Cell-Penetrating Fragments of the Cdk5 Regulatory Subunit Are Protective in Models of Neurodegeneration


Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Experimental Section
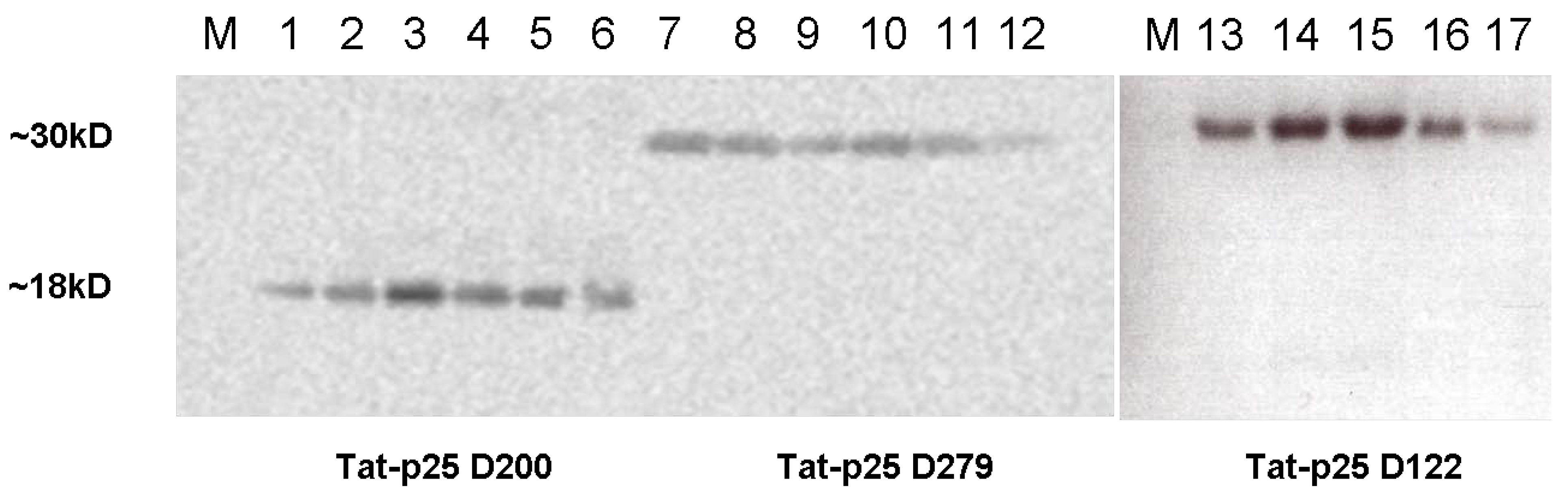
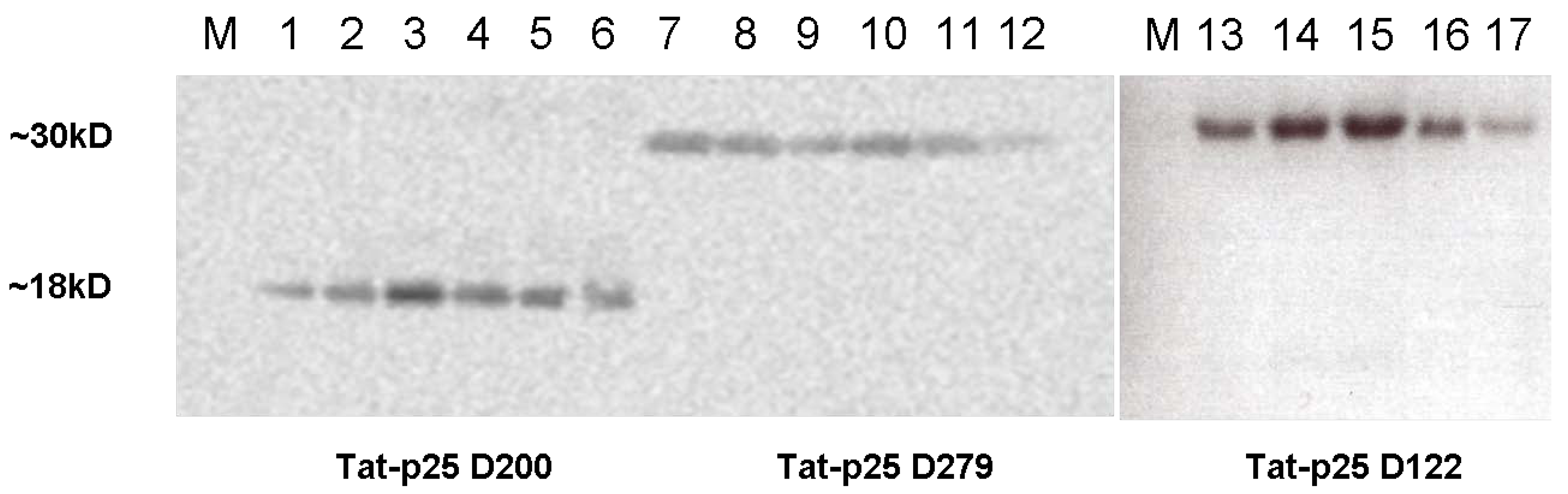
2.1. Cloning of constructs and purification of Tat fusion proteins
2.2. Cell culture and cell death assays
3. Results and Discussion
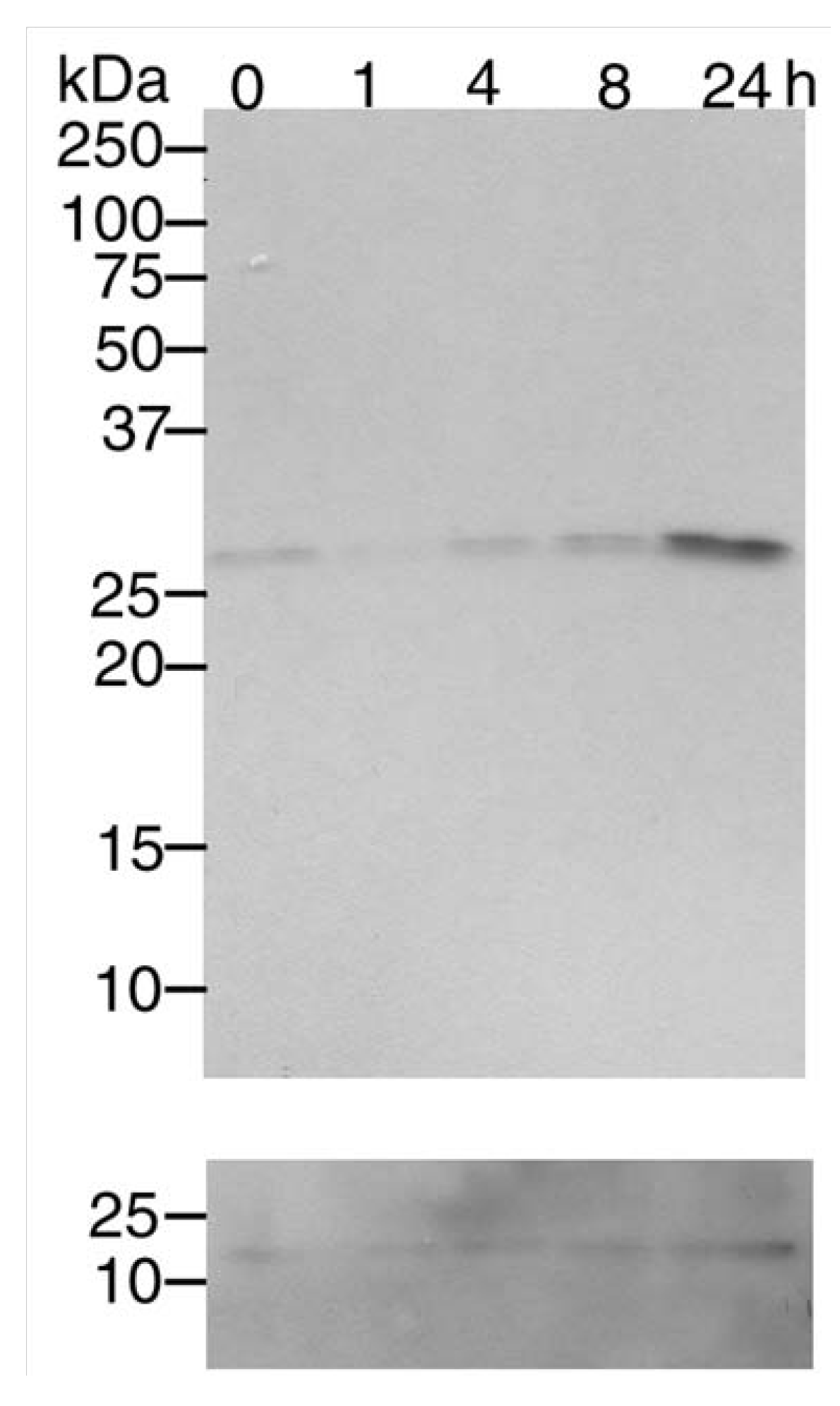
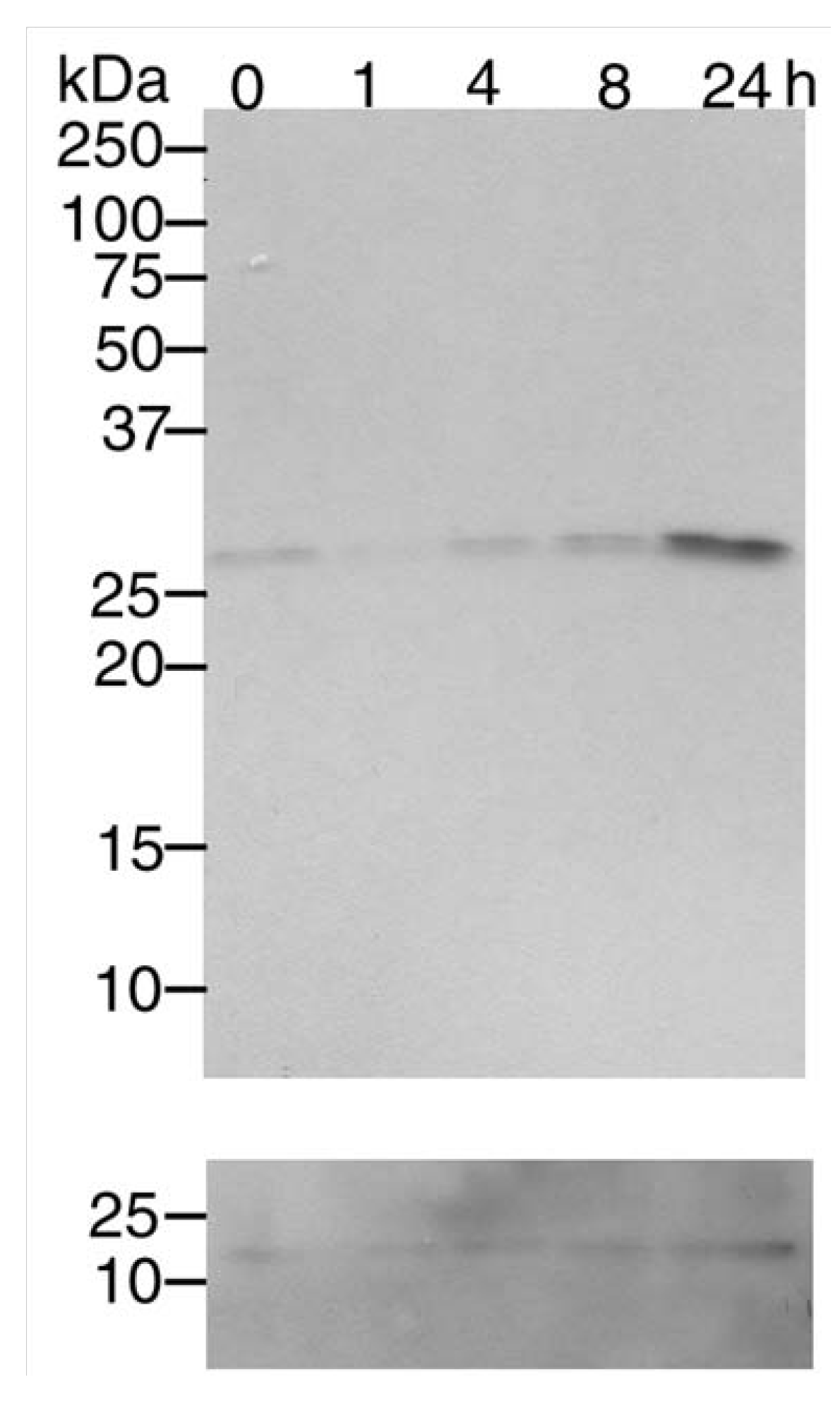
3.1. Mn2+ induces Cdk5 expression
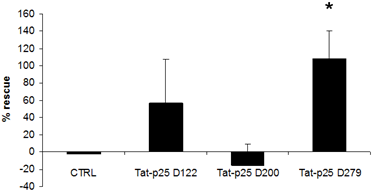
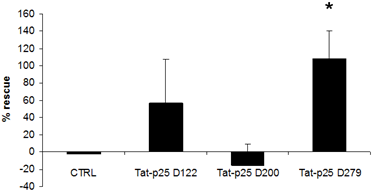
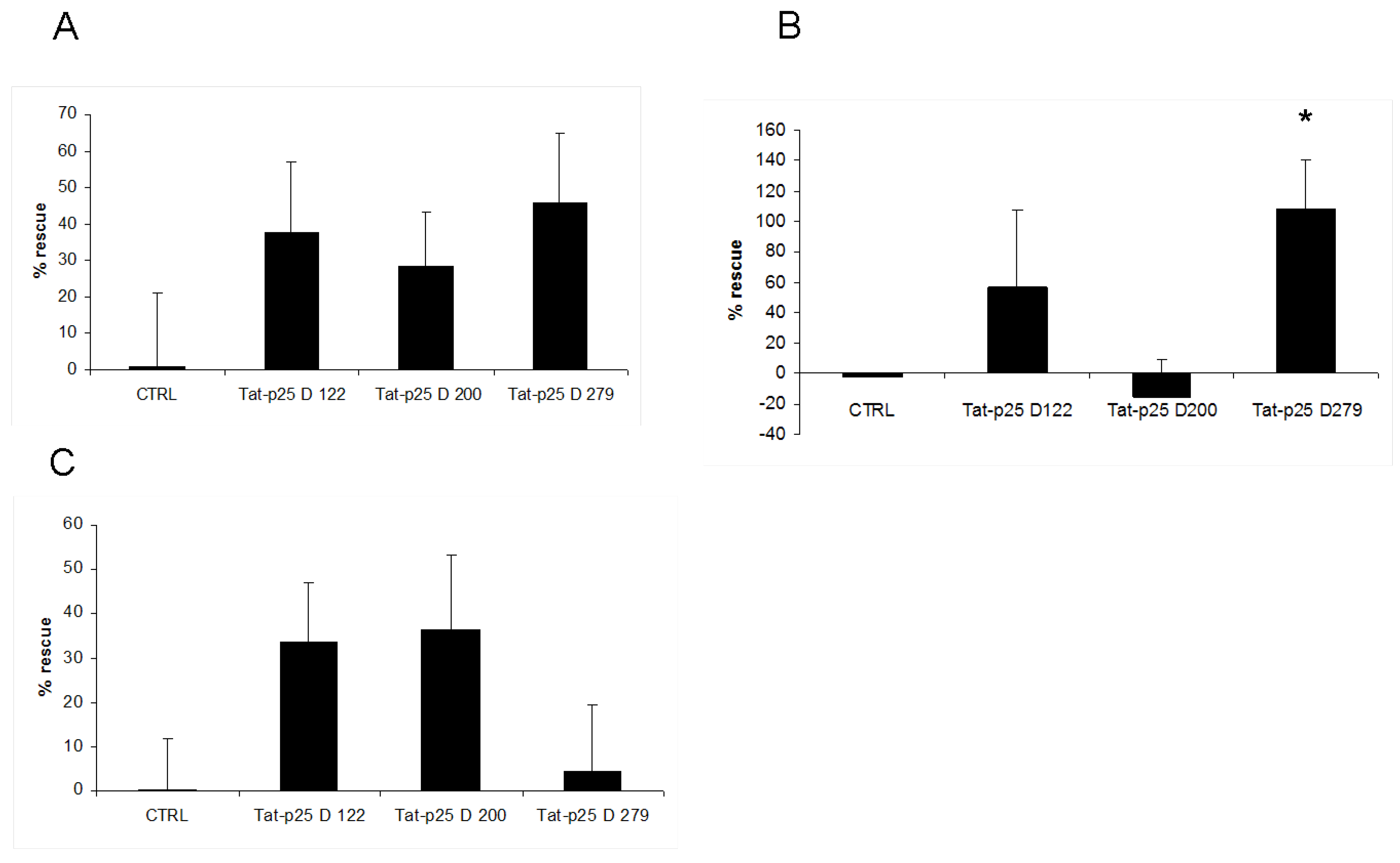
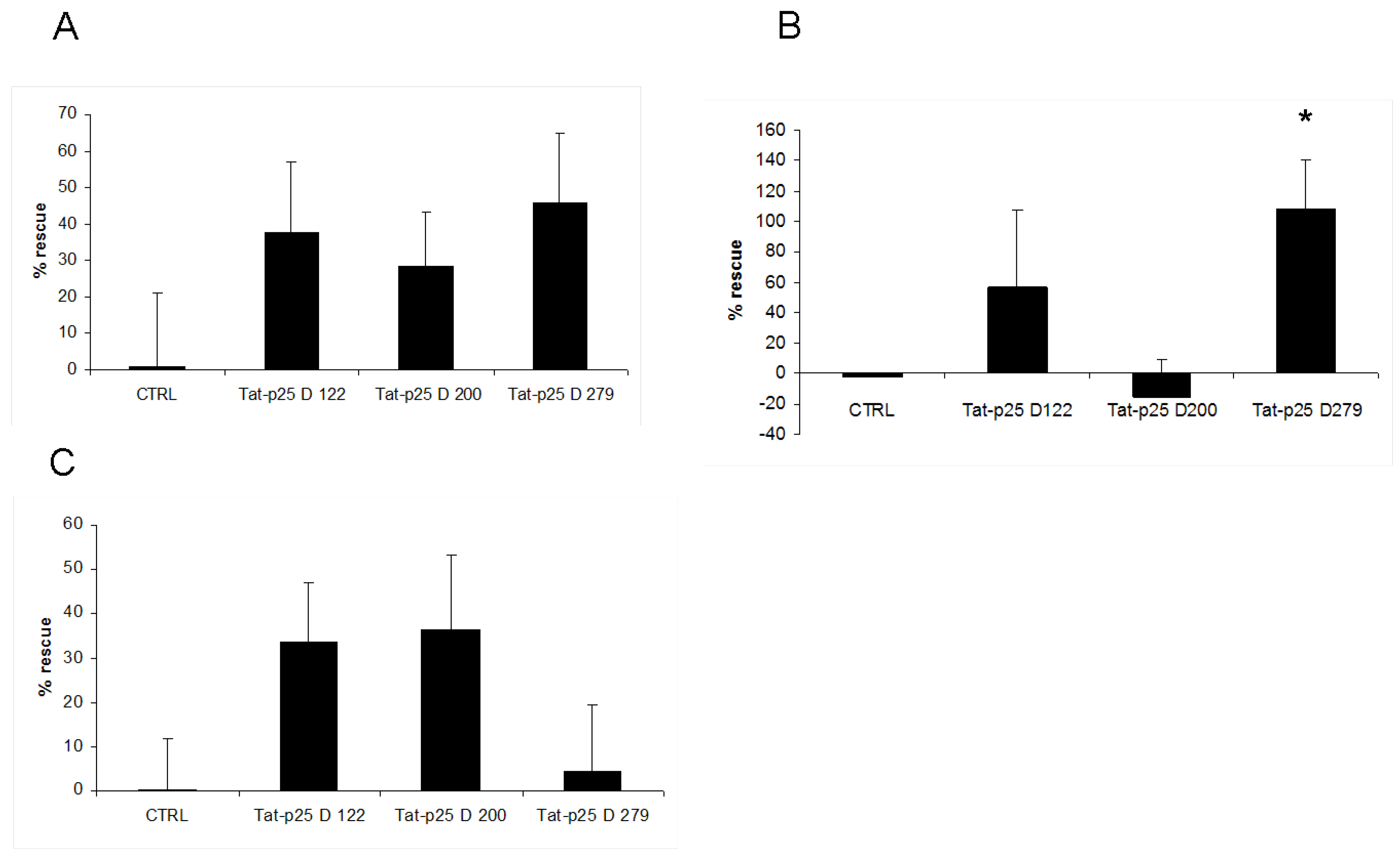
3.2. p25 fragments protect SH-SY5Y cells against cell death stimuli
4. Conclusions
Acknowledgements
References
- Dhavan, R.; Tsai, L.H. A decade of Cdk5. Nat. Rev. Mol. Cell. Biol. 2001, 2749–2759. [Google Scholar]
- Lee, M.S.; Kwon, Y.T.; Mingwei, L.; Peng, J.; Friedlander, R.M.; Tsai, H.T. Neurotoxicity induces cleavage of p35 to p25 by calpain. Nature 2000, 18, 360–364. [Google Scholar]
- Patrick, G.N.; Zukerberg, L.; Nikolic, M.; de la Monte, S.; Dikkes, P.; Tsai, L.H. Conversion of p35 to p25 de-regulates cdk5 activity and promotes neurodegeneration. Nature 1999, 9615–9622. [Google Scholar]
