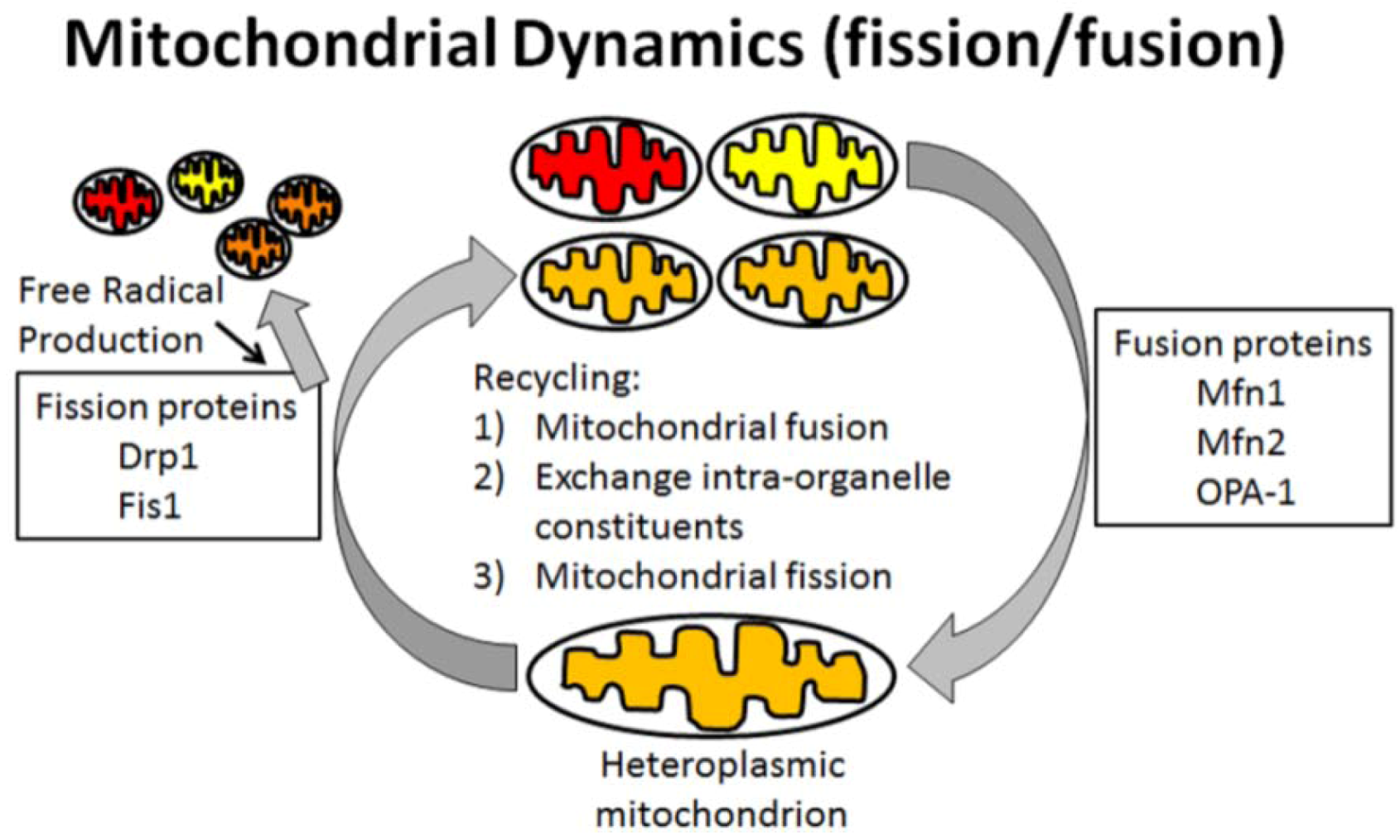
6. Oxidative Stress in Mitochondria Is Associated with AD
7. Mitochondria-Targeted Antioxidants May Be Beneficial in AD
8. Conclusions and Future Directions
Acknowledgements
References
- Selkoe, D.J. Alzheimer’s disease: Genes, proteins, and therapy. Physiol. Rev. 2001, 81, 741–766. [Google Scholar]
- LaFerla, F.M.; Green, K.N.; Oddo, S. Intracellular amyloid-beta in Alzheimer’s disease. Nat. Rev. Neurosci. 2007, 8, 499–509. [Google Scholar] [CrossRef]
- Keller, J.N.; Pang, Z.; Geddes, J.W.; Begley, J.G.; Germeyer, A.; Waeg, G.; Mattson, M.P. Impairment of glucose and glutamate transport and induction of mitochondrial oxidative stress and dysfunction in synaptosomes by amyloid beta-peptide: Role of the lipid peroxidation product 4-hydroxynonenal. J. Neurochem. 1997, 69, 273–284. [Google Scholar]
- Swerdlow, R.H.; Khan, S.M. A “mitochondrial cascade hypothesis” for sporadic Alzheimer’s disease. Med. Hypotheses 2004, 63, 8–20. [Google Scholar] [CrossRef]
- Swerdlow, R.H.; Burns, J.M.; Khan, S.M. The Alzheimer’s disease mitochondrial cascade hypothesis. J. Alzheimers Dis. 2010, 20, S265–S279. [Google Scholar]
- Hirai, K.; Aliev, G.; Nunomura, A.; Fujioka, H.; Russell, R.L.; Atwood, C.S.; Johnson, A.B.; Kress, Y.; Vinters, H.V.; Tabaton, M.; et al. Mitochondrial abnormalities in Alzheimer’s disease. J. Neurosci. 2001, 21, 3017–3023. [Google Scholar]
- Selkoe, D.J. Alzheimer’s disease is a synaptic failure. Science 2002, 298, 789–791. [Google Scholar] [CrossRef]
- Wyss-Coray, T. Inflammation in Alzheimer disease: Driving force, bystander or beneficial response? Nat. Med. 2006, 12, 1005–1015. [Google Scholar]
- Barsoum, M.J.; Yuan, H.; Gerencser, A.A.; Liot, G.; Kushnareva, Y.; Graber, S.; Kovacs, I.; Lee, W.D.; Waggoner, J.; Cui, J.; et al. Nitric oxide-induced mitochondrial fission is regulated by dynamin-related GTPases in neurons. EMBO J. 2006, 25, 3900–3911. [Google Scholar] [CrossRef]
- Greig, N.H.; Utsuki, T.; Ingram, D.K.; Wang, Y.; Pepeu, G.; Scali, C.; Yu, Q.S.; Mamczarz, J.; Holloway, H.W.; Giordano, T.; et al. Selective butyrylcholinesterase inhibition elevates brain acetylcholine, augments learning and lowers Alzheimer beta-amyloid peptide in rodent. Proc. Natl. Acad. Sci. USA 2005, 102, 17213–17218. [Google Scholar]
- Bertoni-Freddari, C.; Fattoretti, P.; Casoli, T.; Meier-Ruge, W.; Ulrich, J. Morphological adaptive response of the synaptic junctional zones in the human dentate gyrus during aging and Alzheimer’s disease. Brain Res. 1990, 517, 69–75. [Google Scholar] [CrossRef]
- DeKosky, S.T.; Scheff, S.W.; Styren, S.D. Structural correlates of cognition in dementia: Quantification and assessment of synapse change. Neurodegeneration 1996, 5, 417–421. [Google Scholar] [CrossRef]
- Tampellini, D.; Gouras, G.K. Synapses, synaptic activity and intraneuronal abeta in Alzheimer’s disease. Front. Aging Neurosci. 2010, 2, 13. [Google Scholar]
- Hardy, J.; Selkoe, D.J. The amyloid hypothesis of Alzheimer’s disease: Progress and problems on the road to therapeutics. Science 2002, 297, 353–356. [Google Scholar] [CrossRef]
- Walsh, D.M.; Selkoe, D.J. A beta oligomers-A decade of discovery. J. Neurochem. 2007, 101, 1172–1184. [Google Scholar] [CrossRef]
- Klein, W.L.; Krafft, G.A.; Finch, C.E. Targeting small Abeta oligomers: The solution to an Alzheimer's disease conundrum? Trends Neurosci. 2001, 24, 219–224. [Google Scholar] [CrossRef]
