New Trends in Cancer Therapy: Targeting Ion Channels and Transporters

Abstract
:1. Introduction
2. Channel Expression in Tumour Cells: An Update
Ion transporters and the control of pH
3. Possible Approaches for Ion Channel Targeting in Oncology

{kind=link}
| Channel type | Function | References |
|---|---|---|
| K+ channels: | Cell proliferation | [1,2,3,28,29,30,31,32,34] |
| KV | Cell invasiveness | |
| Chemoresistance | ||
| Angiogenesis | ||
| Chemical cancerogenesis | ||
| KCa | Cell proliferation | |
| Cell volume | ||
| Cell migration | ||
| KIR | Cell proliferation | |
| K2p | Cell proliferation | |
| Ca2+ channels: | Cell proliferation | [3,11,12,13] |
| CaV | Cell proliferation | |
| SOC | Apoptosis | |
| Na+ channels: | Cell migration | [3,35,36,37,38,39] |
| NaV | Cell invasiveness | |
| TRP channels | Cell proliferation | [3,14,15,16,17,18,19] |
| Apoptosis | ||
| Cell volume | ||
| Angiogenesis | ||
| Cl- channels | Cell volume | [3,20,21,22,23,24,25] |
| Cell migration | ||
| nAChR | Cell proliferation | [40,41,42,43,44,45,46,47] |
| Apoptosis | ||
| Angiogenesis | ||
| Cell invasiveness | ||
| Aquaporins | Cell cycle control | [48,49,50,51,52,53,54,55] |
| Angiogenesis | ||
| Sodium pump | Cell migration | [56,57,58,59,60,61] |
| Cell invasiveness | ||
| Chemoresistance | ||
| Na+/H+ exchanger (NHE1) | Tumor cell metabolism | [26,27,63,66,67,68] |
| Cell invasiveness | ||
| Chemoresistance | ||
| Carbonic Anhydrases (CA-IX, CA-XII) | Tumor cell metabolism | [65,71,72,73] |
| Cell growth | ||
| Cell invasiveness |
4. The Case of Kv11.1 (hERG1 or KCNH2): Specificity and Side Effects
4.1. Not all Kv11.1 blockers produce arrhythmias
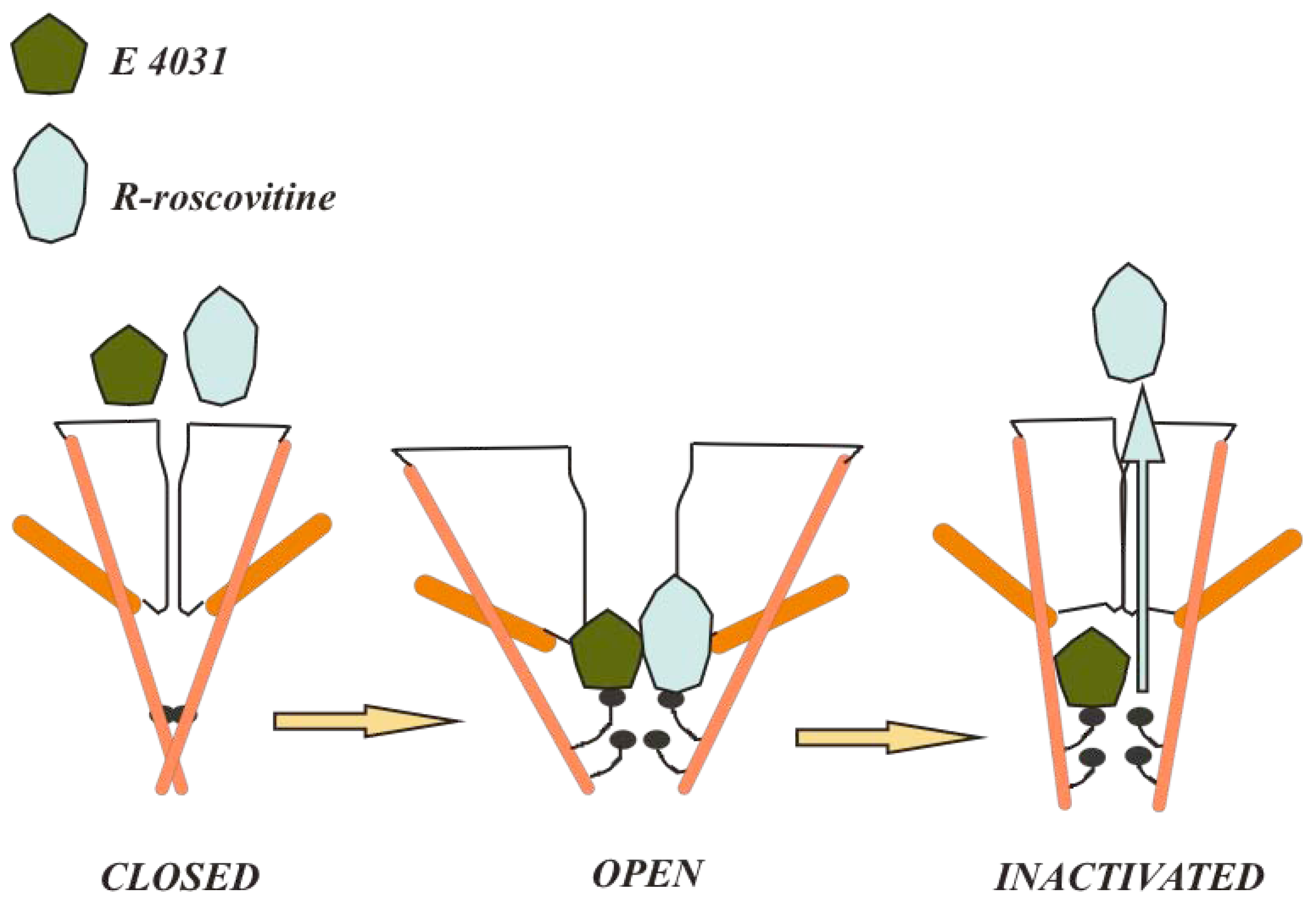
4.2. Compounds that bind different conformational states or different channel regions
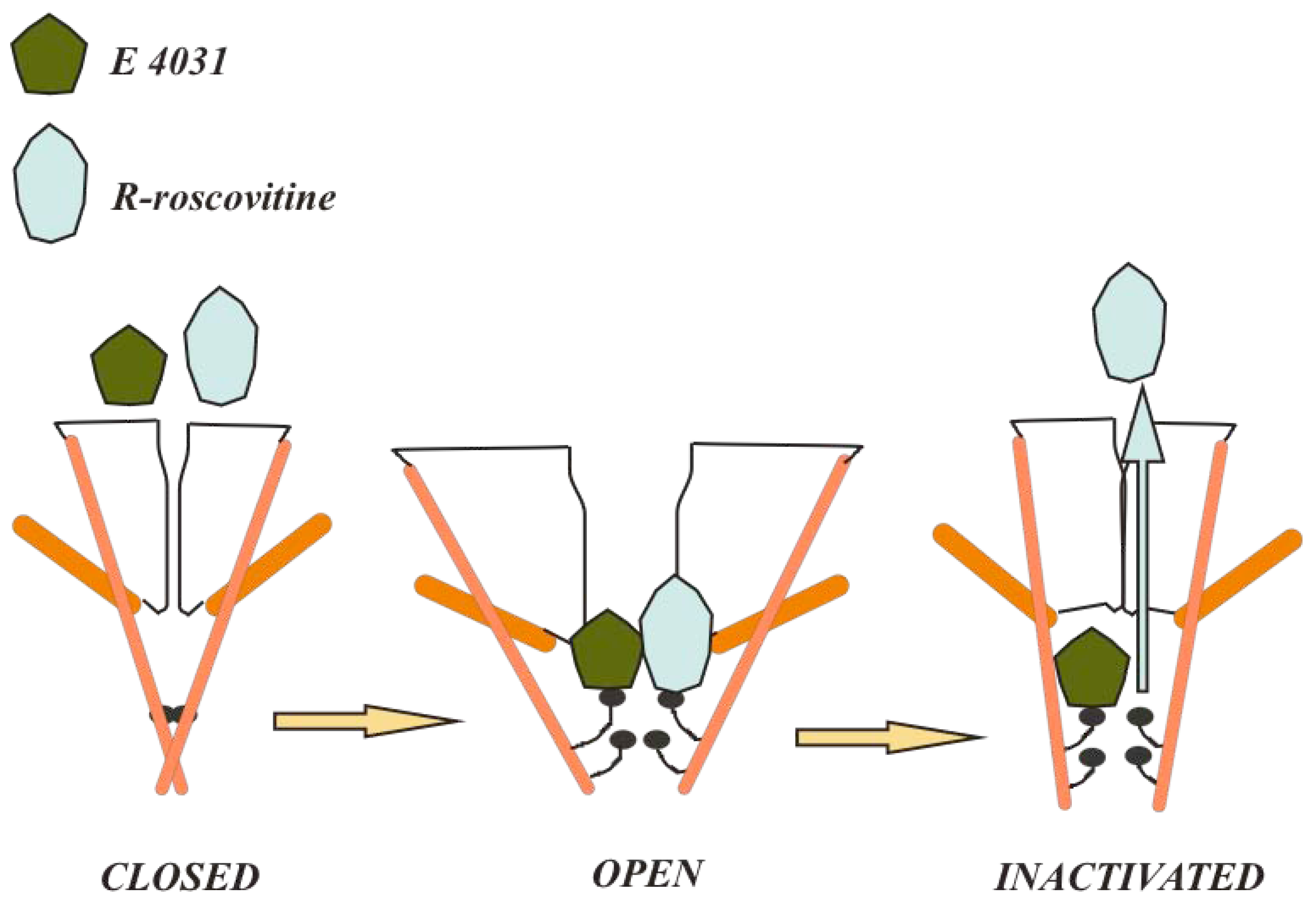
4.3. Peptide toxins: accessibility from the extracellular side
5. Recent Oncological Applications of Ion Channel and Transporter Blockade
5.1. Specific targeting of ion channels with different methods
| Approach | Examples | References |
|---|---|---|
| Specific non-peptide inhibitors | Kv11.1 | [6,7] |
| VGNCs | [128,129,130] | |
| Na+ /K+ ATPase | [134,135], | |
| Targeting channel states | VGNCs | [76] |
| Kv11.1 | [109] | |
| Use of peptide toxins | Kv11.1 | [119] |
| Kv1.3 | [122] | |
| nAChRs | [126,127] | |
| Blocking antibodies | Kv10.1 | [136] |
| Antisense oligonucleotides / siRNAs | CLIC4 | [133] |
| nAChRs | [127] | |
| Delivering cytotoxic compounds | Kv10.1 | [136] |
| ClC-3 | [137] |
5.2. Using channel-specific toxins and antibodies to deliver cytotoxic compounds
5.3. Na+/K+ ATPase
6. Conclusions
References
- Pardo, L.A. Voltage-gated potassium channels in cell proliferation. Physiology 2004, 19, 285–292. [Google Scholar]
- Kunzelmann, K. Ion channels and cancer. J. Memb. Biol. 2005, 205, 159–173. [Google Scholar]
- Arcangeli, A.; Crociani, O.; Lastraioli, E.; Masi, A.; Pillozzi, S.; Becchetti, A. Targeting ion channels in cancer: a novel frontier in antineoplastic therapy. Curr. Med. Chem. 2009, 16, 66–93. [Google Scholar]
- Fiske, J.L.; Fomin, V.P.; Brown, M.L.; Duncan, R.L.; Sikes, R.A. Voltage-sensitive ion channels in cancer. Cancer Metastasis Rev. 2006, 25, 439–500. [Google Scholar]
- Gόmez-Varela, D.; Zwick-Wallasch, E.R.; Knötgen, H.; Sanchez, A.; Hettmann, T.; Ossipov, O.; Weseloh, R.; Contreras-Jurado, C.; Rothe, M.; Stühmer, W.; Pardo, L.A. Monoclonal antibody blockade of the human Eag1 potassium channel function exerts antitumor activity. Cancer Res. 2007, 67, 7343–7349. [Google Scholar]
- Pillozzi, S; Accordi, B.; Veltroni, M.; Masselli, M.; Pancrazzi, E.; Galpa, G.; Lippi, A.; Bernini, G.; Basso, G.; Arcangeli, A. Expression and role of hERG1 channels in pediatric acute lymphoblastic leukaemias : shortcoming of drug resistance by hERG1 channel inhibitors in stroma-supported leukaemia cell cultures in vitro. Blood 2007, 110, 222A. [Google Scholar]
- Pillozzi, S.; Masselli, M.; De Lorenzo, E.; Cilia, E.; Crociani, O.; Amedei, A.; Accordi, B.; Veltroni, M.; Basso, G.; Campana, D.; Becchetti, A.; Arcangeli, A. Overcoming chemotherapy resistance in childhood acute lymphoblastic leukemia by targeting ion channels. Blood 2009, 114, 3085. [Google Scholar]