- Nguyen, M.D.; Lariviere, R.C.; Juien, J.P. Deregulation of Cdk5 in a mouse model of ALS: toxicity alleviated by perikaryal neurofilament inclusions. Neuron 2001, 30, 135–147. [Google Scholar]
- Tseng, H.C.; Zhou, Y.; Shen, Y.; Tsai, L.H. A survey of Cdk5 activator p35 and p25 levels in Alzheimer’s disease brains. FEBS Lett. 2002, 523, 58–62. [Google Scholar] [PubMed]
- Cruz, J.C.; Kim, D.; Moy, L.Y.; Dobbin, M.M.; Sun, X.; Bronson, R.T.; Tsai, L.H. p25/cyclin-dependent kinase 5 induces production and intraneuronal accumulation of amyloid beta in vivo. J. Neurosci. 2006, 26, 10536–10541. [Google Scholar] [PubMed]
- Ahlijanian, M.K.; Barrezueta, N.X.; Williams, R.D.; Jakowski, A.; Kowsz, K.P.; McCarthy, S.; Coskran, T.; Carlo, A.; Seymour, P.A.; Burkhardt, J.E.; Nelson, R.B.; McNeish, J.D. Hyperphosphorylated tau and neurofilament and cytoskeletal disruptions in mice overexpressing human p25, an activator of cdk5. Proc. Nat. Acad. Sci. USA 2000, 97, 2910–2915. [Google Scholar]
