The Soluble Epoxide Hydrolase Inhibitor AR9281 Decreases Blood Pressure, Ameliorates Renal Injury and Improves Vascular Function in Hypertension

Abstract
:1. Introduction
2. Results and Discussion
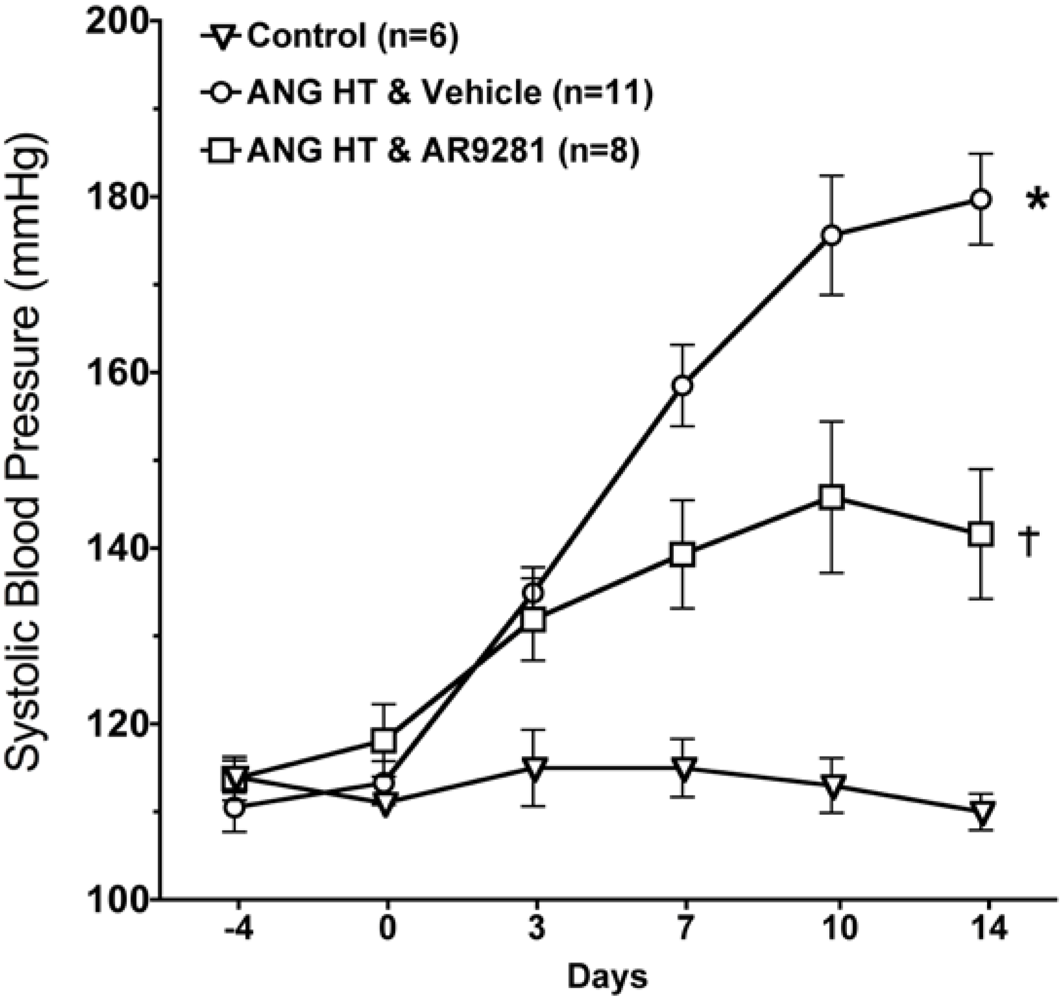
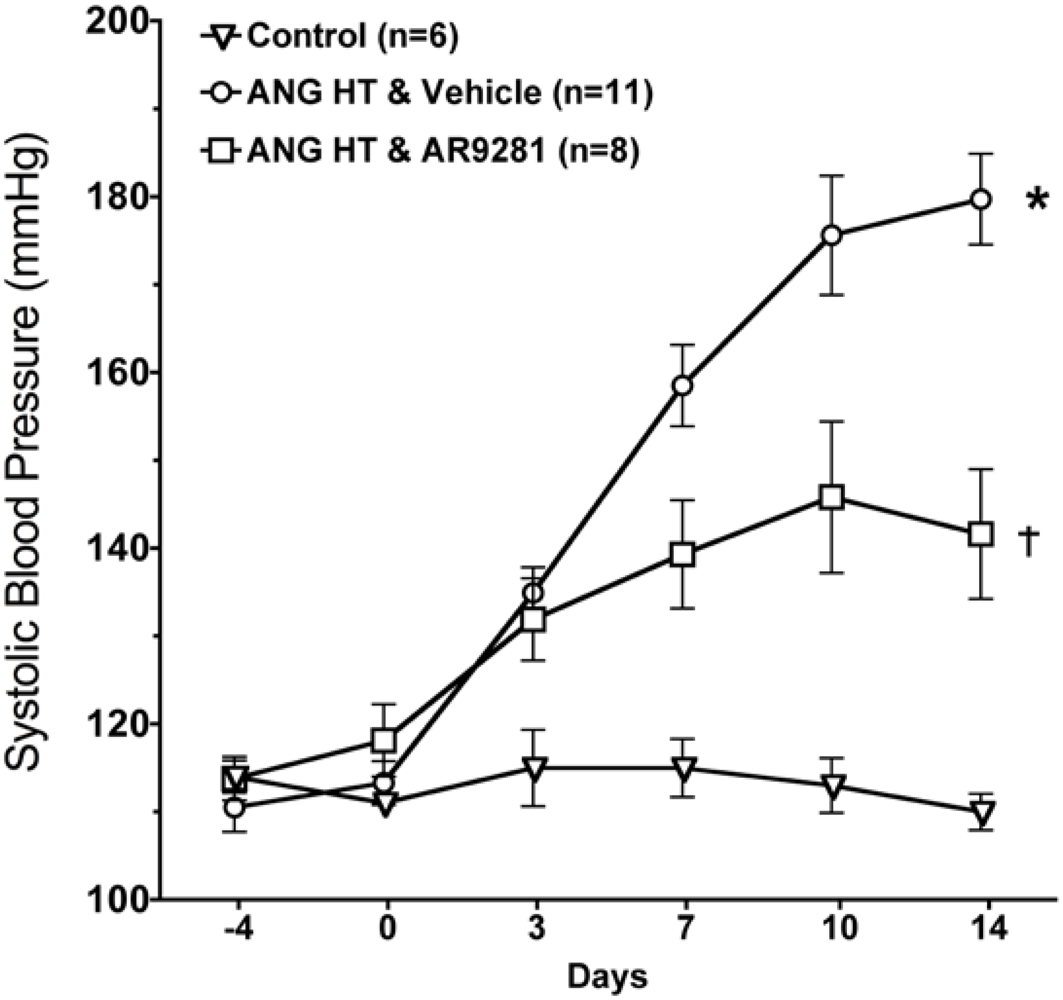
2.1. Blood Pressure