- Lambert, M.P.; Barlow, A.K.; Chromy, B.A.; Edwards, C.; Freed, R.; Liosatos, M.; Morgan, T.E.; Rozovsky, I.; Trommer, B.; Viola, K.L.; et al. Diffusible, nonfibrillar ligands derived from Abeta1-42 are potent central nervous system neurotoxins. Proc. Natl. Acad. Sci. USA 1998, 95, 6448–6453. [Google Scholar]
- Lacor, P.N.; Buniel, M.C.; Chang, L.; Fernandez, S.J.; Gong, Y.; Viola, K.L.; Lambert, M.P.; Velasco, P.T.; Bigio, E.H.; Finch, C.E.; et al. Synaptic targeting by Alzheimer’s-related amyloid beta oligomers. J. Neurosci. 2004, 24, 10191–10200. [Google Scholar]
- Manczak, M.; Calkins, M.J.; Reddy, P.H. Impaired mitochondrial dynamics and abnormal interaction of amyloid beta with mitochondrial protein Drp1 in neurons from patients with Alzheimer’s disease: Implications for neuronal damage. Hum. Mol. Genet. 2011, 20, 2495–2509. [Google Scholar] [CrossRef]
- Broersen, K.; Rousseau, F.; Schymkowitz, J. The culprit behind amyloid beta peptide related neurotoxicity in Alzheimer’s disease: Oligomer size or conformation? Alzheimers Res. Ther. 2010, 2, 12. [Google Scholar] [CrossRef]
- Reddy, P.H.; Manczak, M.; Mao, P.; Calkins, M.J.; Reddy, A.P.; Shirendeb, U. Amyloid-beta and mitochondria in aging and Alzheimer’s disease: Implications for synaptic damage and cognitive decline. J. Alzheimers Dis. 2010, 20, S499–S512. [Google Scholar]
- Calkins, M.J.; Manczak, M.; Mao, P.; Shirendeb, U.; Reddy, P.H. Impaired mitochondrial biogenesis, defective axonal transport of mitochondria, abnormal mitochondrial dynamics and synaptic degeneration in a mouse model of Alzheimer’s disease. Hum. Mol. Genet. 2011, 20, 4515–4529. [Google Scholar] [CrossRef]
- Wang, X.; Su, B.; Siedlak, S.L.; Moreira, P.I.; Fujioka, H.; Wang, Y.; Casadesus, G.; Zhu, X. Amyloid-beta overproduction causes abnormal mitochondrial dynamics via differential modulation of mitochondrial fission/fusion proteins. Proc. Natl. Acad. Sci. USA 2008, 105, 19318–19323. [Google Scholar]
- Wang, X.; Su, B.; Lee, H.G.; Li, X.; Perry, G.; Smith, M.A.; Zhu, X. Impaired balance of mitochondrial fission and fusion in Alzheimer’s disease. J. Neurosci. 2009, 29, 9090–9103. [Google Scholar]
- Du, H.; Guo, L.; Yan, S.; Sosunov, A.A.; McKhann, G.M.; Yan, S.S. Early deficits in synaptic mitochondria in an Alzheimer’s disease mouse model. Proc. Natl. Acad. Sci. USA 2010, 107, 18670–18675. [Google Scholar]
- Manczak, M.; Mao, P.; Calkins, M.J.; Cornea, A.; Reddy, A.P.; Murphy, M.P.; Szeto, H.H.; Park, B.; Reddy, P.H. Mitochondria-targeted antioxidants protect against amyloid-beta toxicity in Alzheimer's disease neurons. J. Alzheimers Dis. 2010, 20, S609–S631. [Google Scholar]
- Reddy, P.H.; Beal, M.F. Amyloid beta, mitochondrial dysfunction and synaptic damage: Implications for cognitive decline in aging and Alzheimer’s disease. Trends Mol. Med. 2008, 14, 45–53. [Google Scholar] [CrossRef]
- Manczak, M.; Anekonda, T.S.; Henson, E.; Park, B.S.; Quinn, J.; Reddy, P.H. Mitochondria are a direct site of A beta accumulation in Alzheimer’s disease neurons: Implications for free radical generation and oxidative damage in disease progression. Hum. Mol. Genet. 2006, 15, 1437–1449. [Google Scholar] [CrossRef]