- Hung, K.S.; Hwang, S.L.; Liang, C.L.; Chen, Y.J.; Lee, T.H.; Liu, J.K.; Howng, S.L.; Wang, C.H. Calpain inhibitor inhibits p35-p25-Cdk5 activation, decreases tau hyperphosphorylation, and improves neurological function after spinal cord hemisection in rats. J. Neuropathol. Exp. Neuro. 2005, 64, 15–26. [Google Scholar]
- Smith, P.D.; Mount, M.P.; Shree, R.; Callaghan, S.; Slack, R.S.; Ansiman, H.; Vincent, I.; Wang, X.; Mao, Z.; Park, D.S. Calpain regulated p35/cdk5 plays a central role in dopaminergic neuron death through modulation of the transcription factor myocyte enhancer factor 2. J. Neurosci. 2006, 26, 440–447. [Google Scholar] [PubMed]
- Saito, T.; Konno, T.; Hosokawa, T.; Asada, A.; Ishiguro, K.; Hisanaga, S. p25/cyclin-dependent kinase 5 promotes the progression of cell death in nucleus of endoplasmic reticulum-stressed neurons. J. Neurochem. 2007, 102, 133–140. [Google Scholar]
- Dietz, G.P.; Bähr, M. Synthesis of cell-penetrating peptides and their application in neurobiology. Methods Mol. Biol. 2007, 399, 181–198. [Google Scholar]
- Dietz, G.P.; Bähr, M. Peptide-enhanced cellular internalization of proteins in neuroscience. Brain Res. Bull. 2005, 68, 103–114. [Google Scholar]
- Schwarze, S.R.; Ho, A.; Vocero-Akbani, A.; Dowdy, S.F. In vivo protein transduction: delivery of a biologically active protein into the mouse. Science 1999, 285, 1569–1572. [Google Scholar] [PubMed]