- Lefranc, F.; Mijatovic, T.; Kondo, Y.; Sauvage, S.; Roland, I.; Debeir, O.; Krstic, D.; Vasic, V.; Gailly, P.; Kondo, S.; Blanco, G.; Kiss, R. Targeting the α1 subunit of the sodium pump to combat glioblastoma cells. Neurosurgery 2008, 62, 211–222. [Google Scholar]
- Matheu, M.P.; Beeton, C.; Garcia, A.; Chi, V.; Rangaraju, S.; Safrina, O.; Monaghan, K.; Uemura, M.I.; Li, D.; Pal, S.; de la Maza, L.M.; Monuku, E.; Flügel, A.; Pennington, M.W.; Parker, I.; Chandy, K.G.; Cahalan, M.D. Imaging of effector memory T cells during a delayed-type hypersensitivity reaction and suppression by Kv1.3 channel block. Immunity 2008, 29, 602–614. [Google Scholar] [CrossRef] [PubMed]
- Bianchi, L.; Wible, B.; Arcangeli, A.; Taglialatela, M.; Morra, F.; Castaldo, P.; Crociani, O.; Rosati, B.; Faravelli, L.; Olivotto, M.; Wanke, E. Herg encodes a K+ current highly conserved in tumors of different histogenesis: a selective advantage for cancer cells? Cancer Res. 1998, 58, 815–822. [Google Scholar] [PubMed]
- Munaron, L.; Antoniotti, S.; Fiorio Pla, A.; Lovisolo, D. Blocking Ca2+ entry: a way to control cell proliferation. Curr. Med. Chem. 2004, 11, 1533–1543. [Google Scholar]
- Panner, A.; Wurster, R.D. T-type calcium channels and tumor proliferation. Cell Calcium 2006, 40, 253–259. [Google Scholar]
- Monteith, G.R.; McAndrew, D.; Faddy, H.M.; Roberts-Thomson, S.J. Calcium and cancer: targeting Ca2+ transport. Nat. Rev. Cancer 2007, 7, 519–530. [Google Scholar]
- Bödding, M. TRP proteins and cancer. Cell Signal. 2007, 19, 617–624. [Google Scholar]
- Prevarskaya, N.; Zhang, L.; Barritt, G. TRP channels in cancer. Biochem. Biophys. Acta 2007, 1772, 937–946. [Google Scholar]
- Bidaux, G.; Flourakis, M.; Thebault, S.; Zholos, A.; Beck, B.; Gkika, D.; Roudbaraki, M.; Bonnal, J.; Mauroy, B.; Shuba, Y.; Skryma, R.; Prevarskaya, N. Prostate cell differentiation status determines transient receptor potential melastatin member 8 channel subcellular localization and function. J. Clin. Invest. 2007, 117, 1647–1657. [Google Scholar] [Green Version]
- Zhang, L.; Barritt, G.J. TRPM8 in prostate cancer cells: a potential diagnostic and prognostic marker with a secretory function? Endocr. Relat. Cancer 2006, 13, 27–38. [Google Scholar] [CrossRef] [PubMed]
- Bates, D.O.; Hillman, N.J.; Williams, B.; Neal, C.R.; Pocock, T.M. Regulation of microvascular permeability by vascular endothelial growth factors. J. Anat. 2002, 200, 581–597. [Google Scholar]
- Hamdollah Zadeh, M.A.; Glass, C.A.; Magnussen, A.; Hancox, J.C.; Bates, D.O. VEGF-mediated elevated intracellular calcium and angiogenesis in human microvascular endothelial cells in vitro are inhibited by dominant negative TRPC6. Microcirculation 2008, 15, 605–614. [Google Scholar] [PubMed]
- Hoffmann, E.K.; Lambert, I.H.; Pedersen, S.F. Physiology of cell volume regulation in vertebrates. Physiol. Rev. 2009, 89, 193–277. [Google Scholar]
- Sontheimer, H. An unexpected role for ion channels in brain tumor metastasis. Exp. Biol. Med. 2008, 233, 779–791. [Google Scholar]
- Cuddapah, V.A.; Sontheimer, H. Molecular interaction and functional regulation of ClC-3 by Ca2+/calmodulin-dependent protein kinase II (CaMKII) in human malignant glioma. J. Biol. Chem. 2010. [Google Scholar] [CrossRef]
- Mamelak, A.N.; Rosenfeld, S.; Bucholz, R.; Raubitschek, A.; Nabors, L.B.; Fiveash, J.B.; Shen, S.; Khazaeli, M.B.; Colcher, D.; Liu, A.; Osman, M.; Guthrie, B.; Schade-Bijur, S.; Hablitz, D.M.; Alvarez, V.L.; Gonda, M.A. Phase I single-dose study of intracavitary-administered iodine-131-TM-601 in adults with recurrent high-grade glioma. J. Clin. Oncol. 2006, 24, 3644–3650. [Google Scholar]
- Schwab, A.; Nechyporuk-Zloy, V.; Fabian, A.; Stock, C. Cells move when ions and water flow. Pflügers Arch. 2007, 453, 421–432. [Google Scholar] [PubMed]
- Schwab, A.; Wojnowski, L.; Gabriel, K.; Oberleithner, H. Oscillating activity of a Ca2+-sensitive K+ channel - a prerequisite for migration of alkali-transformed Madin-Darby canine kidney (MDCK-F) cells. J. Clin. Invest. 1994, 93, 1631–1636. [Google Scholar] [CrossRef] [PubMed]
- Stock, C.; Müller, M.; Mally, S.; Noël, J.; Eder, C.; Schwab, A. pH nanoenvironment at the surface of single melanoma cells. Cell Physiol. Biochem. 2007, 20, 679–686. [Google Scholar]
- Stock, C.; Schwab, A. Protons make tumor cells move like clockwork. Pflügers Arch. 2009, 458, 981–992. [Google Scholar] [CrossRef] [PubMed]
