Cell-Penetrating Peptides—Mechanisms of Cellular Uptake and Generation of Delivery Systems

{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
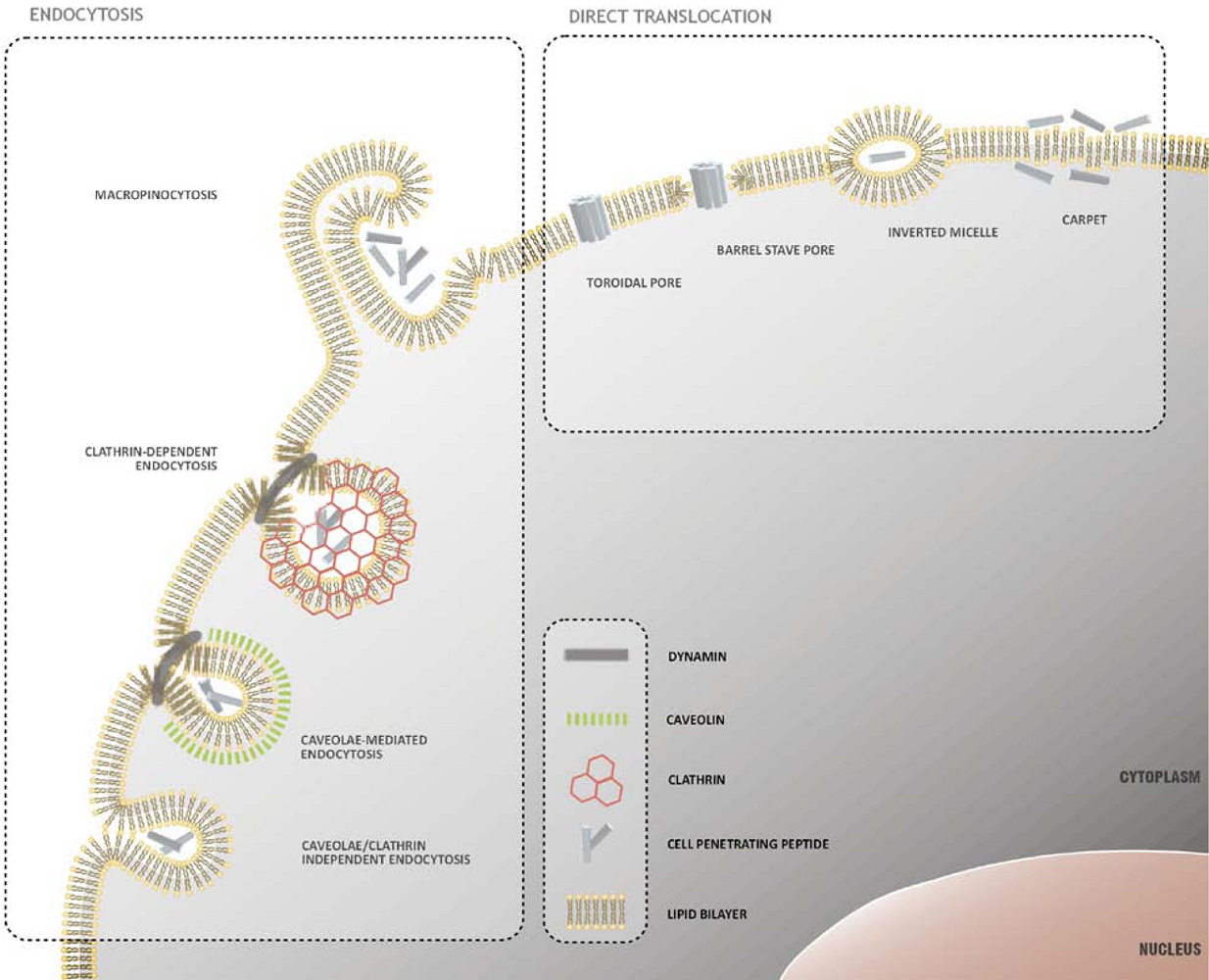
2. Mechanisms of Cellular Internalization of CPPs
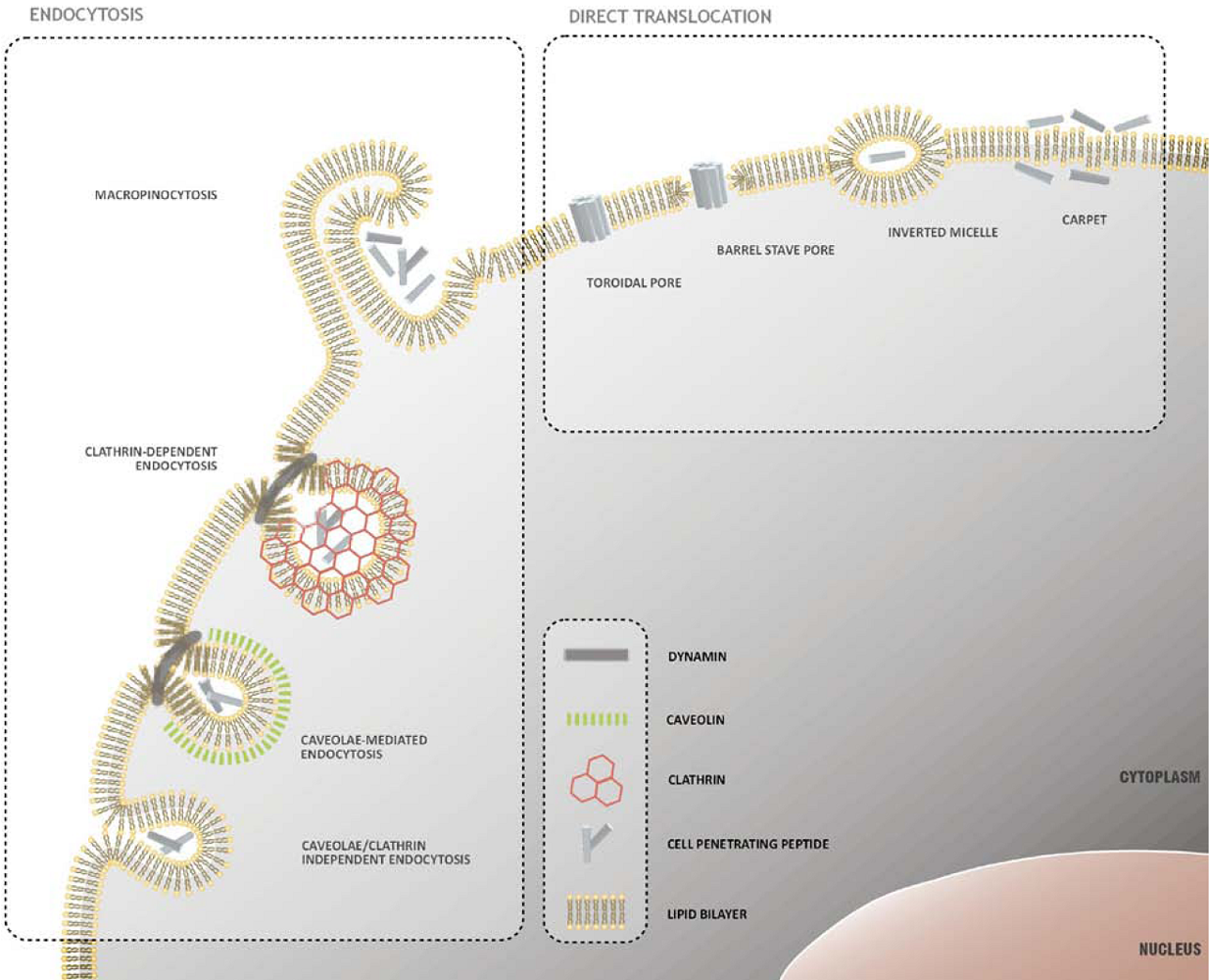
2.1. Direct Translocation of CPPs across Biological Membranes
2.2. Endocytosis as a Pathway for CPP Internalization
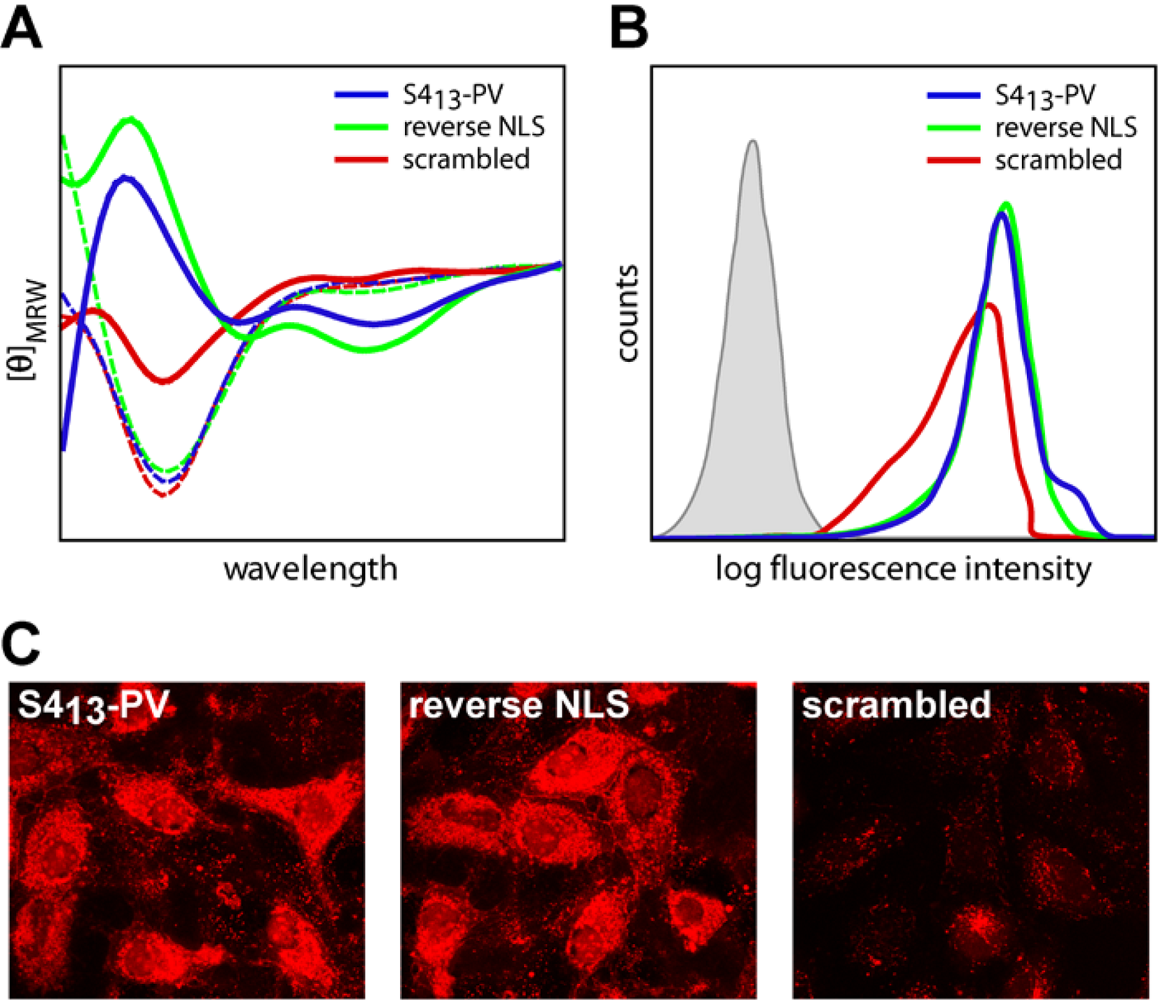
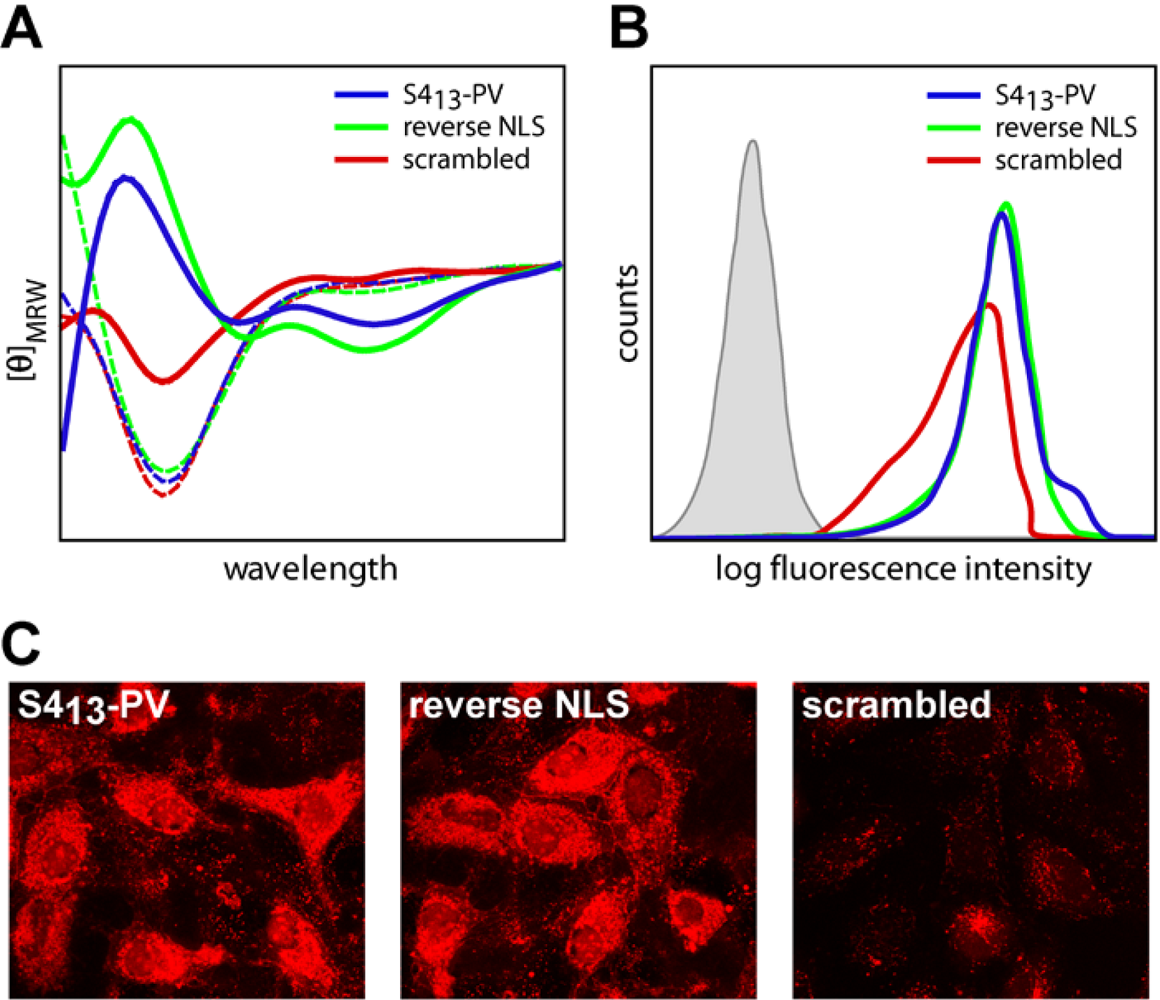
2.3. Mechanisms of Internalization of S413-PV Peptide
3. CPPs-Based Strategies for Delivery of Therapeutic Molecules
3.1. Protein Delivery
3.2. Liposome and Nanoparticle Delivery
3.3. Antisense Oligonucleotide Delivery
3.4. siRNA Delivery
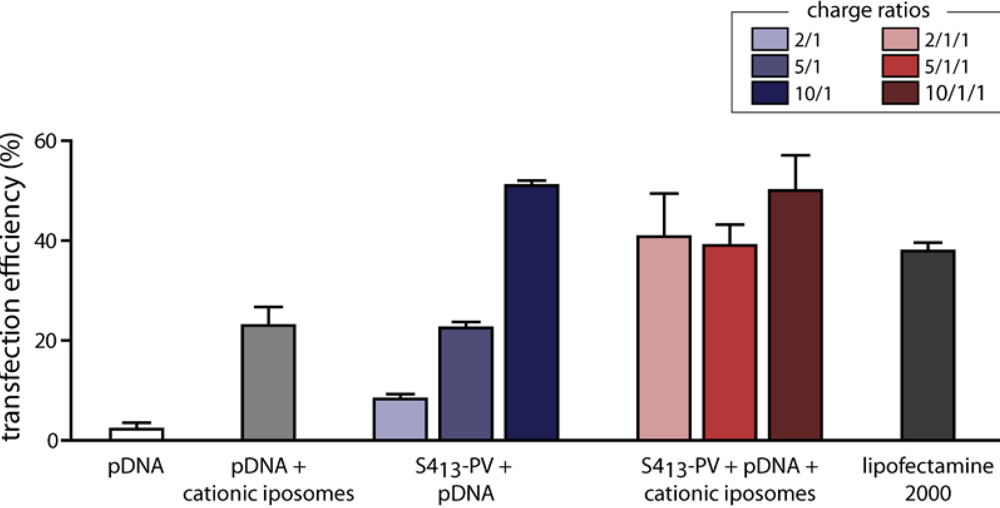
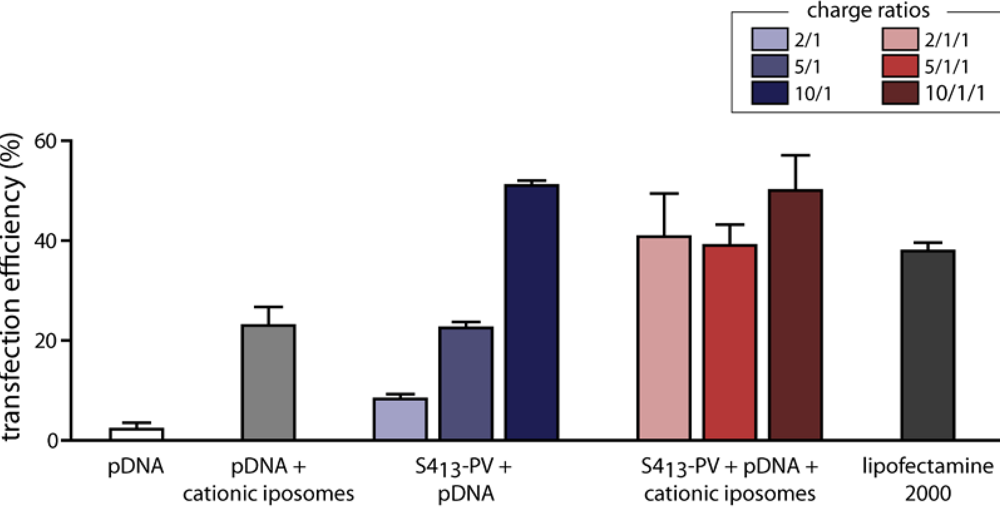
3.5. Gene Delivery
4. Conclusions
Acknowledgments
References
- Lindgren, M.; Hallbrink, M.; Prochiantz, A.; Langel, U. Cell-penetrating peptides. Trends Pharmacol. Sci. 2000, 21, 99–103. [Google Scholar] [CrossRef]
- Frankel, A.D.; Pabo, C.O. Cellular uptake of the tat protein from human immunodeficiency virus. Cell 1988, 55, 1189–1193. [Google Scholar] [CrossRef]
- Green, M.; Loewenstein, P.M. Autonomous functional domains of chemically synthesized human immunodeficiency virus tat trans-activator protein. Cell 1988, 55, 1179–1188. [Google Scholar] [CrossRef]
- Joliot, A.H.; Triller, A.; Volovitch, M.; Pernelle, C.; Prochiantz, A. alpha-2,8-Polysialic acid is the neuronal surface receptor of antennapedia homeobox peptide. New Biol. 1991, 3, 1121–1134. [Google Scholar] [PubMed]
- Derossi, D.; Joliot, A.H.; Chassaing, G.; Prochiantz, A. The third helix of the Antennapedia homeodomain translocates through biological membranes. J. Biol. Chem. 1994, 269, 10444–10450. [Google Scholar] [PubMed]
- Morris, M.C.; Depollier, J.; Mery, J.; Heitz, F.; Divita, G. A peptide carrier for the delivery of biologically active proteins into mammalian cells. Nat. Biotechnol. 2001, 19, 1173–1176. [Google Scholar] [CrossRef]
- Meade, B.R.; Dowdy, S.F. Exogenous siRNA delivery using peptide transduction domains/cell penetrating peptides. Adv. Drug Deliv. Rev. 2007, 59, 134–140. [Google Scholar] [CrossRef]
- Heitz, F.; Morris, M.C.; Divita, G. Twenty years of cell-penetrating peptides: from molecular mechanisms to therapeutics. Br. J. Pharmacol. 2009, 157, 195–206. [Google Scholar] [CrossRef]
- Morris, M.C.; Deshayes, S.; Heitz, F.; Divita, G. Cell-penetrating peptides: from molecular mechanisms to therapeutics. Biol. Cell 2008, 100, 201–217. [Google Scholar]
- Langel, U. Cell Penetrating Peptides: Processes and Applications. 2002; CRC Press: Boca Raton, FL, USA. [Google Scholar]
- Dietz, G.P.; Bahr, M. Delivery of bioactive molecules into the cell: the Trojan horse approach. Mo.l Cell Neurosci. 2004, 27, 85–131. [Google Scholar] [CrossRef]
- Magzoub, M.; Graslund, A. Cell-penetrating peptides: [corrected] from inception to application. Q. Rev. Biophys. 2004, 37, 147–195. [Google Scholar] [CrossRef]
- Joliot, A.; Prochiantz, A. Transduction peptides: from technology to physiology. Nat. Cell Biol. 2004, 6, 189–196. [Google Scholar] [CrossRef]
- Deshayes, S.; Morris, M.C.; Divita, G.; Heitz, F. Cell-penetrating peptides: tools for intracellular delivery of therapeutics. Cell. Mol. Life. Sci. 2005, 62, 1839–1849. [Google Scholar] [CrossRef]
- Vives, E. Present and future of cell-penetrating peptide mediated delivery systems: "is the Trojan horse too wild to go only to Troy?". J. Control. Release 2005, 109, 77–85. [Google Scholar] [CrossRef]
- El-Andaloussi, S.; Holm, T.; Langel, U. Cell-penetrating peptides: mechanisms and applications. Curr. Pharm. Des. 2005, 11, 3597–3611. [Google Scholar] [CrossRef]
- Patel, L.N.; Zaro, J.L.; Shen, W.C. Cell Penetrating Peptides: Intracellular Pathways and Pharmaceutical Perspectives. Pharm. Res. 2007. [Google Scholar]
- Wagstaff, K.M.; Jans, D.A. Protein transduction: cell penetrating peptides and their therapeutic applications. Curr. Med. Chem. 2006, 13, 1371–1387. [Google Scholar] [CrossRef]
- Gupta, B.; Levchenko, T.S.; Torchilin, V.P. Intracellular delivery of large molecules and small particles by cell-penetrating proteins and peptides. Adv. Drug. Deliv. Rev. 2005, 57, 637–651. [Google Scholar] [CrossRef]
- Lundberg, M.; Wikstrom, S.; Johansson, M. Cell surface adherence and endocytosis of protein transduction domains. Mol. Ther. 2003, 8, 143–150. [Google Scholar] [CrossRef]
- Richard, J.P.; Melikov, K.; Vives, E.; Ramos, C.; Verbeure, B.; Gait, M.J.; Chernomordik, L.V.; Lebleu, B. Cell-penetrating peptides. A reevaluation of the mechanism of cellular uptake. J. Biol. Chem. 2003, 278, 585–590. [Google Scholar] [CrossRef]
- Vives, E.; Richard, J.P.; Rispal, C.; Lebleu, B. TAT peptide internalization: seeking the mechanism of entry. Curr. Protein Pept. Sci. 2003, 4, 125–132. [Google Scholar] [CrossRef]
- Hariton-Gazal, E.; Feder, R.; Mor, A.; Graessmann, A.; Brack-Werner, R.; Jans, D.; Gilon, C.; Loyter, A. Targeting of nonkaryophilic cell-permeable peptides into the nuclei of intact cells by covalently attached nuclear localization signals. Biochemistry 2002, 41, 9208–9214. [Google Scholar] [CrossRef]
- Derossi, D.; Calvet, S.; Trembleau, A.; Brunissen, A.; Chassaing, G.; Prochiantz, A. Cell internalization of the third helix of the Antennapedia homeodomain is receptor-independent. J. Biol. Chem. 1996, 271, 18188–18193. [Google Scholar] [CrossRef]