- Kilic, U.; Kilic, E.; Dietz, G.P.; Bähr, M. Intravenous TAT-GDNF is protective after focal cerebral ischemia in mice. Stroke 2003, 34, 1304–1310. [Google Scholar]
- Nagahara, H.; Vocero-Akbani, A.M.; Snyder, E.L.; Ho, A.; Latham, D.G.; Lissy, N.A.; Becker-Hapak, M.; Ezhevsky, S.A.; Dowdy, S.F. Transduction of full-length TAT fusion proteins into mammalian cells: TAT-p27Kip1 induces cell migration. Nat. Med. 1998, 4, 1449–1452. [Google Scholar]
- Poon, R.Y.C.; Lew, J.; Hunter, T. Identification of Functional Domains in the Neuronal Cdk5 Activator Protein. J. Biol. Chem. 1997, 272, 5703–5708. [Google Scholar] [PubMed]
- Nagel, F.; Dohm, C.P.; Bähr, M.; Wouters, F.S.; Dietz, G.P. Quantitative evaluation of chaperone activity by different preparations of a cell-penetrating Hsp70. J. Neurosci. Methods 2008, 171, 226–232. [Google Scholar]
- Zhang, B.F.; Peng, F.F.; Zhang, W.; Shen, H.; Wu, S.B.; Wu, D.C. Involvement of cyclin dependent kinase 5 and its activator p35 in staurosporine-induced apoptosis of cortical neurons. Acta Pharmacol. Sin. 2004, 25, 1105–1111. [Google Scholar]