- Devi, L.; Prabhu, B.M.; Galati, D.F.; Avadhani, N.G.; Anandatheerthavarada, H.K. Accumulation of amyloid precursor protein in the mitochondrial import channels of human Alzheimer’s disease brain is associated with mitochondrial dysfunction. J. Neurosci. 2006, 26, 9057–9068. [Google Scholar] [CrossRef]
- Crouch, P.J.; Blake, R.; Duce, J.A.; Ciccotosto, G.D.; Li, Q.X.; Barnham, K.J.; Curtain, C.C.; Cherny, R.A.; Cappai, R.; Dyrks, T.; et al. Copper-dependent inhibition of human cytochrome c oxidase by a dimeric conformer of amyloid-beta1-42. J. Neurosci. 2005, 25, 672–679. [Google Scholar] [CrossRef]
- Caspersen, C.; Wang, N.; Yao, J.; Sosunov, A.; Chen, X.; Lustbader, J.W.; Xu, H.W.; Stern, D.; McKhann, G.; Yan, S.D. Mitochondrial Abeta: A potential focal point for neuronal metabolic dysfunction in Alzheimer’s disease. FASEB J. 2005, 19, 2040–2041. [Google Scholar]
- Hansson Petersen, C.A.; Alikhani, N.; Behbahani, H.; Wiehager, B.; Pavlov, P.F.; Alafuzoff, I.; Leinonen, V.; Ito, A.; Winblad, B.; Glaser, E.; et al. The amyloid beta-peptide is imported into mitochondria via the TOM import machinery and localized to mitochondrial cristae. Proc. Natl. Acad. Sci. USA 2008, 105, 13145–13150. [Google Scholar]
- Yao, J.; Irwin, R.W.; Zhao, L.; Nilsen, J.; Hamilton, R.T.; Brinton, R.D. Mitochondrial bioenergetic deficit precedes Alzheimer’s pathology in female mouse model of Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2009, 106, 14670–14675. [Google Scholar]
- Lustbader, J.W.; Cirilli, M.; Lin, C.; Xu, H.W.; Takuma, K.; Wang, N.; Caspersen, C.; Chen, X.; Pollak, S.; Chaney, M.; et al. ABAD directly links Abeta to mitochondrial toxicity in Alzheimer’s disease. Science 2004, 304, 448–452. [Google Scholar]
- Du, H.; Guo, L.; Fang, F.; Chen, D.; Sosunov, A.A.; McKhann, G.M.; Yan, Y.; Wang, C.; Zhang, H.; Molkentin, J.D.; et al. Cyclophilin D deficiency attenuates mitochondrial and neuronal perturbation and ameliorates learning and memory in Alzheimer’s disease. Nat. Med. 2008, 14, 1097–1105. [Google Scholar] [CrossRef]
- Reddy, P.H. Amyloid beta, mitochondrial structural and functional dynamics in Alzheimer’s disease. Exp. Neurol. 2009, 218, 286–292. [Google Scholar] [CrossRef]
- Attwell, D.; Laughlin, S.B. An energy budget for signaling in the grey matter of the brain. J. Cereb. Blood Flow Metab. 2001, 21, 1133–1145. [Google Scholar] [CrossRef]
- Ames, A., 3rd. CNS energy metabolism as related to function. Brain Res. Brain Res. Rev. 2000, 34, 42–68. [Google Scholar] [CrossRef]
- Pellerin, L. Food for thought: The importance of glucose and other energy substrates for sustaining brain function under varying levels of activity. Diabetes Metab. 2010, 36, S59–S63. [Google Scholar] [CrossRef]
- Pellerin, L.; Bouzier-Sore, A.K.; Aubert, A.; Serres, S.; Merle, M.; Costalat, R.; Magistretti, P.J. Activity-dependent regulation of energy metabolism by astrocytes: An update. Glia 2007, 55, 1251–1262. [Google Scholar] [CrossRef]
- Wong-Riley, M.T. Cytochrome oxidase: An endogenous metabolic marker for neuronal activity. Trends Neurosci. 1989, 12, 94–101. [Google Scholar] [CrossRef]
- Wong-Riley, M.T. Bigenomic regulation of cytochrome C oxidase in neurons and the tight coupling between neuronal activity and energy metabolism. Adv. Exp. Med. Biol. 2012, 748, 283–304. [Google Scholar]
- Scarpulla, R.C. Nuclear control of respiratory chain expression by nuclear respiratory factors and PGC-1-related coactivator. Ann. NY Acad. Sci. 2008, 1147, 321–334. [Google Scholar]