- Huq, I.; Ping, Y.H.; Tamilarasu, N.; Rana, T.M. Controlling human immunodeficiency virus type 1 gene expression by unnatural peptides. Biochemistry 1999, 38, 5172–5177. [Google Scholar] [CrossRef]
- Wender, P.A.; Mitchell, D.J.; Pattabiraman, K.; Pelkey, E.T.; Steinman, L.; Rothbard, J.B. The design, synthesis, and evaluation of molecules that enable or enhance cellular uptake: peptoid molecular transporters. Proc. Natl. Acad. Sci. U. S. A. 2000, 97, 13003–13008. [Google Scholar] [CrossRef]
- Ferrari, M.E.; Nguyen, C.M.; Zelphati, O.; Tsai, Y.; Felgner, P.L. Analytical methods for the characterization of cationic lipid-nucleic acid complexes. Hum. Gene Ther. 1998, 9, 341–351. [Google Scholar] [CrossRef]
- Fittipaldi, A.; Ferrari, A.; Zoppe, M.; Arcangeli, C.; Pellegrini, V.; Beltram, F.; Giacca, M. Cell membrane lipid rafts mediate caveolar endocytosis of HIV-1 Tat fusion proteins. J. Biol. Chem. 2003, 278, 34141–34149. [Google Scholar] [CrossRef]
- Wadia, J.S.; Stan, R.V.; Dowdy, S.F. Transducible TAT-HA fusogenic peptide enhances escape of TAT-fusion proteins after lipid raft macropinocytosis. Nat. Med. 2004, 10, 310–315. [Google Scholar] [CrossRef]
- Kaplan, I.M.; Wadia, J.S.; Dowdy, S.F. Cationic TAT peptide transduction domain enters cells by macropinocytosis. J. Control. Release 2005, 102, 247–253. [Google Scholar] [CrossRef]
- Richard, J.P.; Melikov, K.; Brooks, H.; Prevot, P.; Lebleu, B.; Chernomordik, L.V. Cellular uptake of unconjugated TAT peptide involves clathrin-dependent endocytosis and heparan sulfate receptors. J. Biol. Chem. 2005, 280, 15300–15306. [Google Scholar] [CrossRef]
- Belting, M. Heparan sulfate proteoglycan as a plasma membrane carrier. Trends Biochem. Sci. 2003, 28, 145–151. [Google Scholar] [CrossRef]
- Fuchs, S.M.; Raines, R.T. Pathway for polyarginine entry into mammalian cells. Biochemistry 2004, 43, 2438–2444. [Google Scholar] [CrossRef]
- Berlose, J.P.; Convert, O.; Derossi, D.; Brunissen, A.; Chassaing, G. Conformational and associative behaviours of the third helix of antennapedia homeodomain in membrane-mimetic environments. Eur. J. Biochem. 1996, 242, 372–386. [Google Scholar] [CrossRef] [PubMed]
- Pouny, Y.; Rapaport, D.; Mor, A.; Nicolas, P.; Shai, Y. Interaction of antimicrobial dermaseptin and its fluorescently labeled analogues with phospholipid membranes. Biochemistry 1992, 31, 12416–12423. [Google Scholar] [CrossRef]
- Shai, Y. Mechanism of the binding, insertion and destabilization of phospholipid bilayer membranes by alpha-helical antimicrobial and cell non-selective membrane-lytic peptides. Biochim. Biophys. Acta 1999, 1462, 55–70. [Google Scholar] [CrossRef]
- Matsuzaki, K.; Sugishita, K.; Miyajima, K. Interactions of an antimicrobial peptide, magainin 2, with lipopolysaccharide-containing liposomes as a model for outer membranes of gram-negative bacteria. FEBS Lett. 1999, 449, 221–224. [Google Scholar] [CrossRef]
- Lundberg, P.; Langel, U. A brief introduction to cell-penetrating peptides. J. Mol. Recognit. 2003, 16, 227–233. [Google Scholar] [CrossRef]
- Yang, L.; Harroun, T.A.; Weiss, T.M.; Ding, L.; Huang, H.W. Barrel-stave model or toroidal model? A case study on melittin pores. Biophys. J. 2001, 81, 1475–1485. [Google Scholar] [CrossRef]
- Conner, S.D.; Schmid, S.L. Regulated portals of entry into the cell. Nature 2003, 422, 37–44. [Google Scholar] [CrossRef]
- Mayor, S.; Pagano, R.E. Pathways of clathrin-independent endocytosis. Nat. Rev. Mol. Cel.l Biol. 2007, 8, 603–612. [Google Scholar] [CrossRef]
- Duchardt, F.; Fotin-Mleczek, M.; Schwarz, H.; Fischer, R.; Brock, R. A comprehensive model for the cellular uptake of cationic cell-penetrating peptides. Traffic 2007, 8, 848–866. [Google Scholar] [CrossRef]
- Fischer, R.; Fotin-Mleczek, M.; Hufnagel, H.; Brock, R. Break on through to the other side-biophysics and cell biology shed light on cell-penetrating peptides. Chembiochem 2005, 6, 2126–2142. [Google Scholar] [CrossRef]
- Ferrari, A.; Pellegrini, V.; Arcangeli, C.; Fittipaldi, A.; Giacca, M.; Beltram, F. Caveolae-mediated internalization of extracellular HIV-1 tat fusion proteins visualized in real time. Mol. Ther. 2003, 8, 284–294. [Google Scholar] [CrossRef]
- Nakase, I.; Niwa, M.; Takeuchi, T.; Sonomura, K.; Kawabata, N.; Koike, Y.; Takehashi, M.; Tanaka, S.; Ueda, K.; Simpson, J.C.; Jones, A.T.; Sugiura, Y.; Futaki, S. Cellular uptake of arginine-rich peptides: roles for macropinocytosis and actin rearrangement. Mol. Ther. 2004, 10, 1011–1022. [Google Scholar] [CrossRef]
- Gump, J.M.; June, R.K.; Dowdy, S.F. Revised role of glycosaminoglycans in TAT PTD-mediated cellular transduction. J. Biol. Chem. 2009. [Google Scholar]
- Ter-Avetisyan, G.; Tunnemann, G.; Nowak, D.; Nitschke, M.; Herrmann, A.; Drab, M.; Cardoso, M.C. Cell entry of arginine-rich peptides is independent of endocytosis. J. Biol. Chem. 2009, 284, 3370–3378. [Google Scholar] [CrossRef]
- Jones, A.T. Gateways and tools for drug delivery: endocytic pathways and the cellular dynamics of cell penetrating peptides. Int. J. Pharm. 2008, 354, 34–38. [Google Scholar] [CrossRef] [PubMed]
- Lundin, P.; Johansson, H.; Guterstam, P.; Holm, T.; Hansen, M.; Langel, U.; S, E.L.A. Distinct uptake routes of cell-penetrating peptide conjugates. Bioconjug. Chem. 2008, 19, 2535–2542. [Google Scholar] [CrossRef]
