3. Results and Discussion
For all ten series of glycine site antagonists of NMDA receptors (
I–VIII), as listed in
Table 1,
Table 2,
Table 3,
Table 4,
Table 5,
Table 6,
Table 7,
Table 8, the best correlations (Equations 1–10) obtained are summarized below along with externally validated Equations (1a–10a):
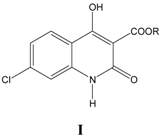
i. Inhibition of glycine/NMDA site by I (Table 1) [24]
− logIC50 = 0.157(0.100)CMR + 1.139 (0.363)I1 + 4.000(0.852)
n = 13, r=0.950, s=0.18, F2,10 = 45.93(7.56)
r2 = 0.90, r2A = 0.88, Q2 = 0.83, Qy = 5.28
CMR range = 6.21-10.16, outliers = 20, 22
r2(CMR vs. ClogP) = 0.63 (1)
− logIC50 = 0.131(0.124)CMR + 1.119(0.394)I1 + 4.277(1.049)
n = 10, r = 0.960, s = 0.17, F2,7 = 41.07(9.55)
r2 = 0.92, r2A = 0.90, Q2 = 0.84, Qy = 5.75
Excluded compounds = 8,14,17 (1a)
The activity in Equation (1) is largely governed by CMR, which reveals that there is an overall dispersion interaction of the molecule with the target receptor. Indicator variable I1 takes a value of 1 for compounds with conformational constraints in the form of triple bond and zero, if otherwise. A positive I1 coefficient indicates high electronic density moieties in the form of triple bond add to the potency of the molecule by strengthening dispersive forces involved in ligand-receptor interaction. Interestingly, CMR is a very good measure of dispersive force. A small correlation between CMR and ClogP presumably indicates that lipophilicity of molecules is also important but we could not obtain a good correlation with ClogP. Compounds 20 and 22 were not included in the equation as they exhibited inconsistent activities.
Table 1.
A series of 4-hydroxyquinolin-2(1
H)-ones, 3-esters I, with their structural parameters and binding affinities.
![Pharmaceuticals 03 03167 i001]()
Table 1.
A series of 4-hydroxyquinolin-2(1H)-ones, 3-esters I, with their structural parameters and binding affinities. ![Pharmaceuticals 03 03167 i001]()
| C.N. | R | I1 | CMR | −logIC50 |
|---|
| Obs. | Cal. | Δ | Ext. Pred. | Δ |
|---|
| 8 | Et | 0.00 | 6.68 | 4.78 | 5.05 | −0.27 | 5.15 | −0.37 |
| 10 | Me | 0.00 | 6.21 | 5.19 | 4.98 | 0.21 | | |
| 12 | Allyl | 0.00 | 7.09 | 5.19 | 5.12 | 0.07 | | |
| 13 | Propargyl | 0.00 | 6.94 | 5.04 | 5.09 | −0.05 | | |
| 14 | CH2-3-(phenol) | 0.00 | 8.78 | 5.21 | 5.38 | −0.17 | 5.43 | −0.22 |
| 15 | (CH2)2-3-(phenol) | 0.00 | 9.24 | 5.74 | 5.45 | 0.29 | | |
| 16 | (CH2)3-3-(phenol) | 0.00 | 9.70 | 5.58 | 5.52 | 0.06 | | |
| 17 | (CH2)4-3-(phenol) | 0.00 | 10.16 | 5.58 | 5.60 | −0.02 | 5.43 | −0.03 |
| 18 | (CH2)2-2-(phenol) | 0.00 | 9.24 | 5.15 | 5.45 | −0.15 | | |
| 19 | (CH2)2-4-(phenol) | 0.00 | 9.24 | 5.53 | 5.45 | 0.08 | | |
| 20 | (CH2)2-2-(pyridine) | 0.00 | 9.02 | 4.56a | 5.42 | −0.86 | | |
| 21 | (CH2)2-2-(thiophene) | 0.00 | 9.01 | 5.52 | 5.42 | 0.10 | | |
| 22 | (CH2)2-3-(indole) | 0.00 | 10.14 | 6.37a | 5.59 | 0.78 | | |
| 23 | CH2C≡C-4-(anisole) | 1.00 | 9.97 | 6.57 | 6.71 | −0.14 | | |
| 24 | CH2C≡C-4-(phenol) | 1.00 | 9.50 | 6.77 | 6.63 | 0.14 | | |
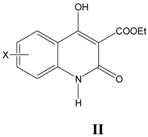
ii. Inhibition of glycine/NMDA site by II (Table 2) [24]
− logIC50 = − 0.440(0.219)ClogP + 0.230(0.070)ClogP2 + 4.771(0.161)
n = 5, r = 0.997, s = 0.04, F1,3 = 165.81(34.12)
r2 = 0.99, r2A = 0.99, Q2 = 0.83, Qy = 26.24
Inversion point = 0.96(0.65 − 1.16)
r2(CMR vs. ClogP) = 0.032 (2)
In the generation of Equation (2), activity seems to be dependent upon ClogP in an allosteric way. Activity decreases with ClogP upto inversion point and then increases. For good activity, ClogP of molecules atleast should be above inversion point (0.96). The observation gets support from the derivation of Equation (3), which indicates a positive linear relationship of biological activity with ClogP in arange above inversion point (1.22–2.70).