- Meuer, K.; Suppanz, I.E.; Lingor, P.; Planchamp, V.; Göricke, B.; Fichtner, L.; Braus, G.H.; Dietz, G.P.; Jakobs, S.; Bähr, M.; Weishaupt, J.H. Cyclin-dependent kinase 5 is an upstream regulator of mitochondrial fission during neuronal apoptosis. Cell Death Differ. 2007, 14, 651–661. [Google Scholar]
- Alvira, D.; Tajes, M.; Verdaguer, E.; Acuña-Castroviejo, D.; Folch, J.; Camins, A.; Pallas, M. Inhibition of the cdk5/p25 fragment formation may explain the antiapoptotic effects of melatonin in an experimental model of Parkinson's disease. J. Pineal. Res. 2006, 40, 251–258. [Google Scholar]
- Calne, D.B.; Chu, N.S.; Luz, C.C.; Huang, C.S.; Olanow, W. Manganism and idiopathic parkinsonism: Similarities and differences. Neurology 1994, 44, 1583–1586. [Google Scholar]
- Cook, D.G.; Fahn, S.; Brait, K.A. Chronic manganese intoxication. Arch. Neurol. 1974, 30, 59–64. [Google Scholar]
- Higashi, Y.; Asanuma, M.; Miyazaki, I.; Hattori, N.; Mizuno, Y.; Ogawa, N. Parkin attenuates manganese-induced dopaminergic cell death. J. Neurochem. 2004, 89, 1490–1497. [Google Scholar]
- Roth, J.A.; Singleton, S.; Feng, J.; Garrick, M.; Paradkar, P.N. Parkin regulates metal transport via proteasomal degradation of the 1B isoforms of divalent metal transporter 1. J. Neurochem. 2010, 113, 454–464. [Google Scholar]