- Soomets, U.; Lindgren, M.; Gallet, X.; Hallbrink, M.; Elmquist, A.; Balaspiri, L.; Zorko, M.; Pooga, M.; Brasseur, R.; Langel, U. Deletion analogues of transportan. Biochim. Biophys. Acta 2000, 1467, 165–176. [Google Scholar] [CrossRef]
- Caron, N.J.; Quenneville, S.P.; Tremblay, J.P. Endosome disruption enhances the functional nuclear delivery of Tat-fusion proteins. Biochem. Biophys. Res. Commun. 2004, 319, 12–20. [Google Scholar] [CrossRef]
- Seglen, P.O.; Grinde, B.; Solheim, A.E. Inhibition of the lysosomal pathway of protein degradation in isolated rat hepatocytes by ammonia, methylamine, chloroquine and leupeptin. Eur. J. Biochem. 1979, 95, 215–225. [Google Scholar] [CrossRef]
- Maiolo 3rd, J.R.; Ottinger, E.A.; Ferrer, M. Specific redistribution of cell-penetrating peptides from endosomes to the cytoplasm and nucleus upon laser illumination. J. Am. Chem. Soc. 2004, 126, 15376–15377. [Google Scholar] [CrossRef]
- Matsushita, M.; Noguchi, H.; Lu, Y.F.; Tomizawa, K.; Michiue, H.; Li, S.T.; Hirose, K.; Bonner-Weir, S.; Matsui, H. Photo-acceleration of protein release from endosome in the protein transduction system. FEBS Lett. 2004, 572, 221–226. [Google Scholar] [CrossRef]
- Veldhoen, S.; Laufer, S.D.; Trampe, A.; Restle, T. Cellular delivery of small interfering RNA by a non-covalently attached cell-penetrating peptide: quantitative analysis of uptake and biological effect. Nucleic Acids Res. 2006, 34, 6561–6573. [Google Scholar] [CrossRef]
- Endoh, T.; Sisido, M.; Ohtsuki, T. Cellular siRNA delivery mediated by a cell-permeant RNA-binding protein and photoinduced RNA interference. Bioconjug. Chem. 2008, 19, 1017–1024. [Google Scholar] [CrossRef]
- Mano, M.; Teodosio, C.; Paiva, A.; Simoes, S.; Pedroso de Lima, M.C. On the mechanisms of the internalization of S4(13)-PV cell-penetrating peptide. Biochem. J. 2005, 390, 603–612. [Google Scholar] [CrossRef]
- Mueller, J.; Kretzschmar, I.; Volkmer, R.; Boisguerin, P. Comparison of cellular uptake using 22 CPPs in 4 different cell lines. Bioconjug. Chem. 2008, 19, 2363–2374. [Google Scholar] [CrossRef]
- Trabulo, S.; Mano, M.; Faneca, H.; Cardoso, A.L.; Duarte, S.; Henriques, A.; Paiva, A.; Gomes, P.; Simoes, S.; de Lima, M.C. S4(13)-PV cell penetrating peptide and cationic liposomes act synergistically to mediate intracellular delivery of plasmid DNA. J. Gene Med. 2008, 10, 1210–1222. [Google Scholar] [CrossRef]
- Mano, M.; Henriques, A.; Paiva, A.; Prieto, M.; Gavilanes, F.; Simoes, S.; de Lima, M.C. Interaction of S413-PV cell penetrating peptide with model membranes: relevance to peptide translocation across biological membranes. J. Pept. Sci. 2007, 13, 301–313. [Google Scholar] [CrossRef]
- Mano, M.; Henriques, A.; Paiva, A.; Prieto, M.; Gavilanes, F.; Simoes, S.; Pedroso de Lima, M.C. Cellular uptake of S413-PV peptide occurs upon conformational changes induced by peptide-membrane interactions. Biochim. Biophys. Acta 2006, 1758, 336–346. [Google Scholar] [CrossRef]
- Magzoub, M.; Kilk, K.; Eriksson, L.E.; Langel, U.; Graslund, A. Interaction and structure induction of cell-penetrating peptides in the presence of phospholipid vesicles. Biochim. Biophys. Acta 2001, 1512, 77–89. [Google Scholar] [CrossRef]
- Magzoub, M.; Eriksson, L.E.; Graslund, A. Comparison of the interaction, positioning, structure induction and membrane perturbation of cell-penetrating peptides and non-translocating variants with phospholipid vesicles. Biophys. Chem. 2003, 103, 271–288. [Google Scholar] [CrossRef]
- Thoren, P.E.; Persson, D.; Esbjorner, E.K.; Goksor, M.; Lincoln, P.; Norden, B. Membrane binding and translocation of cell-penetrating peptides. Biochemistry 2004, 43, 3471–3489. [Google Scholar] [CrossRef]
- Deshayes, S.; Heitz, A.; Morris, M.C.; Charnet, P.; Divita, G.; Heitz, F. Insight into the mechanism of internalization of the cell-penetrating carrier peptide Pep-1 through conformational analysis. Biochemistry 2004, 43, 1449–1457. [Google Scholar] [CrossRef]
- Magzoub, M.; Eriksson, L.E.; Graslund, A. Conformational states of the cell-penetrating peptide penetratin when interacting with phospholipid vesicles: effects of surface charge and peptide concentration. Biochim. Biophys. Acta 2002, 1563, 53–63. [Google Scholar] [CrossRef]
- Joanne, P.; Galanth, C.; Goasdoue, N.; Nicolas, P.; Sagan, S.; Lavielle, S.; Chassaing, G.; El Amri, C.; Alves, I.D. Lipid reorganization induced by membrane-active peptides probed using differential scanning calorimetry. Biochim. Biophys. Acta 2009, 1788, 1772–1781. [Google Scholar] [CrossRef]
- Deshayes, S.; Gerbal-Chaloin, S.; Morris, M.C.; Aldrian-Herrada, G.; Charnet, P.; Divita, G.; Heitz, F. On the mechanism of non-endosomial peptide-mediated cellular delivery of nucleic acids. Biochim. Biophys. Acta 2004, 1667, 141–147. [Google Scholar] [CrossRef]
- Ziegler, A.; Blatter, X.L.; Seelig, A.; Seelig, J. Protein transduction domains of HIV-1 and SIV TAT interact with charged lipid vesicles. Binding mechanism and thermodynamic analysis. Biochemistry 2003, 42, 9185–9194. [Google Scholar] [CrossRef]
- Opalinska, J.B.; Gewirtz, A.M. Nucleic-acid therapeutics: basic principles and recent applications. Nat. Rev. Drug Discov. 2002, 1, 503–514. [Google Scholar] [CrossRef]