Table 2.
A series of 3-(ethoxycarbonyl)-4-hydroxyquinolin-2(1
H)-ones II, aromatic substitutions with their structural parameters and binding affinities.
![Pharmaceuticals 03 03167 i002]()
Table 2.
A series of 3-(ethoxycarbonyl)-4-hydroxyquinolin-2(1H)-ones II, aromatic substitutions with their structural parameters and binding affinities. ![Pharmaceuticals 03 03167 i002]()
| C.N | R | ClogP | −logIC50 |
|---|
| Obs. | Cal. | Δ |
|---|
| 29 | 7-CN | 0.85 | 4.58 | 4.56 | 0.02 |
| 30 | 7-CF3 | 2.30 | 4.91 | 4.97 | −0.06 |
| 31 | 7-NO2 | 1.16 | 4.60 | 4.57 | 0.03 |
| 32 | 5-I, 7-Cl | 3.25 | 5.79 | 5.76 | 0.03 |
| 33 | 6,7-(NO2)2 | −0.42 | 4.98 | 4.99 | −0.01 |
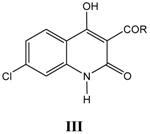
iii. Inhibition of glycine/NMDA site by III (Table 3) [24]
−logIC50 = 0.601(0.369)ClogP + 4.448(0.788)
n = 5, r = 0.948, s = 0.11, F1,3 = 26.85(34.12)
r 2 = 0.99, r2A = 0.99, Q2 = 0.80, Qy = 8.62
outliers: 9, 42 r2(CMR vs. ClogP) = 0.352 (3)
Table 3.
A series of 4-hydroxyquinolin-2(1
H)-ones, 3-ketones III, with their structural parameters and binding affinities.
![Pharmaceuticals 03 03167 i003]()
Table 3.
A series of 4-hydroxyquinolin-2(1H)-ones, 3-ketones III, with their structural parameters and binding affinities. ![Pharmaceuticals 03 03167 i003]()
| C.N. | R | ClogP | −logIC50 |
|---|
| Obs. | Cal. | Δ |
|---|
| 9 | C6H5 | 2.70 | 5.50a | 6.07 | −0.57 |
| 35 | 3-thiophene | 2.34 | 5.70 | 5.86 | −0.16 |
| 36 | 3-furan | 1.90 | 5.62 | 5.91 | −0.29 |
| 37 | 3-pyridine | 1.22 | 5.16 | 5.18 | −0.02 |
| 39 | CH2C6H5 | 2.64 | 6.03 | 6.03 | 0.00 |
| 40 | CH2-3-(thiophene) | 2.28 | 5.97 | 5.82 | 0.15 |
| 42 | cyclopropane | 1.51 | 6.38a | 5.36 | 1.02 |
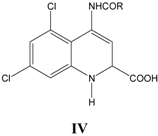
iv. Inhibition of glycine/NMDA site by IV (Table 4) [25]
− logIC50 = − 0.235(0.076)L1 + 3.853(0.909)CMR
− 0.176(0.045)CMR2 − 0.549(0.260)I2 – 12.124(4.378)
n = 18, r = 0.960, s = 0.14, F3,14 = 38.44(5.56)
r2 = 0.92, r2A = 0.90, Q2 = 0.85, Qy =7 .06
CMRo = 10.9(10.7-11.3), outlier = 22
r2(CMR vs. ClogP) = 0.37 (4)
− logIC50 = − 0.239(0.121)L1 + 3.895(0.719)CMR
− 0.179(0.035)CMR2 − 0.524(0.175)I2 – 12.311(3.306)
n = 12, r = 0.98, s = 0.07, F3,8 = 81.95(7.59)
r2= 0.98, r2A= 0.97, Q2= 0.94, Qy= 13.36
CMRo = 11.00(10.80−11.30)
Excluded compounds = 21, 14, 29, 30, 31 (4a)
In Equation (4), again CMR emerges as an important parameter with normal allosteric effect. The CMRo obtained is 10.9 (10.7-11.3). This equation is particularly interesting as it provides clues for further synthesis of promising molecules with an optimum CMR range. The negative coefficient of L1 indicates that probably a size limited lipophilic pocket is present near to substituent R in receptor, which does not allow lengthy substituents for efficient binding. Indicator variable I2 stands with a value of unity for compounds with saturated ring as substituent at R and zero, if otherwise. A negative I2 in equation reveals that saturated rings are not preferred probably because of their thin delocalized electron cloud, which renders them weak to participate in dispersive force interaction between molecule and receptor for binding.
Table 4.
A series of trans-4-amido-2-carboxytetrahydroquinolines IV, with their structural parameters and binding affinities.
![Pharmaceuticals 03 03167 i004]()
Table 4.