- Lastraioli, E.; Guasti, L.; Crociani, O.; Polvani, S.; Hofmann, G.; Witchel, H.; Bencini, L.; Calistri, M.; Messerini, L.; Scatizzi, M.; Moretti, R.; Wanke, E.; Olivotto, M.; Mugnai, G.; Arcangeli, A. Herg1 gene and HERG1 protein are overexpressed in colorectal cancers and regulate cell invasion of tumor cells. Cancer Res. 2004, 64, 606–611. [Google Scholar]
- Pillozzi, S.; Brizzi, M.F.; Bernabei, P.A.; Bartolozzi, B.; Caporale, R.; Basile, V.; Boddi, V.; Pegoraro, L.; Becchetti, A.; Arcangeli, A. VEGFR-1 (FLT-1), beta1 integrin, and hERG K+ channel for a macromolecular signaling complex in acute myeloid leukemia: role in cell migration and clinical outcom. Blood 2007, 110, 1238–1250. [Google Scholar]
- Afrasiabi, E.; Hietamäki, M.; Viitanen, T.; Sukumaran, P.; Bergelin, N.; Törnquist, K. Expression and significance of HERG (KCNH2) potassium channels in the regulation of MDA-MB-435S melanoma cell proliferation and migration. Cell Signal. 2010, 22, 57–64. [Google Scholar]
- Pillozzi, S.; Brizzi, M.F.; Balzi, M.; Crociani, O.; Cherubini, A.; Guasti, L.; Bartolozzi, B.; Becchetti, A.; Wanke, E.; Bernabei, P.A.; Olivotto, M.; Pegoraro, L.; Arcangeli, A. HERG potassium channels are constitutively espressed in primary human acute myeloid leukemia and regulate cell proliferation of normal and leukemic hemopoietic progenitors. Leukemia 2002, 16, 1791–1798. [Google Scholar]
- Crociani, O.; Guasti, L.; Balzi, M.; Becchetti, A.; Wanke, E.; Olivotto, M.; Wymore, R.S.; Arcangeli, A. Cell cycle-dependent expression of HERG1 and HERG1B isoforms in tumor cells. J. Biol. Chem. 2003, 278, 2947–2955. [Google Scholar]
- Arcangeli, A.; Becchetti, A. Complex functional interaction between integrin receptors and ion channels. Trends Cell Biol. 2006, 16, 631–639. [Google Scholar]
- Ousingsawat, J.; Spitzner, M.; Puntheeranurak, S.; Terracciano, L.; Tornillo, L.; Bubendorf, L.; Kunzelmann, K.; Schreiber, R. Expression of voltage-gated potassium channels in human and mouse colonic carcinoma. Clin. Cancer Res. 2007, 13, 824–831. [Google Scholar]
- Fulgenzi, G.; Graciotti, L.; Faronato, M.; Soldovieri, M.V.; Miceli, F.; Amoroso, S.; Annunziato, L.; Procopio, A.; Taglialatela, M. Human neoplastic mesothelial cells express voltage-gated sodium channels involved in cell motility. Int. J. Biochem. Cell Biol. 2006, 38, 1146–1159. [Google Scholar]
- Diss, J.K.; Fraser, S.P.; Djamgoz, M.B. Voltage-gated Na+ channels: multiplicity of expression, plasticity, functional implications and pathophysiological aspects. Eur. Biophys. J. 2004, 33, 180–193. [Google Scholar] [PubMed]
- Gillet, L.; Roger, S.; Besson, P.; Lecaille, F.; Gore, J.; Bougnoux, P.; Lalmanach, G.; Le Guennec, J.Y. Voltage-gated Sodium Channel Activity Promotes Cysteine Cathepsin-dependent Invasiveness and Colony Growth of Human Cancer Cells. J. Biol. Chem. 2009, 284, 8680–8691. [Google Scholar]
- Chioni, A.M.; Brackenbury, W.J.; Calhoun, J.D.; Isom, L.L.; Djamgoz, M.B. A novel adhesion molecule in human breast cancer cells: voltage-gated Na+ channel beta1 subunit. Int. J. Biochem. Cell Biol. 2009, 41, 1216–1227. [Google Scholar]
- Diss, J.K.; Stewart, D.; Pani, F.; Foster, C.S.; Walker, M.M.; Patel, A.; Djamgoz, M.B. A potential novel marker for human prostate cancer: voltage-gated sodium channel expression in vivo. Prostate Cancer Prostatic Dis. 2005, 8, 266–273. [Google Scholar] [CrossRef]
- Maneckjee, R.; Minna, J.D. Opioid and nicotine receptors affect growth regulation of human lung cancer cell lines. Proc. Natl. Acad. Sci. USA 1990, 87, 3294–3298. [Google Scholar]
- Dasgupta, P.; Rastogi, S.; Pillai, S.; Ordonez-Ercan, D.; Morris, M.; Haura, E.; Chellappan, S. Nicotine induces cell proliferation by β-arrestin-mediated activation of Src and Rb-Raf1 pathways. J. Clin. Invest. 2006, 111, 31–33. [Google Scholar]
- Lam, D.C.; Girard, L.; Ramirez, R.; Chau, W.; Suen, W.; Sheridan, S.; Tin, V.P.C.; Chung, L.; Wong, M.P.; Shay, J.W.; Gazdar, A.F.; Lam, W.; Minna, J.D. Expression of nicotinic acetylcholine receptor subunit genes in non-small-cell lung cancer reveals differences between smokers and nonsmokers. Cancer Res. 2007, 67, 4638–4647. [Google Scholar]
- Egleton, R.D.; Brown, K.C.; Dasgupta, P. Nicotinic acetylcholine receptors in cancer: multiple roles in proliferation and inhibition of apoptosis. Trends Pharmacol. Sci. 2008, 29, 151–158. [Google Scholar]
- Schuller, H.M. Is cancer triggered by altered signalling of nicotinic acetylcholine receptors? Nat. Rev. Cancer 2009, 9, 195–205. [Google Scholar] [CrossRef]