- Kelly, D.P.; Scarpulla, R.C. Transcriptional regulatory circuits controlling mitochondrial biogenesis and function. Genes Dev. 2004, 18, 357–368. [Google Scholar] [CrossRef]
- Scarpulla, R.C. Metabolic control of mitochondrial biogenesis through the PGC-1 family regulatory network. Biochim. Biophys. Acta 2011, 1813, 1269–1278. [Google Scholar]
- Fernandez-Marcos, P.J.; Auwerx, J. Regulation of PGC-1alpha, a nodal regulator of mitochondrial biogenesis. Am. J. Clin. Nutr. 2011, 93, S884–S890. [Google Scholar] [CrossRef]
- Ventura-Clapier, R.; Garnier, A.; Veksler, V. Transcriptional control of mitochondrial biogenesis: The central role of PGC-1alpha. Cardiovasc. Res. 2008, 79, 208–217. [Google Scholar] [CrossRef]
- Kann, O.; Kovacs, R. Mitochondria and neuronal activity. Am. J. Physiol. Cell Physiol. 2007, 292, C641–C657. [Google Scholar] [CrossRef]
- Kann, O.; Schuchmann, S.; Buchheim, K.; Heinemann, U. Coupling of neuronal activity and mitochondrial metabolism as revealed by NAD(P)H fluorescence signals in organotypic hippocampal slice cultures of the rat. Neuroscience 2003, 119, 87–100. [Google Scholar] [CrossRef]
- Bindokas, V.P.; Lee, C.C.; Colmers, W.F.; Miller, R.J. Changes in mitochondrial function resulting from synaptic activity in the rat hippocampal slice. J. Neurosci. 1998, 18, 4570–4587. [Google Scholar]
- Pivovarova, N.B.; Pozzo-Miller, L.D.; Hongpaisan, J.; Andrews, S.B. Correlated calcium uptake and release by mitochondria and endoplasmic reticulum of CA3 hippocampal dendrites after afferent synaptic stimulation. J. Neurosci. 2002, 22, 10653–10661. [Google Scholar]
- David, G.; Barrett, E.F. Mitochondrial Ca2+ uptake prevents desynchronization of quantal release and minimizes depletion during repetitive stimulation of mouse motor nerve terminals. J. Physiol. 2003, 548, 425–438. [Google Scholar] [CrossRef]
- Garcia-Chacon, L.E.; Nguyen, K.T.; David, G.; Barrett, E.F. Extrusion of Ca2+ from mouse motor terminal mitochondria via a Na+-Ca2+ exchanger increases post-tetanic evoked release. J. Physiol. 2006, 574, 663–675. [Google Scholar] [CrossRef]
- Medler, K.; Gleason, E.L. Mitochondrial Ca2+ buffering regulates synaptic transmission between retinal amacrine cells. J. Neurophysiol. 2002, 87, 1426–1439. [Google Scholar]
- Calupca, M.A.; Prior, C.; Merriam, L.A.; Hendricks, G.M.; Parsons, R.L. Presynaptic function is altered in snake K+-depolarized motor nerve terminals containing compromised mitochondria. J. Physiol. 2001, 532, 217–227. [Google Scholar] [CrossRef]
- Scotti, A.L.; Chatton, J.Y.; Reuter, H. Roles of Na+-Ca2+ exchange and of mitochondria in the regulation of presynaptic Ca2+ and spontaneous glutamate release. Philos. Trans. R. Soc. Lond. B Biol. Sci. 1999, 354, 357–364. [Google Scholar] [CrossRef]
- Malli, R.; Graier, W.F. Mitochondrial Ca2+ channels: Great unknowns with important functions. FEBS Lett. 2010, 584, 1942–1947. [Google Scholar] [CrossRef]
- Ohsawa, S.; Hamada, S.; Kuida, K.; Yoshida, H.; Igaki, T.; Miura, M. Maturation of the olfactory sensory neurons by Apaf-1/caspase-9-mediated caspase activity. Proc. Natl. Acad. Sci. USA 2010, 107, 13366–13371. [Google Scholar]
- Hyman, B.T.; Yuan, J. Apoptotic and non-apoptotic roles of caspases in neuronal physiology and pathophysiology. Nat. Rev. Neurosci. 2012, 13, 395–406. [Google Scholar] [CrossRef]