A series of trans-4-amido-2-carboxytetrahydroquinolines IV, with their structural parameters and binding affinities. ![Pharmaceuticals 03 03167 i004]()
| C.N. | R | CMR | L1 | I2 | −logIC50 |
|---|
| Obs. | Cal. | Δ | Ext. Pred. | Δ |
|---|
| 17 | CH3 | 7.41 | 3.08 | 0.00 | 6.04 | 6.03 | 0.01 | | |
| 18 | n-Pr | 8.33 | 5.28 | 0.00 | 6.43 | 6.50 | −0.07 | | |
| 19 | c-hexyl | 9.50 | 5.23 | 1.00 | 6.77 | 6.80 | −0.03 | | |
| 20 | CH2-c-hexyl | 9.96 | 5.50 | 1.00 | 6.96 | 6.93 | 0.03 | | |
| 9 | Ph | 9.36 | 6.27 | 0.00 | 7.00 | 7.03 | −0.03 | | |
| 21 | 2-furyl | 8.65 | 5.26 | 0.00 | 6.82 | 6.79 | 0.03 | 6.74 | 0.08 |
| 22 | 4-pyridyl | 9.29 | 5.19 | 0.00 | 6.20a | 7.26 | −1.06 | | |
| 14 | CH2Ph | 9.82 | 4.59 | 0.00 | 7.92 | 7.64 | 0.28 | 7.58 | 0.34 |
| 23 | CH2(3-thienyl) | 9.74 | 4.95 | 0.00 | 7.52 | 7.53 | −0.01 | | |
| 24 | CH2(2-thienyl) | 9.74 | 4.56 | 0.00 | 7.52 | 7.62 | −0.10 | | |
| 25 | CH2C6H4NH2-4 | 10.16 | 5.28 | 0.00 | 7.70 | 7.60 | 0.10 | | |
| 26 | CH2C6H4OH-4 | 9.97 | 5.22 | 0.00 | 7.52 | 7.55 | −0.03 | | |
| 27 | CH2C6H4CH3-4 | 10.28 | 6.00 | 0.00 | 7.52 | 7.46 | 0.06 | | |
| 28 | CH2C6H4OCH3-4 | 10.45 | 5.63 | 0.00 | 7.40 | 7.58 | −0.18 | | |
| 29 | CH2C6H4Cl-4 | 10.31 | 5.62 | 0.00 | 7.30 | 7.55 | −0.25 | 7.48 | −0.18 |
| 30 | (CH2)2Ph | 10.28 | 8.59 | 0.00 | 7.10 | 6.85 | 0.25 | 6.76 | 0.34 |
| 31 | (CH2)3Ph | 10.74 | 8.54 | 0.00 | 6.77 | 6.93 | −0.16 | 6.83 | −0.06 |
| 32 | CHPh2 | 12.23 | 5.80 | 0.00 | 7.22 | 7.28 | −0.06 | | |
| 33 | 9-fluorenyl | 12.01 | 4.94 | 0.00 | 7.70 | 7.58 | 0.12 | | |
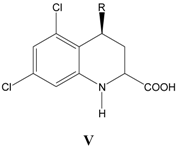
v. Inhibition of glycine/NMDA site by V (Table 5) [25]
− logIC50 = 0.430(0.193)CMR + 2.499(1.874)
n = 6, r = 0.952, s = 0.11, F1,4 = 38.40(21.20)
r2 = 0.90, r2A = 0.88, Q2 = 0.67, Qy = 8.42
CMR range = 8.14-10.49, outlier = 57
r2(CMR vs. ClogP) = 0.31 (5)
− logIC50 = 0.412(0.222)CMR + 2.649(2.142)
n = 5, r = 0.960, s = 0.11, F1,3 = 34.91(34.12)
r2 = 0.92, r2A = 0.89, Q2 = 0.68, Qy = 8.97
Excluded compound = 54 (5a)
Similarly CMR dominates Equation (5) with a positive regression coefficient in the range 8.14-10.49, which is a subset of CMR range witnessed in Equation (1), indicating both series I and V bind to the glycine site in a similar way.
Table 5.
A series of conformationally restricted 4-substituents
V, with their structural parameters and binding affinities.
![Pharmaceuticals 03 03167 i005]()
Table 5.
A series of conformationally restricted 4-substituents V, with their structural parameters and binding affinities. ![Pharmaceuticals 03 03167 i005]()
| C.N. | R | CMR | −logIC50 |
|---|
| Obs. | Cal. | Δ | Ext. Pred. | Δ |
|---|
| 51 | 1-pyrrolidin-2-one | 8.14 | 6.04 | 6.00 | 0.04 | | |
| 52 | 1-isoindolin-1-one | 9.63 | 6.43 | 6.64 | −0.21 | | |
| 53 | 2-isoindoline-1,3-dione | 9.80 | 6.77 | 6.72 | 0.05 | | |
| 54 | 2-(1,2-dihydroiso-quinolin-3-(4H) one) | 10.09 | 6.96 | 6.84 | 0.12 | 6.81 | 0.15 |
| 55 | 1-(3-phenylimidazolidin-2-one) | 10.49 | 7.00 | 7.01 | −0.01 | | |
| 56 | 3-(3,4-dihydroquinazolin-2-(1H)-one) | 10.02 | 6.82 | 6.81 | 0.01 | | |
| 57 | -N-indoline-1-carboxamide | 10.45 | 6.20a | 6.99 | −0.79 | | |
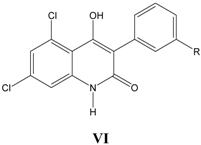
vi. Inhibition of glycine/NMDA site by VI (Table 6) [26]
− logIC50 = −13.60 (9.66)CMR + 0.633 (0.45)CMR2 + 81.45 (51.4)
n = 6, r = 0.940, s = 0.08, F1,4 = 11.42(21.20)
r2 = 0.91, r2A = 0.89, Q2 = 0.64, Qy = 11.190
CMR range = 7.37−11.49, outlier = 6
Inversion point = 10.8(10.6−11.1), r2(CMR vs. ClogP) = 0.61
(6)
− logIC50 = −14.61 (10.6)CMR + 0.681(0.50)CMR2 + 86.71 (56.5)
n = 5, r = 0.975, s = 0.07, F1,3 = 19.49(34.12)
r2 = 0.95, r2A = 0.93, Q2 = 0.80, Qy = 14.55
Excluded compound = 12
(6a)
In Equation (6), a negative allosteric effect with CMR is observed. Activity decreases with CMR upto inversion point (10.8) and then increases with further increase in CMR.