- Said Hassane, F.; Saleh, A.F.; Abes, R.; Gait, M.J.; Lebleu, B. Cell penetrating peptides: overview and applications to the delivery of oligonucleotides. Cell. Mol. Life Sci. 2009. [Google Scholar]
- Veldhoen, S.; Laufer, S.D.; Restle, T. Recent developments in Peptide-based nucleic Acid delivery. Int. J. Mol. Sci. 2008, 9, 1276–1320. [Google Scholar] [CrossRef]
- Torchilin, V.P. Cell penetrating peptide-modified pharmaceutical nanocarriers for intracellular drug and gene delivery. Biopolymers 2008, 90, 604–610. [Google Scholar] [CrossRef]
- Temsamani, J.; Vidal, P. The use of cell-penetrating peptides for drug delivery. Drug Discov. Today 2004, 9, 1012–1019. [Google Scholar] [CrossRef]
- Kilk, K.; El-Andaloussi, S.; Jarver, P.; Meikas, A.; Valkna, A.; Bartfai, T.; Kogerman, P.; Metsis, M.; Langel, U. Evaluation of transportan 10 in PEI mediated plasmid delivery assay. J. Control. Release 2005, 103, 511–523. [Google Scholar] [CrossRef]
- Rudolph, C.; Plank, C.; Lausier, J.; Schillinger, U.; Muller, R.H.; Rosenecker, J. Oligomers of the arginine-rich motif of the HIV-1 TAT protein are capable of transferring plasmid DNA into cells. J. Biol. Chem. 2003, 278, 11411–11418. [Google Scholar] [CrossRef]
- Ignatovich, I.A.; Dizhe, E.B.; Pavlotskaya, A.V.; Akifiev, B.N.; Burov, S.V.; Orlov, S.V.; Perevozchikov, A.P. Complexes of plasmid DNA with basic domain 47-57 of the HIV-1 Tat protein are transferred to mammalian cells by endocytosis-mediated pathways. J. Biol. Chem. 2003, 278, 42625–42636. [Google Scholar] [CrossRef]
- Kogure, K.; Moriguchi, R.; Sasaki, K.; Ueno, M.; Futaki, S.; Harashima, H. Development of a non-viral multifunctional envelope-type nano device by a novel lipid film hydration method. J. Control. Release 2004, 98, 317–323. [Google Scholar] [CrossRef]
- Kogure, K.; Akita, H.; Harashima, H. Multifunctional envelope-type nano device for non-viral gene delivery: concept and application of Programmed Packaging. J. Control. Release 2007, 122, 246–251. [Google Scholar] [CrossRef]
- Khalil, I.A.; Kogure, K.; Futaki, S.; Hama, S.; Akita, H.; Ueno, M.; Kishida, H.; Kudoh, M.; Mishina, Y.; Kataoka, K.; Yamada, M.; Harashima, H. Octaarginine-modified multifunctional envelope-type nanoparticles for gene delivery. Gene Ther. 2007, 14, 682–689. [Google Scholar] [CrossRef]
- Tung, C.H.; Mueller, S.; Weissleder, R. Novel branching membrane translocational peptide as gene delivery vector. Bioorg. Med. Chem. 2002, 10, 3609–3614. [Google Scholar] [CrossRef]
- Vandenbroucke, R.E.; De Smedt, S.C.; Demeester, J.; Sanders, N.N. Cellular entry pathway and gene transfer capacity of TAT-modified lipoplexes. Biochim. Biophys. Acta 2007, 1768, 571–579. [Google Scholar] [CrossRef]
- Zhang, C.; Tang, N.; Liu, X.; Liang, W.; Xu, W.; Torchilin, V.P. siRNA-containing liposomes modified with polyarginine effectively silence the targeted gene. J. Control. Release 2006, 112, 229–239. [Google Scholar] [CrossRef]
- Kumar, P.; Wu, H.; McBride, J.L.; Jung, K.E.; Kim, M.H.; Davidson, B.L.; Lee, S.K.; Shankar, P.; Manjunath, N. Transvascular delivery of small interfering RNA to the central nervous system. Nature 2007, 448, 39–43. [Google Scholar] [CrossRef]
- Tan, M.L.; Choong, P.F.; Dass, C.R. Recent developments in liposomes, microparticles and nanoparticles for protein and peptide drug delivery. Peptides 2009, 31, 184–193. [Google Scholar] [CrossRef]
- Wadia, J.S.; Dowdy, S.F. Transmembrane delivery of protein and peptide drugs by TAT-mediated transduction in the treatment of cancer. Adv. Drug Deliv. Rev. 2005, 57, 579–596. [Google Scholar] [CrossRef]
- Mi, Z.; Mai, J.; Lu, X.; Robbins, P.D. Characterization of a class of cationic peptides able to facilitate efficient protein transduction in vitro and in vivo. Mol. Ther. 2000, 2, 339–347. [Google Scholar] [CrossRef]
- Fawell, S.; Seery, J.; Daikh, Y.; Moore, C.; Chen, L.L.; Pepinsky, B.; Barsoum, J. Tat-mediated delivery of heterologous proteins into cells. Proc. Natl. Acad. Sci. USA 1994, 91, 664–668. [Google Scholar] [CrossRef]
- Schwarze, S.R.; Ho, A.; Vocero-Akbani, A.; Dowdy, S.F. In vivo protein transduction: delivery of a biologically active protein into the mouse. Science 1999, 285, 1569–1572. [Google Scholar] [CrossRef]
- Caron, N.J.; Torrente, Y.; Camirand, G.; Bujold, M.; Chapdelaine, P.; Leriche, K.; Bresolin, N.; Tremblay, J.P. Intracellular delivery of a Tat-eGFP fusion protein into muscle cells. Mol. Ther. 2001, 3, 310–318. [Google Scholar] [CrossRef]
- Cao, G.; Pei, W.; Ge, H.; Liang, Q.; Luo, Y.; Sharp, F.R.; Lu, A.; Ran, R.; Graham, S.H.; Chen, J. In vivo Delivery of a Bcl-xL Fusion Protein Containing the TAT Protein Transduction Domain Protects against Ischemic Brain Injury and Neuronal Apoptosis. J. Neurosci. 2002, 22, 5423–5431. [Google Scholar] [PubMed]
- Embury, J.; Klein, D.; Pileggi, A.; Ribeiro, M.; Jayaraman, S.; Molano, R.D.; Fraker, C.; Kenyon, N.; Ricordi, C.; Inverardi, L.; Pastori, R.L. Proteins linked to a protein transduction domain efficiently transduce pancreatic islets. Diabetes 2001, 50, 1706–1713. [Google Scholar] [CrossRef]
- Jin, L.H.; Bahn, J.H.; Eum, W.S.; Kwon, H.Y.; Jang, S.H.; Han, K.H.; Kang, T.C.; Won, M.H.; Kang, J.H.; Cho, S.W.; Park, J.; Choi, S.Y. Transduction of human catalase mediated by an HIV-1 TAT protein basic domain and arginine-rich peptides into mammalian cells. Free Radic. Biol. Med. 2001, 31, 1509–1519. [Google Scholar] [CrossRef]