- D’Amelio, M.; Cavallucci, V.; Middei, S.; Marchetti, C.; Pacioni, S.; Ferri, A.; Diamantini, A.; de Zio, D.; Carrara, P.; Battistini, L.; et al. Caspase-3 triggers early synaptic dysfunction in a mouse model of Alzheimer’s disease. Nat. Neurosci. 2011, 14, 69–76. [Google Scholar]
- Park, K.J.; Grosso, C.A.; Aubert, I.; Kaplan, D.R.; Miller, F.D. p75NTR-dependent, myelin-mediated axonal degeneration regulates neural connectivity in the adult brain. Nat. Neurosci. 2010, 13, 559–566. [Google Scholar] [CrossRef]
- Li, Z.; Sheng, M. Caspases in synaptic plasticity. Mol. Brain 2012, 5, 15. [Google Scholar] [CrossRef]
- Nagerl, U.V.; Eberhorn, N.; Cambridge, S.B.; Bonhoeffer, T. Bidirectional activity-dependent morphological plasticity in hippocampal neurons. Neuron 2004, 44, 759–767. [Google Scholar] [CrossRef]
- Zhou, Q.; Homma, K.J.; Poo, M.M. Shrinkage of dendritic spines associated with long-term depression of hippocampal synapses. Neuron 2004, 44, 749–757. [Google Scholar] [CrossRef]
- Okamoto, K.; Nagai, T.; Miyawaki, A.; Hayashi, Y. Rapid and persistent modulation of actin dynamics regulates postsynaptic reorganization underlying bidirectional plasticity. Nat. Neurosci. 2004, 7, 1104–1112. [Google Scholar] [CrossRef]
- Li, Z.; Jo, J.; Jia, J.M.; Lo, S.C.; Whitcomb, D.J.; Jiao, S.; Cho, K.; Sheng, M. Caspase-3 activation via mitochondria is required for long-term depression and AMPA receptor internalization. Cell 2010, 141, 859–871. [Google Scholar] [CrossRef]
- Jiao, S.; Li, Z. Nonapoptotic function of BAD and BAX in long-term depression of synaptic transmission. Neuron 2011, 70, 758–772. [Google Scholar] [CrossRef]
- Hsieh, H.; Boehm, J.; Sato, C.; Iwatsubo, T.; Tomita, T.; Sisodia, S.; Malinow, R. AMPAR removal underlies Abeta-induced synaptic depression and dendritic spine loss. Neuron 2006, 52, 831–843. [Google Scholar] [CrossRef]
- Ting, J.T.; Kelley, B.G.; Lambert, T.J.; Cook, D.G.; Sullivan, J.M. Amyloid precursor protein overexpression depresses excitatory transmission through both presynaptic and postsynaptic mechanisms. Proc. Natl. Acad. Sci. USA 2007, 104, 353–358. [Google Scholar]
- Almeida, C.G.; Tampellini, D.; Takahashi, R.H.; Greengard, P.; Lin, M.T.; Snyder, E.M.; Gouras, G.K. Beta-amyloid accumulation in APP mutant neurons reduces PSD-95 and GluR1 in synapses. Neurobiol. Dis. 2005, 20, 187–198. [Google Scholar] [CrossRef]
- Rui, Y.; Gu, J.; Yu, K.; Hartzell, H.C.; Zheng, J.Q. Inhibition of AMPA receptor trafficking at hippocampal synapses by beta-amyloid oligomers: The mitochondrial contribution. Mol. Brain 2010, 3, 10. [Google Scholar] [CrossRef]
- Chan, S.L.; Griffin, W.S.; Mattson, M.P. Evidence for caspase-mediated cleavage of AMPA receptor subunits in neuronal apoptosis and Alzheimer's disease. J. Neurosci. Res. 1999, 57, 315–323. [Google Scholar] [CrossRef]
- Davis, A.F.; Clayton, D.A. In situ localization of mitochondrial DNA replication in intact mammalian cells. J. Cell Biol. 1996, 135, 883–893. [Google Scholar] [CrossRef]
- Davis, A.F.; Ropp, P.A.; Clayton, D.A.; Copeland, W.C. Mitochondrial DNA polymerase gamma is expressed and translated in the absence of mitochondrial DNA maintenance and replication. Nucleic Acids Res. 1996, 24, 2753–2759. [Google Scholar] [CrossRef]
- Koenig, E.; Giuditta, A. Protein-synthesizing machinery in the axon compartment. Neuroscience 1999, 89, 5–15. [Google Scholar] [CrossRef]