- Grozio, A.; Paleari, L.; Catassi, A.; Servent, D.; Cilli, M.; Piccardi, F.; Paganuzzi, M.; Cesario, A.; Granone, P.; Mourier, G.; Russo, P. Natural agents targeting the alpha7-nicotinic-receptor in NSCLC: A promising prospective in anti-cancer drug development. Int. J. Cancer 2008, 122, 1911–1915. [Google Scholar]
- Schuller, H.M. Nitrosamines as nicotinic receptor ligands. Life Sci. 2007, 80, 2274–2280. [Google Scholar]
- Zheng, Y.; Ritzenthaler, J.D.; Roman, J.; Han, S. Nicotine stimulates human lung cancer cell growth by inducing fibronectin expression. Am. J. Respir. Cell Mol. Biol. 2007, 37, 681–690. [Google Scholar]
- Chae, Y.K.; Kang, S.K.; Kim, M.S.; Woo, J.; Lee, J.; Chang, S.; Kim, D.W.; Kim, M.; Park, S.; Kim, I.; Keam, B.; Rhee, J.; Koo, N.H.; Park, G.; Kim, S.H.; Jang, S.E.; Kweon, I.Y.; Sidransky, D.; Moon, C. Human AQP5 plays a role in the progression of chronic myelogenous leukemia (CML). PLoS One 2008, 3, 2594. [Google Scholar]
- Kang, S.K.; Chae, Y.K.; Woo, J.; Kim, M.S.; Park, J.C.; Lee, J.; Soria, J.C.; Jang, S.J.; Sidransky, D.; Moon, C. Role of human aquaporin 5 in colorectal carcinogenesis. Am. J. Pathol. 2008, 173, 518–525. [Google Scholar]
- Chae, Y.K.; Woo, J.; Kim, M.J.; Kang, S.K.; Kim, M.S.; Lee, J.; Lee, S.K.; Gong, G.; Kim, Y.H.; Soria, J.C.; Jang, S.J.; Sidransky, D.; Moon, C. Expression of aquaporin 5 (AQP5) promotes tumor invasion in human non small cell lung cancer. PLoS One 2008, 3, e2162. [Google Scholar]
- Vacca, A.; Frigeri, A.; Ribatti, D.; Nicchia, G.P.; Nico, B.; Ria, R.; Svelto, M.; Dammacco, F. Microvessel overexpression of aquaporin 1 parallels bone marrow angiogenesis in patients with active multiple myeloma. Br. J. Haematol. 2001, 113, 415–421. [Google Scholar] [CrossRef]
- Saadoun, S.; Papadopoulos, M.C.; Davies, D.C.; Bell, B.A.; Krishna, S. Increased aquaporin 1 water channel expression in human brain tumours. Br. J. Cancer 2002, 87, 621–623. [Google Scholar]
- Moon, C.; Soria, J.C.; Jang, S.J.; Lee, J.; Hoque, M.O.; Sibony, M.; Trink, B.; Chang, Y.S.; Sidransky, D.; Mao, L. Involvement of aquaporins in colorectal carcinogenesis. Oncogene 2003, 22, 6699–6703. [Google Scholar]
- Woo, J.; Lee, J.; Chae, Y.K.; Kim, M.S.; Baek, J.H.; Park, J.C.; Park, M.J.; Smith, I.M.; Trink, B.; Ratovitski, E.; Lee, T.; Park, B.; Jang, S.J.; Soria, J.C.; Califano, J.A.; Sidransky, D.; Moon, C. Overexpression of AQP5, a putative oncogene, promotes cell growth and transformation. Cancer Lett. 2008, 264, 54–62. [Google Scholar] [CrossRef] [PubMed]
- Woo, J.; Lee, J.; Kim, M.S.; Jang, S.J.; Sidransky, D.; Moon, C. The effect of aquaporin 5 overexpression on the Ras signaling pathway. Biochem. Biophys. Res. Commun. 2008, 367, 291–298. [Google Scholar]
- Mijatovic, T.; Van Quaquebeke, E.; Delest, B.; Debeir, O.; Darro, F.; Kiss, R. Cardiotonic steroids on the road to anti-cancer therapy. Biochim. Biophys. Acta 2007, 1776, 32–57. [Google Scholar] [PubMed]
- Mijatovic, T; Roland, I.; Van Quaquebeke, E.; Nilsson, B.; Mathieu, A.; van Vynckt, F.; Darro, F.; Blanco, G.; Facchini, V.; Kiss, R. The alpha1 subunit of the sodium pump could represent a novel target to combat non-small cell lung cancers. J. Pathol. 2007, 212, 170–179. [Google Scholar] [CrossRef] [PubMed]
- Senner, V.; Schmidtpeter, S.; Braune, S.; Puttmann, S.; Thanos, S.; Bartsch, U.; Schachner, M.; Paulus, W. AMOG/beta2 and glioma invasion: Does loss of AMOG make tumour cells run amok? Neuropathol. Appl. Neurobiol. 2003, 29, 370–377. [Google Scholar] [CrossRef] [PubMed]
- Lefranc, F.; Brotchi, J.; Kiss, R. Present and future issues in the treatment of malignant gliomas, with a special emphasis on cell migration and the resistance of migrating glioma cells to apoptosis. J. Clin. Oncol. 2005, 23, 2411–2422. [Google Scholar]
- Lefranc, F.; Kiss, R. The sodium pump α1 subunit as a potential target to combat apoptosis-resistant glioblastomas. Neoplasia 2008, 10, 198–206. [Google Scholar]
- Lefranc, F.; Rynkowski, M.; Dewitte, O.; Kiss, R. Present and potential future adjuvant issues in high-grade astrocytic glioma treatment. In Advances and Technical Standards in Neurosurgery; Pickard, J.D., Ed.; Springer-Verlag: Wien, Austria, 2009. [Google Scholar]
- Warburg, O. On the origins of cancer cells. Science 1956, 123, 309–314. [Google Scholar]