- Li, Y.; Sun, L.; Cai, T.; Zhang, Y.; Lv, S.; Wang, Y.; Ye, L. Alpha-Synuclein overexpression during manganese-induced apoptosis in SH-SY5Y neuroblastoma cells. Brain Res. Bull. 2010, 81, 428–433. [Google Scholar]
- Hirata, Y. Manganese-induced apoptosis in PC12 cells. Neurotoxicol. Teratol. 2002, 24, 639–653. [Google Scholar]
- Paoletti, P.; Vila, I.; Rife, M.; Lizcano, J.M.; Alberch, J.; Gines, S. Dopaminergic and Glutaminergic Signaling Crosstalk in Huntington’s Disease Neurodegeneration: The Role of p25/Cyclin-Dependent Kinase 5. Neurobiol. Dis. 2008, 28, 10090–10101. [Google Scholar]
- Brouilet, E.P.; Shinobu, L.; McGarvey, U.; Hochberg, F.; Beal, M.F. Manganese injection into the Rat Striatum Produces Excitotoxic Lesions by Impairing Energy Metabolism. Exp. Neurol. 1993, 120, 89–94. [Google Scholar]
- Kojima, H.; Abiru, Y.; Sakajiri, K.; Watabe, K.; Ohishi, N.; Takamori, M.; Hatanaka, H.; Yagi, K. Adenovirus-mediated transduction with human glial cell line-derived neurotrophic factor gene prevents 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-induced dopamine depletion in striatum of mouse brain. Biochem. Biophys. Res. Commun. 1997, 238, 569–573. [Google Scholar]