- Alvarez, J.; Giuditta, A.; Koenig, E. Protein synthesis in axons and terminals: Significance for maintenance, plasticity and regulation of phenotype. With a critique of slow transport theory. Prog. Neurobiol. 2000, 62, 1–62. [Google Scholar] [CrossRef]
- Twiss, J.L.; van Minnen, J. New insights into neuronal regeneration: The role of axonal protein synthesis in pathfinding and axonal extension. J. Neurotrauma 2006, 23, 295–308. [Google Scholar] [CrossRef]
- Gioio, A.E.; Eyman, M.; Zhang, H.; Lavina, Z.S.; Giuditta, A.; Kaplan, B.B. Local synthesis of nuclear-encoded mitochondrial proteins in the presynaptic nerve terminal. J. Neurosci. Res. 2001, 64, 447–453. [Google Scholar] [CrossRef]
- Sotelo-Silveira, J.R.; Calliari, A.; Kun, A.; Koenig, E.; Sotelo, J.R. RNA trafficking in axons. Traffic 2006, 7, 508–515. [Google Scholar] [CrossRef]
- Magnusson, J.; Orth, M.; Lestienne, P.; Taanman, J.W. Replication of mitochondrial DNA occurs throughout the mitochondria of cultured human cells. Exp. Cell Res. 2003, 289, 133–142. [Google Scholar] [CrossRef]
- Amiri, M.; Hollenbeck, P.J. Mitochondrial biogenesis in the axons of vertebrate peripheral neurons. Dev. Neurobiol. 2008, 68, 1348–1361. [Google Scholar] [CrossRef]
- Calkins, M.J.; Reddy, P.H. Assessment of newly synthesized mitochondrial DNA using BrdU labeling in primary neurons from Alzheimer’s disease mice: Implications for impaired mitochondrial biogenesis and synaptic damage. Biochim. Biophys. Acta 2011, 1812, 1182–1189. [Google Scholar]
- Sheng, Z.H.; Cai, Q. Mitochondrial transport in neurons: Impact on synaptic homeostasis and neurodegeneration. Nat. Rev. Neurosci. 2012, 13, 77–93. [Google Scholar] [CrossRef]
- Miller, K.E.; Sheetz, M.P. Axonal mitochondrial transport and potential are correlated. J. Cell Sci. 2004, 117, 2791–2804. [Google Scholar] [CrossRef]
- Verburg, J.; Hollenbeck, P.J. Mitochondrial membrane potential in axons increases with local nerve growth factor or semaphorin signaling. J. Neurosci. 2008, 28, 8306–8315. [Google Scholar] [CrossRef]
- Schon, E.A.; Przedborski, S. Mitochondria: The next (neurode)generation. Neuron 2011, 70, 1033–1053. [Google Scholar] [CrossRef]
- Reddy, P.H.; Shirendeb, U.P. Mutant huntingtin, abnormal mitochondrial dynamics, defective axonal transport of mitochondria, and selective synaptic degeneration in Huntington’s disease. Biochim. Biophys. Acta 2012, 1822, 101–110. [Google Scholar]
- Martin, L.J. Mitochondrial pathobiology in ALS. J. Bioenerg. Biomembr. 2011, 43, 569–579. [Google Scholar] [CrossRef]
- Calkins, M.J.; Reddy, P.H. Amyloid beta impairs mitochondrial anterograde transport and degenerates synapses in Alzheimer’s disease neurons. Biochim. Biophys. Acta 2011, 1812, 507–513. [Google Scholar]
- Wang, X.; Perry, G.; Smith, M.A.; Zhu, X. Amyloid-beta-derived diffusible ligands cause impaired axonal transport of mitochondria in neurons. Neurodegener. Dis. 2010, 7, 56–59. [Google Scholar] [CrossRef]
- Rui, Y.; Tiwari, P.; Xie, Z.; Zheng, J.Q. Acute impairment of mitochondrial trafficking by beta-amyloid peptides in hippocampal neurons. J. Neurosci. 2006, 26, 10480–10487. [Google Scholar] [CrossRef]
- Fang, C.; Bourdette, D.; Banker, G. Oxidative stress inhibits axonal transport: Implications for neurodegenerative diseases. Mol. Neurodegener. 2012, 7, 29. [Google Scholar] [CrossRef]