- Cardone, R.A.; Casavola, V.; Reshkin, S.J. The role of disturbed pH dynamics and the Na+/H+ exchanger in metastasis. Nat. Rev. Cancer 2005, 5, 786–795. [Google Scholar]
- Perona, R.; Serrano, R. Increased pH and tumorigenicity of fibroblasts expressing a yeast proton pump. Nature 1988, 334, 438–440. [Google Scholar]
- Swietach, P.; Vaughan-Jones, R.D.; Harris, A.L. Regulation of tumor pH and the role of carbonic anhydrase 9. Cancer Metastasis Rev. 2007, 26, 299–310. [Google Scholar]
- Cardone, R.A.; Bellizzi, A.; Busco, G.; Weinmann, E.J.; Dell’Aquila, M.E.; Casavola, V.; Azzariti, A.; Mangia, A.; Paradiso, A.; Reshkin, S.J. The NHERF1 PDZ2 domain regulates PKA-RhoA-p38 mediated NHE1 activation and invasion in breast tumor cells. Mol. Biol. Cell 2007, 18, 1768–1780. [Google Scholar]
- Cardone, R.A.; Busco, G.; Greco, M.R.; Bellizzi, A.; Accardi, R.; Cafarelli, A.; Monterisi, S.; Carratù, P.; Casavola, V.; Paradiso, A.; Tommasino, M.; Reshkin, S.J. HPV16 E7-dependent transformation activates NHE1 through a PKA-RhoA-induced inhibition of p38alpha. PLoS One 2008, 3, e3529. [Google Scholar]
- Mangia, A.; Chiriatti, A.; Bellizzi, A.; Malfettone, A.; Stea, B.; Zito, F.A.; Reshkin, S.J.; Simone, G.; Paradiso, A. Biological role of NHERF1 protein expression in breast cancer. Histopathology 2009, 55, 600–608. [Google Scholar]
- Harguindey, S.; Arranz, J.L.; Wahl, M.L.; Orive, G.; Reshkin, S.J. Proton transport inhibitors as potentially selective anticancer drugs. Anticancer Res. 2009, 29, 2127–2136. [Google Scholar]
- Favia, A.; Guerra, L.; Fanelli, T.; Cardone, R.A.; Monterisi, S.; Di Sole, F.; Castellani, S.; Chen, M.; Seidler, U.; Reshkin, S.J.; Conese, M.; Casavola, V. Na+/H+ exchanger regulatory factor 1 overexpression-dependent increase of cytoskeleton organization is fundamental in the rescue of F508del cystic fibrosis trans membrane conductance regulator in human air way CFBE41o-cells. Mol. Biol. Cell 2010, 21, 73–86. [Google Scholar]
- Kivela, A.J.; Parkkila, S.; Saarnio, J.; Karttunen, T.J.; Kivela, J.; Parkkila, A.K.; Pastorekova, S.; Pastorek, J.; Waheed, A.; Sly, W.S.; Rajaniemi, H. Expression of transmembrane carbonic anhydrase isoenzymes IX and XII in normal human pancreas and pancreatic tumours. Histochem. Cell Biol. 2000, 114, 197–204. [Google Scholar]
- Ivanov, S.; Liao, S.Y.; Ivanova, A.; Danilkovitch-Miagkova, A.; Tarasova, N.; Weirich, G.; Merrill, M.J.; Proescholdt, M.A.; Oldfield, E.H.; Lee, J.; Zavada, J.; Waheed, A.; Sly, W.S.; Lerman, M.I.; Stanbridge, E.J. Expression of hypoxia-inducible cell-surface transmembrane carbonic anhydrases in human cancer. Am. J. Pathol. 2001, 158, 905–919. [Google Scholar]
- Swietach, P.; Patiar, S.; Supuran, C.T.; Harris, A.L.; Vaughan-Jones, R.D. The role of carbonic anhydrase 9 in regulating extracellular and intracellular pH in three-dimensional tumor cell growth. J. Biol. Chem. 2009, 284, 20299–20310. [Google Scholar]
- Andersen, O.S. Perspectives on how to drug an ion channel. J. Gen. Physiol. 2008, 131, 395–397. [Google Scholar]
- Becchetti, A.; Arcangeli, A. A comment on ion channels as pharmacological targets in oncology. J. Gen. Physiol. 2008, 132, 313–314. [Google Scholar] [CrossRef] [PubMed]
- Kaczorowski, G.J.; McManus, O.B.; Priest, B.T.; Garcia, M.L. Ion channels as drug targets: the next GPCRs. J. Gen. Physiol. 2008, 131, 399–405. [Google Scholar]
- Cavalli, A.; Poluzzi, E.; De Ponti, F.; Recanatini, M. Toward a pharmacophore for drugs inducing the long QT syndrome: insights from a CoMFA study of HERG K+ channel blockers. J. Med. Chem. 2002, 45, 3844–3853. [Google Scholar]
- Recanatini, M.; Cavalli, A.; Masetti, M. Modeling HERG and its interactions with drugs: recent avances in light of current potassium channel simulations. Chem. Med. Chem. 2008, 3, 523–535. [Google Scholar]
- Doyle, D.A.; Morais Cabral, J.; Pfuetzner, R.A.; Kuo, A.; Gulbis, J.M.; Cohen, S.L.; Chait, B.T.; MacKinnon, R. The structure of the potassium channel: molecular basis of K+ conduction and selectivity. Science 1998, 280, 69–77. [Google Scholar]
- Dutzler, R.; Campbell, E.B.; Cadene, M.; Chait, B.T.; MacKinnon, R. X-ray structure of a ClC chloride channel at 3.0 Å reveals the molecular basis of anion selectivity. Nature 2002, 415, 287–294. [Google Scholar] [PubMed]
- Xie, M.; Holmqvist, M.H.; Hsia, A.Y. Available online: www.currentdrugdiscovery.com April 2004.