Table 6.
A series of substituted 3-phenyl-4-hydroxy-2-quinolones VI, with their structural parameters and binding affinities.
![Pharmaceuticals 03 03167 i006]()
Table 6.
A series of substituted 3-phenyl-4-hydroxy-2-quinolones VI, with their structural parameters and binding affinities. ![Pharmaceuticals 03 03167 i006]()
| C.N. | R | CMR | −logIC50 |
|---|
| Obs. | Cal. | Δ | Ext. Pred. | Δ |
|---|
| 6 | H | 7.37 | 6.77a | 15.56 | −8.83 | | |
| 9 | CH2Ph | 10.24 | 8.39 | 8.49 | −0.10 | | |
| 10 | CH2PhOMe-4 | 10.87 | 8.35 | 8.33 | 0.02 | | |
| 11 | CH2PhOCH2OMe-4 | 11.49 | 8.66 | 8.67 | −0.01 | | |
| 12 | CH2(3-thienyl) | 10.16 | 8.64 | 8.54 | 0.10 | 8.57 | 0.07 |
| 13 | OPh | 9.94 | 8.70 | 8.74 | −0.04 | | |
| 14 | O(3-thienyl) | 9.86 | 8.85 | 8.83 | 0.02 | | |
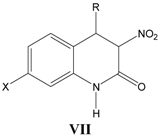
vii. Inhibition of glycine/NMDA site by VII (Table 7) [27]
− logIC50 = −2.748(1.769)CMR + 0.140(0.093)CMR2
+ 1.660(0.766)Ia + 16.958(7.869)
n = 10, r = 0.918, s = 0.21, F2,8 = 10.67(8.65)
r2 = 0.87, r2A = 0.83, Q2 = 0.42, Qy = 4.41
Inversion point = 9.82(9.38-10.9), CMR range = 7.05-11.90
r2(CMR vs. ClogP) = 0.04
(7)
− logIC50 = −3.215(1.984)CMR + 0.162(0.105)CMR2
+ 1.792(0.793)Ia + 19.166(8.815)
n = 7, r = 0.976, s = 0.14, F2,5 = 19.73(13.27)
r2 = 0.95, r2A = 0.92, Q2 = 0.77, Qy = 7.18
Inversion point = 9.93(9.48-11.1)
Excluded compounds = 4, 16, 19
(7a)
Equation (7) involves inverse allosteric effect with an inversion point of 9.82. Indicator variable Ia= 1 for compounds containing acidic moieties and zero for neutral and basic moieties. A positive Ia indicates presence of acidic substituents is preferred which interacts with a proton acceptor site present in the glycine site close to R.
Table 7.
A series of 3-nitro-3,4-dihydro-2(1
H)-quinolones
VII with their structural parameters and binding affinities.
![Pharmaceuticals 03 03167 i007]()
Table 7.
A series of 3-nitro-3,4-dihydro-2(1H)-quinolones VII with their structural parameters and binding affinities. ![Pharmaceuticals 03 03167 i007]()
| C.N. | R | X | Ia | CMR | −logIC50 |
|---|
| Obs. | Cal. | Δ | Ext. Pred. | Δ |
|---|
| 4 | OH | Cl | 1.00 | 7.53 | 5.88 | 5.86 | 0.02 | 5.85 | 0.03 |
| 13 | OCH2COOMe | H | 0.00 | 7.05 | 4.54 | 4.54 | 0.00 | | |
| 14 | OCH2COOH | Cl | 1.00 | 7.51 | 5.65 | 5.88 | −0.23 | | |
| 15 | OCH2CONMe2 | Cl | 1.00 | 9.56 | 5.25 | 5.14 | 0.11 | | |
| 16 | OCH2CONH2 | Cl | 1.00 | 11.44 | 5.74 | 5.50 | 0.24 | 5.39 | 0.35 |
| 17 | OCH2CN | Cl | 1.00 | 10.94 | 5.12 | 5.30 | −0.18 | | |
| 18 | OCH2-Pyr-2 | Cl | 1.00 | 9.00 | 4.91 | 5.22 | −0.31 | | |
| 19 | O(CH2)2NMe2 | Cl | 1.00 | 9.49 | 5.43 | 5.14 | 0.33 | 5.02 | 0.41 |
| 20 | OCH2COMe | Cl | 1.00 | 11.90 | 5.60 | 5.74 | −0.14 | | |
| 21 | OCH2C(Me)=NOH | Cl | 1.00 | 7.06 | 6.38 | 6.19 | 0.19 | | |
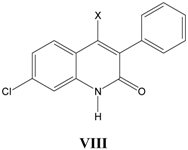
viii. Inhibition of glycine/NMDA site by VIII (Table 8) [28]
− logIC50= 7.965(3.677)CMR − 0.397(0.180)CMR2
+ 1.162(0.319)Ia − 34.426(18.651)
n = 9, r = 0.977, s = 0.13, F2,6 = 34.32(10.92)
r2 = 0.95, r2A = 0.93, Q2 = 0.84, Qy = 7.29
CMRo = 10.0(9.76-10.20) CMR range = 7.56-11.56
Outliers = 3,16, r2(CMR vs. ClogP) = 0.10
(8)
− logIC50 = 9.201(5.698)CMR − 0.457(0.280)CMR2
+ 1.217(0.517)Ia − 40.614(28.777)
n = 7, r = 0.977, s = 0.12, F2,4 = 20.85(18.00)
r2 = 0.96, r2A = 0.94, Q2 = 0.57, Qy = 7.88
CMRo = 10.1(9.69−10.3), Excluded compounds = 6, 12
(8a)
In derivation of Equation (8), where heteroatom N of substituent R1 is connected to 3-phenylquinolin-2(1H)-one nucleus, a positive allosteric effect is observed. Activity increases upto inversion point (CMRo = 10) and then starts decreasing. Preference for acidic substituents is indicated by a positive coefficient of Ia. This equation could provide some clue in proposing molecules falling in optimum CMR range (9.76-10.20).