- Yoon, H.Y.; Lee, S.H.; Cho, S.W.; Lee, J.E.; Yoon, C.S.; Park, J.; Kim, T.U.; Choi, S.Y. TAT-mediated delivery of human glutamate dehydrogenase into PC12 cells. Neurochem. Int. 2002, 41, 37–42. [Google Scholar] [CrossRef]
- Kwon, H.Y.; Eum, W.S.; Jang, H.W.; Kang, J.H.; Ryu, J.; Ryong Lee, B.; Jin, L.H.; Park, J.; Choi, S.Y. Transduction of Cu,Zn-superoxide dismutase mediated by an HIV-1 Tat protein basic domain into mammalian cells. FEBS Lett. 2000, 485, 163–167. [Google Scholar] [CrossRef]
- Kabouridis, P.S.; Hasan, M.; Newson, J.; Gilroy, D.W.; Lawrence, T. Inhibition of NF-kappa B activity by a membrane-transducing mutant of I kappa B alpha. J. Immunol. 2002, 169, 2587–2593. [Google Scholar] [CrossRef] [PubMed]
- Wheeler, D.S.; Dunsmore, K.E.; Wong, H.R. Intracellular delivery of HSP70 using HIV-1 Tat protein transduction domain. Biochem. Biophys. Res. Commun. 2003, 301, 54–59. [Google Scholar] [CrossRef]
- Deshayes, S.; Morris, M.; Heitz, F.; Divita, G. Delivery of proteins and nucleic acids using a non-covalent peptide-based strategy. Adv. Drug Deliv. Rev. 2008, 60, 537–547. [Google Scholar] [CrossRef]
- Gros, E.; Deshayes, S.; Morris, M.C.; Aldrian-Herrada, G.; Depollier, J.; Heitz, F.; Divita, G. A non-covalent peptide-based strategy for protein and peptide nucleic acid transduction. Biochim. Biophys. Acta 2006, 1758, 384–393. [Google Scholar] [CrossRef]
- Torchilin, V.P. Multifunctional nanocarriers. Adv. Drug Deliv. Rev. 2006, 58, 1532–1555. [Google Scholar] [CrossRef]
- McCarthy, J.R.; Kelly, K.A.; Sun, E.Y.; Weissleder, R. Targeted delivery of multifunctional magnetic nanoparticles. Nanomed. 2007, 2, 153–167. [Google Scholar] [CrossRef]
- McCarthy, J.R.; Weissleder, R. Multifunctional magnetic nanoparticles for targeted imaging and therapy. Adv. Drug Deliv. Rev. 2008, 60, 1241–1251. [Google Scholar] [CrossRef]
- Josephson, L.; Tung, C.H.; Moore, A.; Weissleder, R. High-efficiency intracellular magnetic labeling with novel superparamagnetic-Tat peptide conjugates. Bioconjug. Chem. 1999, 10, 186–191. [Google Scholar] [CrossRef]
- Lewin, M.; Carlesso, N.; Tung, C.H.; Tang, X.W.; Cory, D.; Scadden, D.T.; Weissleder, R. Tat peptide-derivatized magnetic nanoparticles allow in vivo tracking and recovery of progenitor cells. Nat. Biotechnol. 2000, 18, 410–414. [Google Scholar] [CrossRef]
- Torchilin, V.P.; Levchenko, T.S.; Rammohan, R.; Volodina, N.; Papahadjopoulos-Sternberg, B.; D'Souza, G.G. Cell transfection in vitro and in vivo with nontoxic TAT peptide-liposome-DNA complexes. Proc. Natl. Acad. Sci. U. S. A. 2003, 100, 1972–1977. [Google Scholar] [CrossRef]
- Tseng, Y.L.; Liu, J.J.; Hong, R.L. Translocation of liposomes into cancer cells by cell-penetrating peptides penetratin and tat: a kinetic and efficacy study. Mol. Pharmacol. 2002, 62, 864–872. [Google Scholar] [CrossRef]
- Jarver, P.; Langel, U. The use of cell-penetrating peptides as a tool for gene regulation. Drug Discov. Today 2004, 9, 395–402. [Google Scholar] [CrossRef]
- Laufer, S.D.; Restle, T. Peptide-mediated cellular delivery of oligonucleotide-based therapeutics in vitro: quantitative evaluation of overall efficacy employing easy to handle reporter systems. Curr. Pharm. Des. 2008, 14, 3637–3655. [Google Scholar] [CrossRef]
- Gleave, M.E.; Monia, B.P. Antisense therapy for cancer. Nat. Rev. Cancer 2005, 5, 468–479. [Google Scholar] [CrossRef]
- Juliano, R.; Bauman, J.; Kang, H.; Ming, X. Biological barriers to therapy with antisense and siRNA oligonucleotides. Mol. Pharm. 2009, 6, 686–695. [Google Scholar] [CrossRef]
- Juliano, R.; Alam, M.R.; Dixit, V.; Kang, H. Mechanisms and strategies for effective delivery of antisense and siRNA oligonucleotides. Nucleic Acids Res. 2008, 36, 4158–4171. [Google Scholar] [CrossRef]
- Kurreck, J. Antisense technologies. Improvement through novel chemical modifications. Eur. J. Biochem. 2003, 270, 1628–1644. [Google Scholar] [CrossRef]
- Lebleu, B.; Moulton, H.M.; Abes, R.; Ivanova, G.D.; Abes, S.; Stein, D.A.; Iversen, P.L.; Arzumanov, A.A.; Gait, M.J. Cell penetrating peptide conjugates of steric block oligonucleotides. Adv. Drug Deliv. Rev. 2008, 60, 517–529. [Google Scholar] [CrossRef]
- Gait, M.J. Peptide-mediated cellular delivery of antisense oligonucleotides and their analogues. Cell. Mol. Life Sci. 2003, 60, 844–853. [Google Scholar] [PubMed]
- Morris, M.C.; Vidal, P.; Chaloin, L.; Heitz, F.; Divita, G. A new peptide vector for efficient delivery of oligonucleotides into mammalian cells. Nucleic Acids Res. 1997, 25, 2730–2736. [Google Scholar] [CrossRef]
- Zatsepin, T.S.; Turner, J.J.; Oretskaya, T.S.; Gait, M.J. Conjugates of oligonucleotides and analogues with cell penetrating peptides as gene silencing agents. Curr. Pharm. Des. 2005, 11, 3639–3654. [Google Scholar] [CrossRef]
- Pooga, M.; Soomets, U.; Hallbrink, M.; Valkna, A.; Saar, K.; Rezaei, K.; Kahl, U.; Hao, J.X.; Xu, X.J.; Wiesenfeld-Hallin, Z.; Hokfelt, T.; Bartfai, T.; Langel, U. Cell penetrating PNA constructs regulate galanin receptor levels and modify pain transmission in vivo. Nat. Biotechnol. 1998, 16, 857–861. [Google Scholar] [CrossRef]
- Abes, S.; Moulton, H.M.; Clair, P.; Prevot, P.; Youngblood, D.S.; Wu, R.P.; Iversen, P.L.; Lebleu, B. Vectorization of morpholino oligomers by the (R-Ahx-R)4 peptide allows efficient splicing correction in the absence of endosomolytic agents. J. Control. Release 2006, 116, 304–313. [Google Scholar] [CrossRef]