- Shi, W.; Wymore, R.S.; Wang, H.-S.; Pan, Z.; Cohen, I.S.; McKinnon, D.; Dixon, J.E. Identification of two nervous system-specific members of the erg potassium channel gene family. J. Neurosci. 1997, 17, 9423–9432. [Google Scholar]
- Schäfer, R.; Wulfsen, I.; Behrens, S.; Weinsberg, F.; Bauer, C.K.; Schwarz, J.R. The erg-like potassium current in rat lactotrophs. J. Physiol. 1999, 518, 410–416. [Google Scholar]
- Rosati, B.; Marchetti, P.; Crociani, O.; Lecchi, M.; Lupi, R.; Arcangeli, A.; Olivotto, M.; Wanke, E. Glucose- and arginine-induced insulin secretion by human pancreatic beta-cells: the role of HERG K(+) channels in firing and release. FASEB J. 2000, 14, 2601–2610. [Google Scholar]
- Gullo, F.; Ales, E.; Rosati, B.; Lecchi, M.; Masi, A.; Guasti, L.; Cano-Abad, M.F.; Arcangeli, A.; Lopez, M.G.; Wanke, E. ERG K+ channel blockade enhances firing and epinephrine secretion in rat chromaffin cells: the missing link to LQT2-related sudden death? FASEB J. 2003, 17, 330–332. [Google Scholar] [PubMed]
- Papa, M.; Boscia, F.; Canitano, A.; Castaldo, P.; Sellitti, S.; Annunziato, L.; Taglialatela, M. Expression pattern of the ether-à-gogo-related (ERG) K+ channel-encoding genes ERG1, ERG2, and ERG3 in the adult rat central nervous system. J. Comp. Neurol. 2003, 466, 119–135. [Google Scholar] [CrossRef] [PubMed]
- Guasti, L.; Cilia, E.; Crociani, O.; Hofmann, G.; Polvani, S.; Becchetti, A.; Wanke, E.; Tempia, F.; Arcangeli, A. Expression pattern of the Ether-à-go-go-related (ERG) family proteins in the adult mouse central nervous system: evidence for coassembly of different subunits. J. Comp. Neurol. 2005, 491, 157–174. [Google Scholar] [CrossRef] [PubMed]
- Sacco, T.; Bruno, A.; Wanke, E.; Tempia, F. Functional roles of an ERG current isolated in cerebellar Purkinje neurons. J. Neurophysiol. 2003, 90, 1817–1828. [Google Scholar]
- Furlan, F.; Guasti, L.; Avossa, D.; Becchetti, A.; Cilia, E.; Ballerini, L.; Arcangeli, A. Interneurons transiently express the ERG K+ channels during development of mouse spinal networks in vitro. Neuroscience 2005, 135, 1179–1192. [Google Scholar] [CrossRef] [PubMed]
- Furlan, F.; Taccola, G.; Grandolfo, M.; Guasti, L.; Arcangeli, A.; Nistri, A.; Ballerini, A. ERG conductance expression modulates the excitability of ventral horn GABAergic interneurons that control rhythmic oscillations in the developing mouse spinal cord. J. Neurosci. 2007, 27, 919–928. [Google Scholar]
- Pessia, M.; Servettini, I.; Panichi, R.; Guasti, L.; Grassi, S.; Arcangeli, A.; Wanke, E.; Pettorossi, V.E. ERG voltage-gated K+ channels regulate excitability and discharge dynamics of the medial vestibular nucleus neurons. J. Physiol. 2008, 586, 4877–4890. [Google Scholar]
- Hirdes, W.; Napp, N.; Wulfsen, I.; Schweizer, M.; Schwarz, J.R.; Bauer, C.K. Erg K+ currents modulate excitability in mouse mitral/tufted neurons. Pflügers Arch. 2009, 459, 55–70. [Google Scholar] [CrossRef] [PubMed]
- Huffaker, S.J.; Chen, J.; Nicodemus, K.K.; Sambataro, F.; Yang, F.; Mattay, V.; Lipska, B.K.; Hyde, T.M.; Song, J.; Rujescu, D.; Giegling, I.; Mayilyan, K.; Proust, M.J.; Soghoyan, A.; Caforio, G.; Callicott, J.H.; Bertolino, A.; Meyer-Lindenberg, A.; Chang, J.; Ji, Y.; Egan, M.F.; Goldberg, T.E.; Kleinman, J.F.; Lu, B.; Weinberger, D.R. A primate-specific, brain isoform of KCNH2 affects cortical physiology, cognition, neuronal repolarization and risk of schizophrenia. Nat. Med. 2009, 15, 509–518. [Google Scholar] [PubMed]
- Sanguinetti, M.C.; Jang, C.; Curran, M.E.; Keating, M.T. A mechanistic link between an inherited and an acquired cardiac arrhythmia : HERG encodes the Ikr potassium channel. Cell 1995, 81, 299–307. [Google Scholar]
- Witchel, H.J.; Hancox, J.C. Familial and acquired long QT syndrome and the cardiac rapid delayed rectifier potassium current. Clin. Exp. Pharmacol. Physiol. 2000, 27, 753–766. [Google Scholar]
- Curran, M.E.; Splawski, I.; Timothy, K.W.; Vincent, G.M.; Green, E.D.; Keating, M.T. A molecular basis for cardiac arrhythmia: HERG mutations cause long QT syndrome. Cell 1995, 80, 795–803. [Google Scholar]
- Raschi, E.; Vasina, V.; Poluzzi, E.; De Ponti, F. The hERG K+ channel: target and antitarget strategies in drug development. Pharmacol. Res. 2008, 57, 181–195. [Google Scholar]
- Smith, G.A.M.; Tsui, H.; Newell, E.W.; Jiang, X.; Zhu, X.; Tsui, F.W.L.; Schlichter, L.C. Functional up-regulation of HERG K+ in neoplastic hematopoietic cells. J. Biol. Chem. 2002, 277, 18528–18534. [Google Scholar]
- Shao, X.; Wu, K.; Hao, Z.; Hong, L.; Zhang, J.; Fan, D. The potent inhibitory effects of cisapride, a specific blocker for humans ether-à-go-go-related gene (HERG) channel, on gastric cancer. Cancer Biol. Ther. 2005, 4, 295–301. [Google Scholar] [CrossRef] [PubMed]
- Shao, X.; Wu, K.; Guo, X.; Xie, M.; Zhang, J.; Fan, D. Expression and significance of HERG protein in gastric cancer. Cancer Biol. Ther. 2008, 7, 45–50. [Google Scholar] [CrossRef] [PubMed]
- Masi, A.; Becchetti, A.; Restano-Cassulini, R.; Polvani, S.; Hofmann, G.; Buccoliero, A.M.; Paglierani, M.; Pollo, B.; Taddei, G.L.; Gallina, P.; Di Lorenzo, M.; Franceschetti, S.; Wanke, E.; Arcangeli, A. hERG1 channels are overexpressed in glioblastoma multiforme and modulate VEGF secretion in glioblastoma cell lines. Br. J. Cancer 2005, 93, 781–792. [Google Scholar]