Table 8.
A series of 4-substituted-3-phenylquinolin-2(1
H)-ones
VIII with their structural parameters and binding affinities.
![Pharmaceuticals 03 03167 i008]()
Table 8.
A series of 4-substituted-3-phenylquinolin-2(1H)-ones VIII with their structural parameters and binding affinities. ![Pharmaceuticals 03 03167 i008]()
| C.N. | X = R1 | Ia | CMR | −logIC50 |
|---|
| Obs. | Cal. | Δ | Ext. Pred. | Δ |
|---|
| 3 | NH2 | 0.00 | 7.56 | 5.17a | 3.10 | 2.07 | | |
| 4 | NHCOMe | 0.00 | 8.67 | 4.83 | 4.81 | 0.02 | | |
| 6 | NHSO2Me | 1.00 | 9.62 | 6.57 | 6.65 | −0.08 | 6.82 | −0.25 |
| 7 | NHSO2Ph | 1.00 | 11.57 | 5.92 | 5.79 | 0.13 | | |
| 10 | NHCH2Ph | 0.00 | 10.45 | 5.70 | 5.49 | 0.21 | | |
| 11 | NH(CH2)2NMe2 | 0.00 | 9.82 | 5.49 | 5.54 | −0.05 | | |
| 12 | NH(CH2)3NMe2 | 0.00 | 10.28 | 5.38 | 5.53 | −0.15 | 5.68 | −0.30 |
| 13 | NHCOCOOH | 1.00 | 9.00 | 6.24 | 6.29 | −0.05 | | |
| 14 | NHCOCOOMe | 0.00 | 9.47 | 5.57 | 5.43 | 0.14 | | |
| 15 | NHCOCONH(CH2)2NMe2 | 0.00 | 11.45 | 4.60 | 4.77 | −0.17 | | |
| 16 | NHCOCH2COOH | 1.00 | 9.46 | 5.96a | 6.56 | −0.60 | | |
ix. Inhibition of glycine/NMDA site by VIII (Table 9) [28]
− logIC50 = 0.647(0.313)Ia − 1.289(0.325)CMR + 17.447(2.970)
n = 9, r = 0.974, s = 0.16, F2,6 = 56.05(10.92)
r2 = 0.95, r2A = 0.932, Q2 = 0.91, Qy = 6.24
outlier = 1, CMR range = 4.94-6.96
r2(CMR vs. ClogP):0.03
(9)
−logIC50 = 0.687(0.279)Ia − 1.237(0.285)CMR + 16.928(2.641)
n = 7, r = 0.990, s = 0.11, F2,4 = 99.79(18.00)
r2 = 0.98, r2A = 0.97, Q2 = 0.94, Qy = 9.25
Excluded compounds = 22,27
(9a)
In Equation (9), where heteroatom O of substituent R2 is connected to 3-phenylquinolin-2(1H)-one nucleus, a negative linear relationship is observed with CMR. The equation also indicates a positive correlation with Ia.
Table 9.
A series of 4-substituted-3-phenylquinolin-2(1H)-ones VIII with their structural parameters and binding affinities.
Table 9.