- Dietz, G.P.; Valbuena, P.C.; Dietz, B.; Meuer, K.; Mueller, P.; Weishaupt, J.H.; Bähr, M. Application of a blood-brain-barrier-penetrating form of GDNF in a mouse model for Parkinson's disease. Brain Res. 2006, 1082, 61–66. [Google Scholar] [CrossRef] [PubMed]
- Meikrantz, W.; Schlegel, R. Suppression of apoptosis by dominant negative mutants of cyclin- dependent protein kinases. J. Biol. Chem. 1996, 271, 10205–102059. [Google Scholar]
- Sandal, T.; Stapnes, C.; Kleivdal, H.; Hedin, L.; Doskeland, S.O. A novel, extraneuronal role for cyclin-dependent protein kinase 5 (CDK5): Modulation of cAMP-induced apoptosis in rat leukemia cells. J. Biol. Chem. 2002, 277, 20783–20793. [Google Scholar]
- Veeranna, Shetty, K.T.; Amin, N.; Grant, P.; Albers, R.W.; Pant, H.C. Inhibition of neuronal cyclin-dependent kinase-5 by staurosporine and purine analogs is independent of activation by Munc-18. Neurochem. Res. 1996, 21, 629–636. [Google Scholar] [CrossRef] [PubMed]
- Kesavapany, S.; Li, B.S.; Amin, N.; Zheng, Y.L.; Grant, P.; Pant, H.C. Neuronal cyclin-dependent kinase 5: Role in nervous system function and its specific inhibition by the Cdk5 inhibitory peptide. Biochemica et Biophysica Acta 2004, 1697, 143–153. [Google Scholar]