- Misko, A.; Jiang, S.; Wegorzewska, I.; Milbrandt, J.; Baloh, R.H. Mitofusin 2 is necessary for transport of axonal mitochondria and interacts with the Miro/Milton complex. J. Neurosci. 2010, 30, 4232–4240. [Google Scholar] [CrossRef]
- Fransson, A.; Ruusala, A.; Aspenstrom, P. Atypical Rho GTPases have roles in mitochondrial homeostasis and apoptosis. J. Biol. Chem. 2003, 278, 6495–6502. [Google Scholar]
- Saotome, M.; Safiulina, D.; Szabadkai, G.; Das, S.; Fransson, A.; Aspenstrom, P.; Rizzuto, R.; Hajnoczky, G. Bidirectional Ca2+-dependent control of mitochondrial dynamics by the Miro GTPase. Proc. Natl. Acad. Sci. USA 2008, 105, 20728–20733. [Google Scholar]
- Reddy, P.H.; Tripathi, R.; Troung, Q.; Tirumala, K.; Reddy, T.P.; Anekonda, V.; Shirendeb, U.P.; Calkins, M.J.; Reddy, A.P.; Mao, P.; et al. Abnormal mitochondrial dynamics and synaptic degeneration as early events in Alzheimer's disease: Implications to mitochondria-targeted antioxidant therapeutics. Biochim. Biophys. Acta 2012, 1822, 639–649. [Google Scholar]
- Reddy, P.H. Mitochondrial oxidative damage in aging and Alzheimer’s disease: Implications for mitochondrially targeted antioxidant therapeutics. J. Biomed. Biotechnol. 2006, 2006, 31372. [Google Scholar]
- Chaturvedi, R.K.; Beal, M.F. Mitochondrial approaches for neuroprotection. Ann. NY Acad. Sci. 2008, 1147, 395–412. [Google Scholar]
- Mecocci, P.; Polidori, M.C. Antioxidant clinical trials in mild cognitive impairment and Alzheimer’s disease. Biochim. Biophys. Acta 2012, 1822, 631–638. [Google Scholar]
- Pocernich, C.B.; Butterfield, D.A. Elevation of glutathione as a therapeutic strategy in Alzheimer disease. Biochim. Biophys. Acta 2012, 1822, 625–630. [Google Scholar]
- Murphy, M.P.; Smith, R.A. Targeting antioxidants to mitochondria by conjugation to lipophilic cations. Annu. Rev. Pharmacol. Toxicol. 2007, 47, 629–656. [Google Scholar]
- Szeto, H.H. Mitochondria-targeted peptide antioxidants: Novel neuroprotective agents. AAPS J. 2006, 8, E521–E531. [Google Scholar] [CrossRef]
- Sheu, S.S.; Nauduri, D.; Anders, M.W. Targeting antioxidants to mitochondria: A new therapeutic direction. Biochim. Biophys. Acta 2006, 1762, 256–265. [Google Scholar]
- Schiller, P.W.; Nguyen, T.M.; Berezowska, I.; Dupuis, S.; Weltrowska, G.; Chung, N.N.; Lemieux, C. Synthesis and in vitro opioid activity profiles of DALDA analogues. Eur. J. Med. Chem. 2000, 35, 895–901. [Google Scholar] [CrossRef]
- Zhao, G.M.; Qian, X.; Schiller, P.W.; Szeto, H.H. Comparison of [Dmt1]DALDA and DAMGO in binding and G protein activation at mu, delta, and kappa opioid receptors. J. Pharmacol. Exp. Ther. 2003, 307, 947–954. [Google Scholar] [CrossRef]
- Zhao, K.; Zhao, G.M.; Wu, D.; Soong, Y.; Birk, A.V.; Schiller, P.W.; Szeto, H.H. Cell-permeable peptide antioxidants targeted to inner mitochondrial membrane inhibit mitochondrial swelling, oxidative cell death, and reperfusion injury. J. Biol. Chem. 2004, 279, 34682–34690. [Google Scholar]
- Thomas, D.A.; Stauffer, C.; Zhao, K.; Yang, H.; Sharma, V.K.; Szeto, H.H.; Suthanthiran, M. Mitochondrial targeting with antioxidant peptide SS-31 prevents mitochondrial depolarization, reduces islet cell apoptosis, increases islet cell yield, and improves posttransplantation function. J. Am. Soc. Nephrol. 2007, 18, 213–222. [Google Scholar] [CrossRef]