- Shah, R.R. Drug-induced QT interval prolongation – regulatory guidance and perspectives on hERG channel studies. Novartis Found Symp. 2005, 266, 251–280. [Google Scholar]
- Stanfield, P.J.; Sutcliffe, M.J.; Mitcheson, J.S. Molecular mechanisms for drug interactions with hERG that cause long QT syndrome. Exp. Opin. Drug Metab. Toxicol. 2008, 2, 81–94. [Google Scholar]
- Mitcheson, J.S. hERG potassium channels and the structural basis of drug-induced arrhythmias. Chem. Res. Toxicol. 2008, 21, 1005–1010. [Google Scholar]
- Wallis, R.M. Integrated risk assessment and predictive value to humans of non-clinical repolarization assays. Br. J. Pharmacol. 2010, 159, 115–121. [Google Scholar]
- Chouabe, C.; Drici, M.D.; Romey, G.; Barhanin, J.; Lazdunski, M. hERG and KvLQT1/IsK, the cardiac K+ channels involved in long QT syndromes, are targets for calcium channel blockers. Mol. Pharmacol. 1998, 54, 695–703. [Google Scholar] [PubMed]
- Rampe, D.; Murawsky, M.K.; Gran, J.; Lewis, E.W. The antipsychotic agent sertindole is a high affinity antagonist of the human cardiac potassium channel HERG. J. Pharmacol. Exp. Ther. 1998, 286, 788–793. [Google Scholar]
- Rogawski, M.A.; Löscher, W. The neurobiology of antiepileptic drugs. Nat. Rev. Neurosci. 2004, 5, 553–564. [Google Scholar]
- Ganapathi, S.B.; Kester, M.; Elmslie, K.S. State-dependent block of HERG potassium channels by R-roscovitine: implications for cancer therapy. Am. J. Physiol. Cell Physiol. 2009, 296, C701–C710. [Google Scholar]
- Stork, D.; Timin, E.N.; Berjukow, S.; Huber, C.; Hohaus, A.; Auer, M.; Hering, S. State dependent dissociation of HERG channel inhibitors. Br. J. Pharmacol. 2007, 151, 1368–1376. [Google Scholar]
- Nguyen, A.; Kath, J.C.; Hanson, D.C.; Biggers, M.S.; Canniff, P.C.; Donovan, C.B.; Mather, R.J.; Bruns, M.J.; Rauer, H.; Aiyar, J.; Lepple-Wienhues, A.; Gutman, G.A.; Grissmer, S.; Cahalan, M.D.; Chandy, K.G. Novel nonpeptide agents potently block the C-type inactivated conformation of Kv 1.3 and suppress T cell activation. Mol. Pharmacol. 1996, 50, 1672–1679. [Google Scholar] [PubMed]
- Ader, C.; Schneider, R.; Hornig, S.; Velisetty, P.; Wilson, E.M.; Lange, A.; Giller, K.; Ohmert, I.; Martin-Eauclaire, M.F.; Trauner, D.; Becker, S.; Pongs, O.; Baldus, M. A structural link between inactivation and block of a K+ channel. Nat. Struct. Mol. Biol. 2008, 15, 605–612. [Google Scholar]
- Milnes, J.T.; Dempsey, C.E.; Ridley, J.M.; Crociani, O.; Arcangeli, A.; Hancox, J.C.; Witchel, H.J. Preferential closed channel blockade of HERG potassium currents by chemically synthesised BeKm-1 scorpion toxin. FEBS Lett. 2003, 547, 20–26. [Google Scholar]
- Zhang, M.; Korolkova, Y.V.; Liu, J.; Jiang, M.; Grishin, E.V.; Tseng, G.-N. BeKm-1 is a HERG-specific toxin that shares the structure with ChTx but the mechanism of action with ErgTx1. Biophys. J. 2003, 84, 3022–3036. [Google Scholar]
- Restano-Cassulini, R.; Korolkova, Y.V.; Diochot, S.; Gurrola, G.; Guasti, L.; Possani, L.D.; Lazdunski, M.; Grishin, E.V.; Arcangeli, A.; Wanke, E. Species diversity and peptide toxins blocking selectivity of ether-à-go-go-related gene subfamily K+ channels in the central nervous system. Mol. Pharmacol. 2006, 69, 1673–1683. [Google Scholar]
- Zhang, M.; Liu, X.S.; Diochot, S.; Lazdunski, M.; Tseng, G.N. APETx1 from sea anemone Anthopleura elegantissima is a gating modifier peptide toxin of the human ether-à-go-go- related potassium channel. Mol. Pharmacol. 2007, 72, 259–268. [Google Scholar]
- Wanke, E.; Restano-Cassulini, R. Toxins interacting with ether-à-go-go-related gene voltage-dependent potassium channels. Toxicon 2007, 49, 239–248. [Google Scholar]
- Restano-Cassulini, R.; Olamendi-Portugal, T.; Zamudio, F.; Becerril, B.; Possani, L.D. Two novel ergtoxins, blockers of K+ channels, purified from the Mexican scorpion Centruroides elegans elegans. Neurochem. Res. 2008, 33, 1525–1533. [Google Scholar] [CrossRef] [PubMed]
- Redaelli, E.; Restano-Cassulini, R.; Fuentes Silva, D.; Clement, H.; Schiavon, E.; Zamudio, F.Z.; Odell, G.; Arcangeli, A.; Clare, J.J.; Alagon, A.; Rodriguez de la Vega, R.C.; Possani, L.D.; Wanke, E. Target promiscuity and heterogeneous effects of Tarantula venom peptides affecting Na+ and K+ ion channels. J. Biol. Chem. 2010, 285, 4130–4142. [Google Scholar]
- Panyi, G.; Possani, L.D.; Rodríguez de la Vega, R.C.; Gáspár, R.; Varga, Z. K+ channel blockers: novel tools to inhibit T cell activation leading to specific immunosuppression. Curr. Pharm. Des. 2006, 12, 219. [Google Scholar]
- Wulff, H.; Pennington, M. Targeting effector memory T-cells with Kv1.3 blockers. Curr. Opin. Drug Discov. Devel. 2007, 10, 438. [Google Scholar] [PubMed]
- Beeton, C.; Wulff, H.; Standifer, N.E.; Azam, P.; Mullen, K.M.; Pennington, M.W.; Kolski-Andreaco, A.; Wei, E.; Grino, A.; Counts, D.R.; Wang, P.H.; LeeHealey, C.J.; S Andrews, B.; Sankaranarayanan, A.; Homerick, D.; Roeck, W.W.; Tehranzadeh, J.; Stanhope, K.L.; Zimin, P.; Havel, P.J.; Griffey, S.; Knaus, H.; Nepom, G.T.; Gutman, G.A.; Calabresi, P.A.; Chandy, K.G. Kv1.3 channels are a therapeutic target for T cell-mediated autoimmune diseases. Proc. Natl. Acad. Sci. USA 2006, 103, 17414–17419. [Google Scholar]
- Pagès, F.; Berger, A.; Camus, M.; Sanchez-Cabo, F.; Costes, A.; Molidor, R.; Mlecnik, B.; Kirilovsky, A.; Nilsson, M.; Damotte, D.; Meatchi, T.; Bruneval, P.; Cugnenc, P.H.; Trajanoski, Z.; Fridman, W.H.; Galon, J. Effector memory T cells, early metastasis, and survival in colorectal cancer. N. Engl. J. Med. 2005, 353, 2654–2666. [Google Scholar] [PubMed]