- Kulich, S.M.; Horbinski, C.; Patel, M.; Chu, C.T. 6-Hydroxydopamine induces mitochondrial ERK activation. Free Radic. Biol. Med. 2007, 43, 372–383. [Google Scholar]
© 2010 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Liman, J.; Weishaupt, J.H.; Bähr, M.; Dietz, G.P.H. Cell-Penetrating Fragments of the Cdk5 Regulatory Subunit Are Protective in Models of Neurodegeneration. Pharmaceuticals 2010, 3, 1232-1240. https://doi.org/10.3390/ph3041232
Liman J, Weishaupt JH, Bähr M, Dietz GPH. Cell-Penetrating Fragments of the Cdk5 Regulatory Subunit Are Protective in Models of Neurodegeneration. Pharmaceuticals. 2010; 3(4):1232-1240. https://doi.org/10.3390/ph3041232
Chicago/Turabian StyleLiman, Jan, Jochen H. Weishaupt, Mathias Bähr, and Gunnar P.H. Dietz. 2010. "Cell-Penetrating Fragments of the Cdk5 Regulatory Subunit Are Protective in Models of Neurodegeneration" Pharmaceuticals 3, no. 4: 1232-1240. https://doi.org/10.3390/ph3041232
APA StyleLiman, J., Weishaupt, J. H., Bähr, M., & Dietz, G. P. H. (2010). Cell-Penetrating Fragments of the Cdk5 Regulatory Subunit Are Protective in Models of Neurodegeneration. Pharmaceuticals, 3(4), 1232-1240. https://doi.org/10.3390/ph3041232