A series of 4-substituted-3-phenylquinolin-2(1H)-ones VIII with their structural parameters and binding affinities.
| C.N. | X = R2 | Ia | CMR | −logIC50 |
|---|
| Obs. | Cal. | Δ | Ext. Pred. | Δ |
|---|
| 1 | OH | 1.00 | 7.37 | 6.77a | 7.30 | −0.53 | | |
| 19 | OCH2COOMe | 0.00 | 9.10 | 5.49 | 5.77 | −0.28 | | |
| 20 | OCH2COOH | 1.00 | 8.63 | 6.96 | 6.17 | 0.79 | | |
| 21 | OCH2CONMe2 | 0.00 | 9.80 | 4.94 | 5.18 | −0.24 | | |
| 22 | OCH2CONH2 | 0.00 | 8.82 | 6.38 | 6.03 | 0.03 | 6.01 | 0.37 |
| 23 | OCH2CN | 0.00 | 8.52 | 6.49 | 6.46 | 0.03 | | |
| 25 | OCH2-Pyr-2 | 1.00 | 9.77 | 5.55 | 5.60 | −0.05 | | |
| 26 | O(CH2)2NMe2 | 0.00 | 9.62 | 4.96 | 5.00 | −0.04 | | |
| 27 | OCH2COMe | 0.00 | 8.94 | 5.77 | 5.87 | −0.10 | 5.87 | −0.10 |
| 28 | OCH2C(Me)=NOH | 1.00 | 9.26 | 6.11 | 5.61 | 0.50 | | |
x. Inhibition of glycine/NMDA site by VIII (Table 10) [28]
− logIC50 = 0.781(0.383)Ia + 12.130(7.142)CMR
− 0.662(0.389)CMR2 − 49.338(32.694)
n = 8, r = 0.959, s = 0.13, F2,5 = 15.37(13.27)
r2 = 0.93, r2A =0.90, Q2 = 0.77, Qy = 8.70
CMRo = 9.15(8.89-9.40), CMR range = 8.00-10.16, outlier = 39
r2(CMR vs. ClogP) = 0.04
(10)
−logIC50 = 0.830(0.422)Ia + 11.264(7.739)CMR
− 0.616(0.422)CMR2 − 45.436(35.382)
n = 7, r = 0.974, s = 0.11, F2,4= 18.53(18.00)
r2 = 0.95, r2A = 0.92, Q2 = 0.66, Qy = 8.70
CMRo = 9.14(8.83-9.43), Excluded compound = 4
(10a)
A normal allosteric effect is again seen in Equation (10) with CMR, where carboxylic acid derivatives are substituted at 4-position in VIII. Activity increases upto inversion point (CMRo=9.15) and then decreases with further increase in CMR. This equation again could provide clues in proposing newer molecules falling in the optimum CMR range. Acidic substituents are preferred as indicated by positive coefficient of Ia.
Table 10.
A series of 4-substituted-3-phenylquinolin-2(1H)-ones VIII with their structural parameters and binding affinities.
Table 10.
A series of 4-substituted-3-phenylquinolin-2(1H)-ones VIII with their structural parameters and binding affinities.
| C.N. | X=R | Ia | CMR | −logIC50 |
|---|
| Obs. | Cal. | Δ | Ext. Pred. | Δ |
|---|
| 31 | COOH | 1.00 | 8.00 | 6.04 | 6.06 | −0.02 | | |
| 33 | CH2COOMe | 0.00 | 8.94 | 5.96 | 6.12 | −0.16 | | |
| 34 | CH2COOH | 1.00 | 8.46 | 6.80 | 6.62 | 0.18 | | |
| 36 | CH2CH2COOCH3 | 0.00 | 9.40 | 6.30 | 6.10 | 0.20 | 6.02 | 0.28 |
| 37 | CH2CH2COOH | 1.00 | 8.93 | 6.77 | 6.90 | −0.13 | | |
| 38 | (CH2)2-5-(3-methyl-1,2,4-oxadiazole) | 0.00 | 10.16 | 5.46 | 5.47 | −0.01 | | |
| 39 | CH2CH2CONH2 | 0.00 | 9.12 | 6.72a | 6.23 | 0.49 | | |
| 40 | CH2CH2CN | 0.00 | 8.82 | 6.12 | 6.08 | 0.04 | | |
| 41 | (CH2)2-5-(1H-tetrazole) | 1.00 | 9.86 | 6.62 | 6.59 | 0.03 | | |
A comparison of these ten equations elicits some very interesting points about the nature of the glycine site and the rersulting mechanistic interpretation of its binding interactions. Out of ten equations CMR was found to be important in eight. All the equations satisfy statistical requirements. In data tables, Δ values in two columns are in close proximity indicating strong correlation between internal and external predictivities.
Two different types of allosterism were observed in these equations: normal and inverse, especially with parameter CMR. To interpret contribution of CMR in equations is quite challenging. This parameter includes not only volume but also polarizability of molecule. Polarizability, in turn, is directional and attractive dispersive forces, arising out of the charge transfer reaction between molecule and receptor, cannot be established until both are of opposite nature.
In normal allosterism, activity increases in a linear fashion with CMR up to the inversion point where the quadratic term takes over and it decreases with further increase in CMR, resulting in a closed parabolic graph. Perhaps bulky molecules with some polarizability interact at an opportune position in receptor site and therefore activity increases. With further increase in bulkiness beyond the inversion point, steric hinderance of molecules affect binding and activity starts decreasing. Looking at Equations (4,8,10) with normal allosteric effects, the coefficient of linear portion of CMR in Equation (10) is thrice than in Equation (4) and twice than in Equation (8), indicating contribution of CMR in activity variation is more in Equation (10). A similar CMRo of 10 (± 10%) and s (0.136, 0.134 and 0.130) in all three equations indicate similar binding mechanism. Lateral validation or comparison of QSARs thus establishes the authenticity of these three equations. In Equations (1,5), a positive linear relationship with CMR in the range 6.21-10.16 and 8.14-10.49, respectively, might show but not necessarily, normal allosterism. This could be revealed if further derivatives in higher CMR range are synthesized and tested. In Equations (2,3) although CMR is not present, still a weakly positive relationship similar to linear portion of Equation (4,8,10) is exhibite, suggestive of normal allosterism.