- Klebanoff, C.A.; Gattinoni, L.; Torabi-Parizi, P.; Kerstann, K.; Cardones, A.R.; Finkelstein, S.E.; Palmer, D.C.; Antony, P.A.; Hwang, S.T.; Rosenberg, S.A.; Waldmann, T.A.; Restifo, N.P. Central memory self/tumor-reactive CD8+ T cells confer superior antitumor immunity compared with effector memory T cells. Proc. Natl. Acad. Sci. USA 2005, 102, 9571–9576. [Google Scholar]
- Galon, J.; Costes, A.; Sanchez-Cabo, F.; Kirilovsky, A.; Mlecnik, B.; Lagorce-Pagès, C.; Tosolini, M.; Camus, M.; Berger, A.; Wind, P.; Zinzindohoué, F.; Bruneval, P.; Cugnenc, P.H.; Trajanoski, Z.; Fridman, W.H.; Pagès, F. Type, density, and location of immune cells within human colorectal tumors predict clinical outcome. Science 2006, 313, 1960–1964. [Google Scholar] [PubMed]
- Paleari, L.; Negri, E.; Catassi, A.; Cilli, M.; Servent, D.; D’Angelillo, R.; Cesario, A.; Russo, P.; Fini, M. Inhibition of nonneuronal alpha7-nicotinic receptor for lung cancer treatment. Am. J. Respir. Crit. Care Med. 2009, 179, 1141–1150. [Google Scholar]
- Paleari, L.; Sessa, F.; Catassi, A.; Servent, D.; Mourier, G.; Doria-Miglietta, G.; Ognio, E.; Cilli, M.; Dominioni, L.; Paolucci, M.; Calcaterra, A.; Cesario, A.; Margaritora, S.; Granone, P.; Russo, P. Inhibition of non-neuronal alpha7-nicotinic receptor reduces tumorigenicity in A549 NSCLC xenografts. Int. J. Cancer 2009, 125, 199–211. [Google Scholar]
- Brown, M.L.; Zha, C.C.; Van Dyke, C.C.; Brown, G.B.; Brouillette, W.J. Comparative molecular field analysis of hydantoin binding to the neuronal voltage-dependent sodium channel. J. Med. Chem. 1999, 42, 1537–1545. [Google Scholar]
- Anderson, J.D.; Hansen, T.P.; Lenkowski, P.W.; Walls, A.M.; Choudhury, I.M.; Schenck, H.A.; Friehling, M.; Höll, G.M.; Patel, M.K.; Sikes, R.A.; Brown, M.L. Voltage-gated sodium channel blockers as cytostatic inhibitors of the androgen-independent prostate cancer cell line PC-3. Mol. Cancer Ther. 2003, 2, 1149–1154. [Google Scholar]
- Sikes, R.A.; Walls, A.M.; Brennen, W.N.; Anderson, J.D.; Choudhury-Mukherjee, I.; Schenck, H.A.; Brown, M.L. Therapeutic approaches targeting prostate cancer progression using novelvoltage-gated ion channel blockers. Clin. Prostate Cancer 2003, 2, 181–187. [Google Scholar]
- Xu, S.Z.; Zeng, F.; Lei, M.; Li, J.; Gao, B.; Xiong, C.; Sivaprasadarao, A.; Beech, D.J. Generation of functional ion channel tools by E3 targeting. Nat. Biotechnol. 2005, 23, 1289–1293. [Google Scholar]
- Martial, S.; Giorgelli, J-L.; Renaudo, A.; Derijard, B.; Soriani, O. SP600125 inhibits Kv channels through a JNK-independent pathway in cancer cells. Biochem. Biophys. Res. Commun. 2008, 366, 944–950. [Google Scholar] [CrossRef] [PubMed]
- Suh, K.S.; Mutoh, M.; Gerdes, M.; Yuspa, S.H. CLIC4, an intracellular chloride channel protein, is a novel molecular target for cancer therapy. J. Investig. Dermatol. Symp. Proc. 2005, 10, 105–109. [Google Scholar] [CrossRef] [PubMed]
- Mathieu, V.; Pirker, C.; de Lassalle, E.M.; Vernier, M.; Mijatovic, T.; Deneve, N.; Gaussin, J.F.; Dehoux, M.; Lefranc, F.; Berger, W.; Kiss, R. The sodium pump α1subunit: a disease progression-related target for metastatic melanoma treatment. J. Cell. Mol. Med. 2009, 3960–3972. [Google Scholar] [PubMed]
- Mijatovic, T.; Jungwirth, U.; Heffeter, P.; Hoda, M.A.; Dornetshuber, R.; Kiss, R.; Berger, W. The Na+/K+ ATPase is the Achilles heel of multi-drug-resistant cancer cells. Cancer Lett. 2009, 282, 30–34. [Google Scholar]
- Gómez-Varela, D.; Zwick-Wallasch, E.; Knötgen, H.; Sánchez, A.; Hettmann, T.; Ossipov, D.; Weseloh, R.; Contreras-Jurado, C.; Rothe, M.; Stühmer, W.; Pardo, L.A. Monoclonal antibody blockade of the human Eag1 potassium channel function exerts antitumor activity. Cancer Res. 2007, 67, 7343–7349. [Google Scholar]
- Deshane, J.; Garner, C.C.; Sontheimer, H. Chlorotoxin inhibits glioma cell invasion via matrix metalloproteinase-2. J. Biol. Chem. 2003, 278, 4135–4144. [Google Scholar] [PubMed]
- Hinman, L.M.; Hannann, P.R.; Wallace, R.; Menendez, A.T.; Durr, M.E.; Upeslacis, J. Preparation and characterization of monoclonal antibody conjugates of the calicheamicins: a novel and potent family of antitumor antibiotics. Cancer Res. 1993, 53, 3336–3342. [Google Scholar]
- Singh, Y.; Palombo, M.; Sinko, P.J. Recent trends in targeted anticancer prodrug and conjugate design. Curr. Med. Chem. 2008, 15, 1802–1826. [Google Scholar]
- Onkal, R; Djamgoz, M.B.A. Molecular pharmacology of voltage-gated sodium channel expression in metastatic disease: clinical potential of neonatal Nav 1.5 in breast cancer. Eur. J. Pharmacol. 2009, 625, 206–219. [Google Scholar] [CrossRef] [PubMed]
© 2010 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Arcangeli, A.; Becchetti, A. New Trends in Cancer Therapy: Targeting Ion Channels and Transporters. Pharmaceuticals 2010, 3, 1202-1224. https://doi.org/10.3390/ph3041202
Arcangeli A, Becchetti A. New Trends in Cancer Therapy: Targeting Ion Channels and Transporters. Pharmaceuticals. 2010; 3(4):1202-1224. https://doi.org/10.3390/ph3041202
Chicago/Turabian StyleArcangeli, Annarosa, and Andrea Becchetti. 2010. "New Trends in Cancer Therapy: Targeting Ion Channels and Transporters" Pharmaceuticals 3, no. 4: 1202-1224. https://doi.org/10.3390/ph3041202