- Abes, R.; Moulton, H.M.; Clair, P.; Yang, S.T.; Abes, S.; Melikov, K.; Prevot, P.; Youngblood, D.S.; Iversen, P.L.; Chernomordik, L.V.; Lebleu, B. Delivery of steric block morpholino oligomers by (R-X-R)4 peptides: structure-activity studies. Nucleic Acids Res. 2008, 36, 6343–6354. [Google Scholar] [CrossRef]
- Laufer, S.D.; Recke, A.L.; Veldhoen, S.; Trampe, A.; Restle, T. Noncovalent Peptide-Mediated Delivery of Chemically Modified Steric Block Oligonucleotides Promotes Splice Correction: Quantitative Analysis of Uptake and Biological Effect. Oligonucleotides 2009, 19, 63–80. [Google Scholar] [CrossRef]
- Lehto, T.; Abes, R.; Oskolkov, N.; Suhorutsenko, J.; Copolovici, D.M.; Mager, I.; Viola, J.R.; Simonson, O.E.; Ezzat, K.; Guterstam, P.; Eriste, E.; Smith, C.I.; Lebleu, B.; Samir El, A.; Langel, U. Delivery of nucleic acids with a stearylated (RxR)(4) peptide using a non-covalent co-incubation strategy. J. Control. Release 2009, 141, 42–51. [Google Scholar] [CrossRef]
- Mae, M.; El Andaloussi, S.; Lundin, P.; Oskolkov, N.; Johansson, H.J.; Guterstam, P.; Langel, U. A stearylated CPP for delivery of splice correcting oligonucleotides using a non-covalent co-incubation strategy. J. Control. Release 2009, 134, 221–227. [Google Scholar] [CrossRef]
- Morris, M.C.; Chaloin, L.; Choob, M.; Archdeacon, J.; Heitz, F.; Divita, G. Combination of a new generation of PNAs with a peptide-based carrier enables efficient targeting of cell cycle progression. Gene Ther. 2004, 11, 757–764. [Google Scholar] [CrossRef]
- Morris, M.C.; Gros, E.; Aldrian-Herrada, G.; Choob, M.; Archdeacon, J.; Heitz, F.; Divita, G. A non-covalent peptide-based carrier for in vivo delivery of DNA mimics. Nucleic Acids Res. 2007, 35, e49. [Google Scholar] [CrossRef]
- Oehlke, J.; Wallukat, G.; Wolf, Y.; Ehrlich, A.; Wiesner, B.; Berger, H.; Bienert, M. Enhancement of intracellular concentration and biological activity of PNA after conjugation with a cell-penetrating synthetic model peptide. Eur. J. Biochem. 2004, 271, 3043–3049. [Google Scholar] [CrossRef]
- Kang, S.H.; Cho, M.J.; Kole, R. Up-regulation of luciferase gene expression with antisense oligonucleotides: implications and applications in functional assay development. Biochemistry 1998, 37, 6235–6239. [Google Scholar] [CrossRef]
- Turner, J.J.; Jones, S.; Fabani, M.M.; Ivanova, G.; Arzumanov, A.A.; Gait, M.J. RNA targeting with peptide conjugates of oligonucleotides, siRNA and PNA. Blood Cells Mol. Dis. 2007, 38, 1–7. [Google Scholar] [CrossRef]
- Moulton, H.M.; Nelson, M.H.; Hatlevig, S.A.; Reddy, M.T.; Iversen, P.L. Cellular uptake of antisense morpholino oligomers conjugated to arginine-rich peptides. Bioconjug. Chem. 2004, 15, 290–299. [Google Scholar] [CrossRef]
- Abes, S.; Williams, D.; Prevot, P.; Thierry, A.; Gait, M.J.; Lebleu, B. Endosome trapping limits the efficiency of splicing correction by PNA-oligolysine conjugates. J. Control. Release 2006, 110, 595–604. [Google Scholar] [CrossRef]
- Shiraishi, T.; Pankratova, S.; Nielsen, P.E. Calcium ions effectively enhance the effect of antisense peptide nucleic acids conjugated to cationic tat and oligoarginine peptides. Chem. Biol. 2005, 12, 923–929. [Google Scholar] [CrossRef]
- Wolf, Y.; Pritz, S.; Abes, S.; Bienert, M.; Lebleu, B.; Oehlke, J. Structural requirements for cellular uptake and antisense activity of peptide nucleic acids conjugated with various peptides. Biochemistry 2006, 45, 14944–14954. [Google Scholar] [CrossRef]
- Abes, S.; Moulton, H.; Turner, J.; Clair, P.; Richard, J.P.; Iversen, P.; Gait, M.J.; Lebleu, B. Peptide-based delivery of nucleic acids: design, mechanism of uptake and applications to splice-correcting oligonucleotides. Biochem. Soc. Trans. 2007, 35, 53–55. [Google Scholar] [CrossRef]
- El-Andaloussi, S.; Johansson, H.J.; Lundberg, P.; Langel, U. Induction of splice correction by cell-penetrating peptide nucleic acids. J. Gene Med. 2006, 8, 1262–1273. [Google Scholar] [CrossRef]
- Berg, K.; Prasmickaite, L.; Selbo, P.K.; Hellum, M.; Bonsted, A.; Hogset, A. Photochemical internalization (PCI)--a novel technology for release of macromolecules from endocytic vesicles. Oftalmologia 2003, 56, 67–71. [Google Scholar] [PubMed]
- Boe, S.; Hovig, E. Photochemically induced gene silencing using PNA-peptide conjugates. Oligonucleotides 2006, 16, 145–157. [Google Scholar] [CrossRef]
- Shiraishi, T.; Bendifallah, N.; Nielsen, P.E. Cellular delivery of polyheteroaromate-peptide nucleic acid conjugates mediated by cationic lipids. Bioconjug. Chem. 2006, 17, 189–194. [Google Scholar] [CrossRef]
- Shiraishi, T.; Nielsen, P.E. Photochemically enhanced cellular delivery of cell penetrating peptide-PNA conjugates. FEBS Lett. 2006, 580, 1451–1456. [Google Scholar] [CrossRef]
- Abes, S.; Turner, J.J.; Ivanova, G.D.; Owen, D.; Williams, D.; Arzumanov, A.; Clair, P.; Gait, M.J.; Lebleu, B. Efficient splicing correction by PNA conjugation to an R6-Penetratin delivery peptide. Nucleic Acids Res. 2007, 35, 4495–4502. [Google Scholar] [CrossRef]
- Jearawiriyapaisarn, N.; Moulton, H.M.; Buckley, B.; Roberts, J.; Sazani, P.; Fucharoen, S.; Iversen, P.L.; Kole, R. Sustained dystrophin expression induced by peptide-conjugated morpholino oligomers in the muscles of mdx mice. Mol. Ther. 2008, 16, 1624–1629. [Google Scholar] [CrossRef]