Interestingly, according to a belief, molecules with lower CMR than 10 (° 10%) not being active could be related to evolution in the receptor structure. Logistic modifications in structure and conformaiton of proteins made them insensitive towards unnecessary stimulation from various endogenous small ligands, which started appearing in cell cytoplasm and nucleoplasm with eons of time [
18].
In inverse allosterism, activity decreases initially up to an inversion point and then increases with further increase in CMR resulting in an open parabolic graph. A negative allosteric effect is seen in Equations (6) and (7) with different coefficients of the linear portion of CMR. The CMR ranges where these equations hold applicable are similar [7.37–11.49 for Equation (6) and 7.05–11.90 for Equation (7)]. The point of inversion is 10.8 for Equation (6) and 9.82 for Equation (7). These two equations support each other’s observation; although, the variation in activity with CMR is different in each equation.
In Equation (9), a negative linear relationship with CMR in the range 4.94-6.96 could show inverse allosterism if some more derivatives are synthesized and tested in higher CMR range.
A bilinear relationship of Kubinyi type was also tested by the
Bilin (
www.kubinyi.de) software for equations where parabolic relationship was found, however the relationship was statistically insignificant.
Maximum effect of
Ia is seen in Equations (7) and (8) with a large coefficient of 1.660 and 1.162 respectively. Since
Ia is present in Equations (7,8,9 and 10), where both allosteric types are exhibited, it is tempting to speculate, that these binding mechanisms do not depend upon presence of anionic functionalities or acidic moieties or any other high electronic density fragments as substituents in a glycine site antagonist. This speculation is supported by a study of Carling
et al. [
28], who inferred that anionic functionality is not absolutely required for good activity and glycine site antagonists with neutral and basic moieties at 4-position of 3-phenylquinolin-2(1
H)-ones performed equally well.
Our results of dual allosteric modes of binding for series
VII and
IV and
V gets support from another observation of Carling
et al. [
27] who studied difference in structure activity relationships of these three series and indicated that they do not bind in an identical manner.
Certain QSAR studies although not conducted exhaustively are noteworthy. McQuaid
et al. [
29] conducted a QSAR analysis on a few 3-phenyl substituted-4-hydroxyquinolin-2(1
H)-one compounds as glycine site antagonists (Equation 11) and observed a negative linear relationship of Hammett constant at para position σ
p with binding affinity. This indicates a positive effect of electron donating substituents at phenyl ring towards potency. A high electronic density in phenyl ring is therefore preferred.
− logKi = 0.95(0.18) – 1.26(0.41)σp
n = 6, r=0.84, F = 9.43, p = 0.04 (11)
Fabio
et al. [
30] synthesized some 3-substituted indole-2-carboxylates and conducted a QSAR study on them. The Equation (12) obtained reveals a negative linear relationship of summed MR and π values of substituents present at
o,
m and
p positions of 3-phenyl ring. A negative linear correlation was also obtained with σ
p in the same equation which reveals presence of high electronic density phenyl ring is preferred.
pKi = − 0.53MRomp – 0.39πomp – 0.82σp + 8.23
n = 25, r2 = 0.84, s = 0.28, F = 37, p < 0.0001, r2cv = 0.76 (12)
The importance of MR and σp in Equations (11,12) indicates an electronic transfer reaction between the substituted 3-phenyl ring and some site on the receptor leading to involvement of dispersive forces. This study supports our results of CMR being an important parameter in most of our equations.
It is assumed that at the inversion point the structure of a receptor is forced to change into a new shape, which results in an altogether different type of interaction. A possibility could be that there is more than one binding site, but then it shouldn’t have been binding with the same parameters defined in the first half of the Equation [
31]. The only way to confirm this allosteric interaction is by doing crystallographic studies of molecules present on both sides of inversion point.
Some outlier(s) were identified while generating equations, which were not considered in deriving the equation because of their arbitrary behaviour. In Equations (6 and 8), compounds 6 and 3 were treated as outliers, as their CMR values were too low than other derivatives in the series. The reason for anomalous behaviour of other outliers could not be attributed to a specific reason. Outlier identification in all equations was also judged by applicability domain estimation through William’s plot (HAT matrix leverage vs.. L-O-O residuals).
A good range of data points was present on both sides of the parabola (positive and negative) and 95% confidence was present on the inversion point. These two checks confirm the quality of our conclusions.
We came across some studies involving 3D-CoMFA (Comparative Molecular Field analysis) methodology [
32,
33,
34,
35,
36]. Although quite routinely reported; observations from such studies are difficult to compare with already established QSAR studies. CoMFA with its current status is semiquantitative in nature and does not qualify for quantitative SAR. Results generally are 3D pictures, which cannot be compared precisely. Moreover, the terms used to generate a regression-based model are based on principle components. These terms will have different composition from dataset to dataset so that comparison is impossible leaving pictures as the only tool to compare and conclude [
15,
18,
22,
31].