- Cho, J.; Won, K.; Wu, D.; Soong, Y.; Liu, S.; Szeto, H.H.; Hong, M.K. Potent mitochondria-targeted peptides reduce myocardial infarction in rats. Coron. Artery Dis. 2007, 18, 215–220. [Google Scholar] [CrossRef]
- Cho, S.; Szeto, H.H.; Kim, E.; Kim, H.; Tolhurst, A.T.; Pinto, J.T. A novel cell-permeable antioxidant peptide, SS31, attenuates ischemic brain injury by down-regulating CD36. J. Biol. Chem. 2007, 282, 4634–4642. [Google Scholar]
- Petri, S.; Kiaei, M.; Damiano, M.; Hiller, A.; Wille, E.; Manfredi, G.; Calingasan, N.Y.; Szeto, H.H.; Beal, M.F. Cell-permeable peptide antioxidants as a novel therapeutic approach in a mouse model of amyotrophic lateral sclerosis. J. Neurochem. 2006, 98, 1141–1148. [Google Scholar] [CrossRef]
- Takahashi, R.H.; Almeida, C.G.; Kearney, P.F.; Yu, F.; Lin, M.T.; Milner, T.A.; Gouras, G.K. Oligomerization of Alzheimer’s beta-amyloid within processes and synapses of cultured neurons and brain. J. Neurosci. 2004, 24, 3592–3599. [Google Scholar] [CrossRef]
- Wu, H.Y.; Hudry, E.; Hashimoto, T.; Kuchibhotla, K.; Rozkalne, A.; Fan, Z.; Spires-Jones, T.; Xie, H.; Arbel-Ornath, M.; Grosskreutz, C.L.; et al. Amyloid beta induces the morphological neurodegenerative triad of spine loss, dendritic simplification, and neuritic dystrophies through calcineurin activation. J. Neurosci. 2010, 30, 2636–2649. [Google Scholar]
- Wu, H.Y.; Hudry, E.; Hashimoto, T.; Uemura, K.; Fan, Z.Y.; Berezovska, O.; Grosskreutz, C.L.; Bacskai, B.J.; Hyman, B.T. Distinct dendritic spine and nuclear phases of calcineurin activation after exposure to amyloid-beta revealed by a novel fluorescence resonance energy transfer assay. J. Neurosci. 2012, 32, 5298–5309. [Google Scholar] [CrossRef]
- Manczak, M.; Reddy, P.H. Abnormal interaction between the mitochondrial fission protein Drp1 and hyperphosphorylated tau in Alzheimer’s disease neurons: Implications for mitochondrial dysfunction and neuronal damage. Hum. Mol. Genet. 2012, 21, 2538–2547. [Google Scholar] [CrossRef]
- Galindo, M.F.; Solesio, M.E.; Atienzar-Aroca, S.; Zamora, M.J.; Jordan Bueso, J. Mitochondrial dynamics and mitophagy in the 6-hydroxydopamine preclinical model of Parkinson’s disease. Parkinsons Dis. 2012, 2012, 131058. [Google Scholar]
- Solesio, M.E.; Prime, T.A.; Logan, A.; Murphy, M.P.; Del Mar Arroyo-Jimenez, M.; Jordan, J.; Galindo, M.F. The mitochondria-targeted anti-oxidant MitoQ reduces aspects of mitochondrial fission in the 6-OHDA cell model of Parkinson’s disease. Biochim. Biophys. Acta 2012. in press.
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Calkins, M.J.; Manczak, M.; Reddy, P.H. Mitochondria-Targeted Antioxidant SS31 Prevents Amyloid Beta-Induced Mitochondrial Abnormalities and Synaptic Degeneration in Alzheimer’s Disease. Pharmaceuticals 2012, 5, 1103-1119. https://doi.org/10.3390/ph5101103
Calkins MJ, Manczak M, Reddy PH. Mitochondria-Targeted Antioxidant SS31 Prevents Amyloid Beta-Induced Mitochondrial Abnormalities and Synaptic Degeneration in Alzheimer’s Disease. Pharmaceuticals. 2012; 5(10):1103-1119. https://doi.org/10.3390/ph5101103
Chicago/Turabian StyleCalkins, Marcus J., Maria Manczak, and P. Hemachandra Reddy. 2012. "Mitochondria-Targeted Antioxidant SS31 Prevents Amyloid Beta-Induced Mitochondrial Abnormalities and Synaptic Degeneration in Alzheimer’s Disease" Pharmaceuticals 5, no. 10: 1103-1119. https://doi.org/10.3390/ph5101103