- Yin, H.; Moulton, H.M.; Betts, C.; Seow, Y.; Boutilier, J.; Iverson, P.L.; Wood, M.J. A fusion peptide directs enhanced systemic dystrophin exon skipping and functional restoration in dystrophin-deficient mdx mice. Hum. Mol. Genet. 2009, 18, 4405–4414. [Google Scholar] [CrossRef]
- Yin, H.; Moulton, H.M.; Seow, Y.; Boyd, C.; Boutilier, J.; Iverson, P.; Wood, M.J. Cell-penetrating peptide-conjugated antisense oligonucleotides restore systemic muscle and cardiac dystrophin expression and function. Hum. Mol. Genet. 2008, 17, 3909–3918. [Google Scholar] [CrossRef]
- Trabulo, S.; Resina, S.; Simões, S.; Lebleu, B.; Pedroso de Lima, M.C. A non-covalent strategy combining cationic lipids and CPPs to enhance the delivery of splice correcting oligonucleotides. J. Control. Release 2010, in press. [Google Scholar]
- Grunweller, A.; Hartmann, R.K. RNA interference as a gene-specific approach for molecular medicine. Curr. Med. Chem. 2005, 12, 3143–3161. [Google Scholar] [CrossRef]
- Lu, P.Y.; Xie, F.; Woodle, M.C. In vivo application of RNA interference: from functional genomics to therapeutics. Adv. Genet. 2005, 54, 117–142. [Google Scholar] [PubMed]
- Hannon, G.J. RNA interference. Nature 2002, 418, 244–251. [Google Scholar] [CrossRef]
- Dorsett, Y.; Tuschl, T. siRNAs: applications in functional genomics and potential as therapeutics. Nat. Rev. Drug Discov. 2004, 3, 318–329. [Google Scholar] [CrossRef]
- de Fougerolles, A.; Vornlocher, H.P.; Maraganore, J.; Lieberman, J. Interfering with disease: a progress report on siRNA-based therapeutics. Nat. Rev. Drug Discov. 2007, 6, 443–453. [Google Scholar] [CrossRef]
- Eguchi, A.; Dowdy, S.F. siRNA delivery using peptide transduction domains. Trends Pharmacol. Sci. 2009, 30, 341–345. [Google Scholar] [CrossRef]
- Endoh, T.; Ohtsuki, T. Cellular siRNA delivery using cell-penetrating peptides modified for endosomal escape. Adv. Drug Deliv. Rev. 2009, 61, 704–709. [Google Scholar] [CrossRef]
- Muratovska, A.; Eccles, M.R. Conjugate for efficient delivery of short interfering RNA (siRNA) into mammalian cells. FEBS Lett. 2004, 558, 63–68. [Google Scholar] [CrossRef]
- Chiu, Y.L.; Ali, A.; Chu, C.Y.; Cao, H.; Rana, T.M. Visualizing a correlation between siRNA localization, cellular uptake, and RNAi in living cells. Chem. Biol. 2004, 11, 1165–1175. [Google Scholar] [CrossRef]
- Davidson, T.J.; Harel, S.; Arboleda, V.A.; Prunell, G.F.; Shelanski, M.L.; Greene, L.A.; Troy, C.M. Highly efficient small interfering RNA delivery to primary mammalian neurons induces MicroRNA-like effects before mRNA degradation. J. Neurosci. 2004, 24, 10040–10046. [Google Scholar] [CrossRef]
- Meade, B.R.; Dowdy, S.F. Enhancing the cellular uptake of siRNA duplexes following noncovalent packaging with protein transduction domain peptides. Adv. Drug Deliv. Rev. 2008, 60, 530–536. [Google Scholar] [CrossRef]
- Turner, J.J.; Ivanova, G.D.; Verbeure, B.; Williams, D.; Arzumanov, A.A.; Abes, S.; Lebleu, B.; Gait, M.J. Cell-penetrating peptide conjugates of peptide nucleic acids (PNA) as inhibitors of HIV-1 Tat-dependent trans-activation in cells. Nucleic Acids Res. 2005, 33, 6837–6849. [Google Scholar] [CrossRef]
- Simeoni, F.; Morris, M.C.; Heitz, F.; Divita, G. Insight into the mechanism of the peptide-based gene delivery system MPG: implications for delivery of siRNA into mammalian cells. Nucleic Acids Res. 2003, 31, 2717–2724. [Google Scholar] [CrossRef]
- Lundberg, P.; El-Andaloussi, S.; Sutlu, T.; Johansson, H.; Langel, U. Delivery of short interfering RNA using endosomolytic cell-penetrating peptides. FASEB J. 2007, 21, 2664–2671. [Google Scholar] [CrossRef]
- Crombez, L.; Charnet, A.; Morris, M.C.; Aldrian-Herrada, G.; Heitz, F.; Divita, G. A non-covalent peptide-based strategy for siRNA delivery. Biochem. Soc. Trans. 2007, 35, 44–46. [Google Scholar] [CrossRef]
- Eguchi, A.; Meade, B.R.; Chang, Y.C.; Fredrickson, C.T.; Willert, K.; Puri, N.; Dowdy, S.F. Efficient siRNA delivery into primary cells by a peptide transduction domain-dsRNA binding domain fusion protein. Nat. Biotechnol. 2009, 27, 567–571. [Google Scholar] [CrossRef]
- Kim, W.J.; Christensen, L.V.; Jo, S.; Yockman, J.W.; Jeong, J.H.; Kim, Y.H.; Kim, S.W. Cholesteryl oligoarginine delivering vascular endothelial growth factor siRNA effectively inhibits tumor growth in colon adenocarcinoma. Mol. Ther. 2006, 14, 343–350. [Google Scholar] [CrossRef]
- Zeineddine, D.; Papadimou, E.; Chebli, K.; Gineste, M.; Liu, J.; Grey, C.; Thurig, S.; Behfar, A.; Wallace, V.A.; Skerjanc, I.S.; Puceat, M. Oct-3/4 dose dependently regulates specification of embryonic stem cells toward a cardiac lineage and early heart development. Dev. Cell 2006, 11, 535–546. [Google Scholar] [CrossRef]
- Crombez, L.; Morris, M.C.; Dufort, S.; Aldrian-Herrada, G.; Nguyen, Q.; Mc Master, G.; Coll, J.L.; Heitz, F.; Divita, G. Targeting cyclin B1 through peptide-based delivery of siRNA prevents tumour growth. Nucleic Acids Res. 2009, 37, 4559–4569. [Google Scholar] [CrossRef]
- Li, S.; H., L. Lipidic supramolecular Assemblies for Gene Transfer. J. Liposome Res. 1996, 6, 589–608. [Google Scholar] [CrossRef]