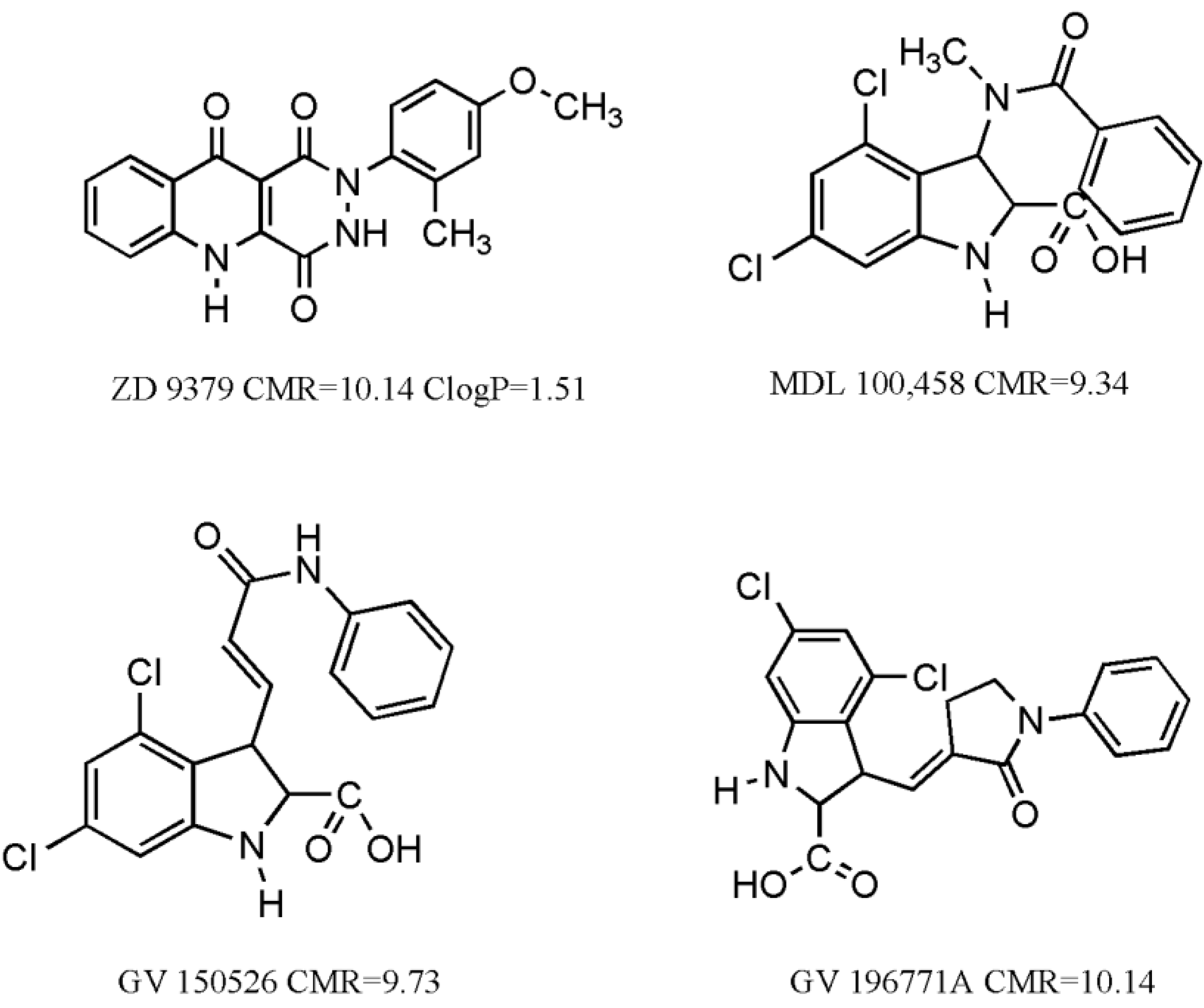
We have also considered some molecules, which have undergone clinical trials [
14] (
Figure 1). Interestingly, their CMR values are close to CMR
o (10 ± 10%). Such results indicate the quality of experimental work and validate the authenticity of equations. Clog
P value was calculated for unionic ZD 9379 at pH = 7, which is in alignment with results from Equation (2) that lipophilicity of molecules should be above 0.96 for good activity. The rest have an ionizable carboxylic moiety in their structure and therefore would ionize at physiological pH = 7.4. Clog
P values would therefore be different at two pH values. In such cases it is better to calculate log
D values, if necessary.
Allosteric interaction has been inferred in previous studies by Bender
et al. [
37] and Hansch
et al. [
38]. Their results yield a normal and inverse parabolic relationship with molecular volume and CMR, respectively. It is unknown where our results fit in the allosteric models first proposed by Monod, Wyman and Changeux [
39] and later reviewed by Changeux and Edelstein [
40]. Koshland
et al. also proposed their pioneering work on protein allosterism [
41]. These models depict changes in protein/receptor containing subunits. In drug discovery, allosteric interaction could be very advantageous in designing newer more efficacious molecules [
42].
Figure 1.
Some molecules having undergone clinical trials for treatment of stroke and/or neuropathic pain.
Figure 1.
Some molecules having undergone clinical trials for treatment of stroke and/or neuropathic pain.


{kind=link}