- Simoes, S.; Slepushkin, V.; Pires, P.; Gaspar, R.; de Lima, M.P.; Duzgunes, N. Mechanisms of gene transfer mediated by lipoplexes associated with targeting ligands or pH-sensitive peptides. Gene Ther. 1999, 6, 1798–1807. [Google Scholar] [CrossRef]
- de Fougerolles, A.R. Delivery vehicles for small interfering RNA in vivo . Hum. Gene Ther. 2008, 19, 125–132. [Google Scholar] [CrossRef]
- Glover, D.J.; Lipps, H.J.; Jans, D.A. Towards safe, non-viral therapeutic gene expression in humans. Nat. Rev. Genet. 2005, 6, 299–310. [Google Scholar] [CrossRef]
- Simoes, S.; Slepushkin, V.; Gaspar, R.; de Lima, M.C.; Duzgunes, N. Gene delivery by negatively charged ternary complexes of DNA, cationic liposomes and transferrin or fusigenic peptides. Gene Ther. 1998, 5, 955–964. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Huang, L. Nonviral gene therapy: promises and challenges. Gene Ther. 2000, 7, 31–34. [Google Scholar] [CrossRef]
- Martin, M.E.; Rice, K.G. Peptide-guided gene delivery. AAPS J. 2007, 9, E18–E29. [Google Scholar] [CrossRef]
- Ogris, M.; Wagner, E. Targeting tumors with non-viral gene delivery systems. Drug Discov. Today 2002, 7, 479–485. [Google Scholar] [CrossRef]
- Morris, M.C.; Chaloin, L.; Mery, J.; Heitz, F.; Divita, G. A novel potent strategy for gene delivery using a single peptide vector as a carrier. Nucleic Acids Res. 1999, 27, 3510–3517. [Google Scholar] [CrossRef]
- Rittner, K.; Benavente, A.; Bompard-Sorlet, A.; Heitz, F.; Divita, G.; Brasseur, R.; Jacobs, E. New basic membrane-destabilizing peptides for plasmid-based gene delivery in vitro and in vivo . Mol. Ther. 2002, 5, 104–114. [Google Scholar] [CrossRef]
- Futaki, S.; Ohashi, W.; Suzuki, T.; Niwa, M.; Tanaka, S.; Ueda, K.; Harashima, H.; Sugiura, Y. Stearylated arginine-rich peptides: a new class of transfection systems. Bioconjug. Chem. 2001, 12, 1005–1011. [Google Scholar] [CrossRef]
- Khalil, I.A.; Futaki, S.; Niwa, M.; Baba, Y.; Kaji, N.; Kamiya, H.; Harashima, H. Mechanism of improved gene transfer by the N-terminal stearylation of octaarginine: enhanced cellular association by hydrophobic core formation. Gene Ther. 2004, 11, 636–644. [Google Scholar] [CrossRef]
- Sandgren, S.; Cheng, F.; Belting, M. Nuclear targeting of macromolecular polyanions by an HIV-Tat derived peptide. Role for cell-surface proteoglycans. J. Biol. Chem. 2002, 277, 38877–38883. [Google Scholar] [CrossRef]
- Lo, S.L.; Wang, S. An endosomolytic Tat peptide produced by incorporation of histidine and cysteine residues as a nonviral vector for DNA transfection. Biomaterials 2008, 29, 2408–2414. [Google Scholar] [CrossRef]
- Torchilin, V.P. Tat peptide-mediated intracellular delivery of pharmaceutical nanocarriers. Adv. Drug Deliv. Rev. 2008, 60, 548–558. [Google Scholar] [CrossRef]
- Vives, E.; Schmidt, J.; Pelegrin, A. Cell-penetrating and cell-targeting peptides in drug delivery. Biochim. Biophys. Acta 2008, 1786, 126–138. [Google Scholar] [PubMed]
- Branden, L.J.; Mohamed, A.J.; Smith, C.I. A peptide nucleic acid-nuclear localization signal fusion that mediates nuclear transport of DNA. Nat. Biotechnol. 1999, 17, 784–787. [Google Scholar] [CrossRef]
- Kleemann, E.; Neu, M.; Jekel, N.; Fink, L.; Schmehl, T.; Gessler, T.; Seeger, W.; Kissel, T. Nano-carriers for DNA delivery to the lung based upon a TAT-derived peptide covalently coupled to PEG-PEI. J. Control. Release 2005, 109, 299–316. [Google Scholar] [CrossRef]
- Torchilin, V.P.; Rammohan, R.; Weissig, V.; Levchenko, T.S. TAT peptide on the surface of liposomes affords their efficient intracellular delivery even at low temperature and in the presence of metabolic inhibitors. Proc. Natl. Acad. Sci. USA 2001, 98, 8786–8791. [Google Scholar] [CrossRef]
- MacKay, J.A.; Li, W.; Huang, Z.; Dy, E.E.; Huynh, G.; Tihan, T.; Collins, R.; Deen, D.F.; Szoka Jr., F.C. HIV TAT peptide modifies the distribution of DNA nanolipoparticles following convection-enhanced delivery. Mol. Ther. 2008, 16, 893–900. [Google Scholar] [CrossRef]
- Hyndman, L.; Lemoine, J.L.; Huang, L.; Porteous, D.J.; Boyd, A.C.; Nan, X. HIV-1 Tat protein transduction domain peptide facilitates gene transfer in combination with cationic liposomes. J. Control. Release 2004, 99, 435–444. [Google Scholar] [CrossRef]
- El-Sayed, A.; Futaki, S.; Harashima, H. Delivery of macromolecules using arginine-rich cell-penetrating peptides: ways to overcome endosomal entrapment. AAPS J. 2009, 11, 13–22. [Google Scholar] [CrossRef]
- El-Sayed, A.; Khalil, I.A.; Kogure, K.; Futaki, S.; Harashima, H. Octaarginine- and octalysine-modified nanoparticles have different modes of endosomal escape. J. Biol. Chem. 2008, 283, 23450–23461. [Google Scholar] [CrossRef]
© 2010 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Share and Cite
Trabulo, S.; Cardoso, A.L.; Mano, M.; De Lima, M.C.P. Cell-Penetrating Peptides—Mechanisms of Cellular Uptake and Generation of Delivery Systems. Pharmaceuticals 2010, 3, 961-993. https://doi.org/10.3390/ph3040961
Trabulo S, Cardoso AL, Mano M, De Lima MCP. Cell-Penetrating Peptides—Mechanisms of Cellular Uptake and Generation of Delivery Systems. Pharmaceuticals. 2010; 3(4):961-993. https://doi.org/10.3390/ph3040961
Chicago/Turabian StyleTrabulo, Sara, Ana Luísa Cardoso, Miguel Mano, and Maria C. Pedroso De Lima. 2010. "Cell-Penetrating Peptides—Mechanisms of Cellular Uptake and Generation of Delivery Systems" Pharmaceuticals 3, no. 4: 961-993. https://doi.org/10.3390/ph3040961