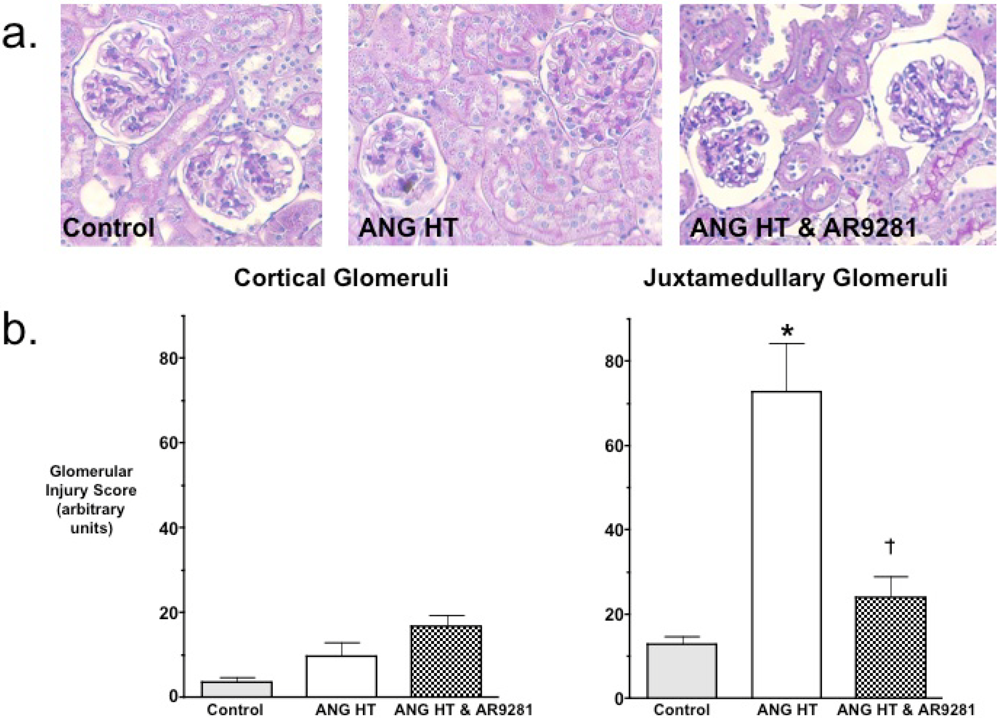
2.2. Glomerular Injury
2.3. Renal Inflammation

{kind=link}
{kind=link}
{kind=link}
 |
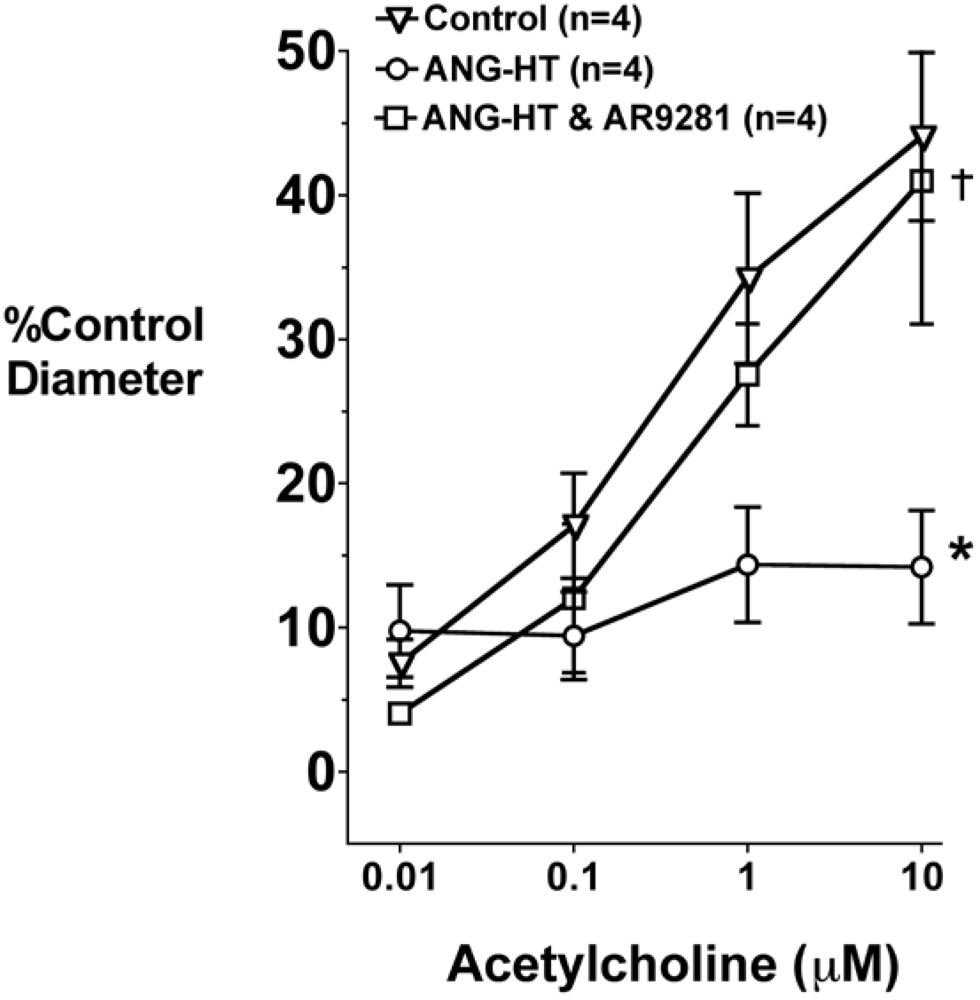
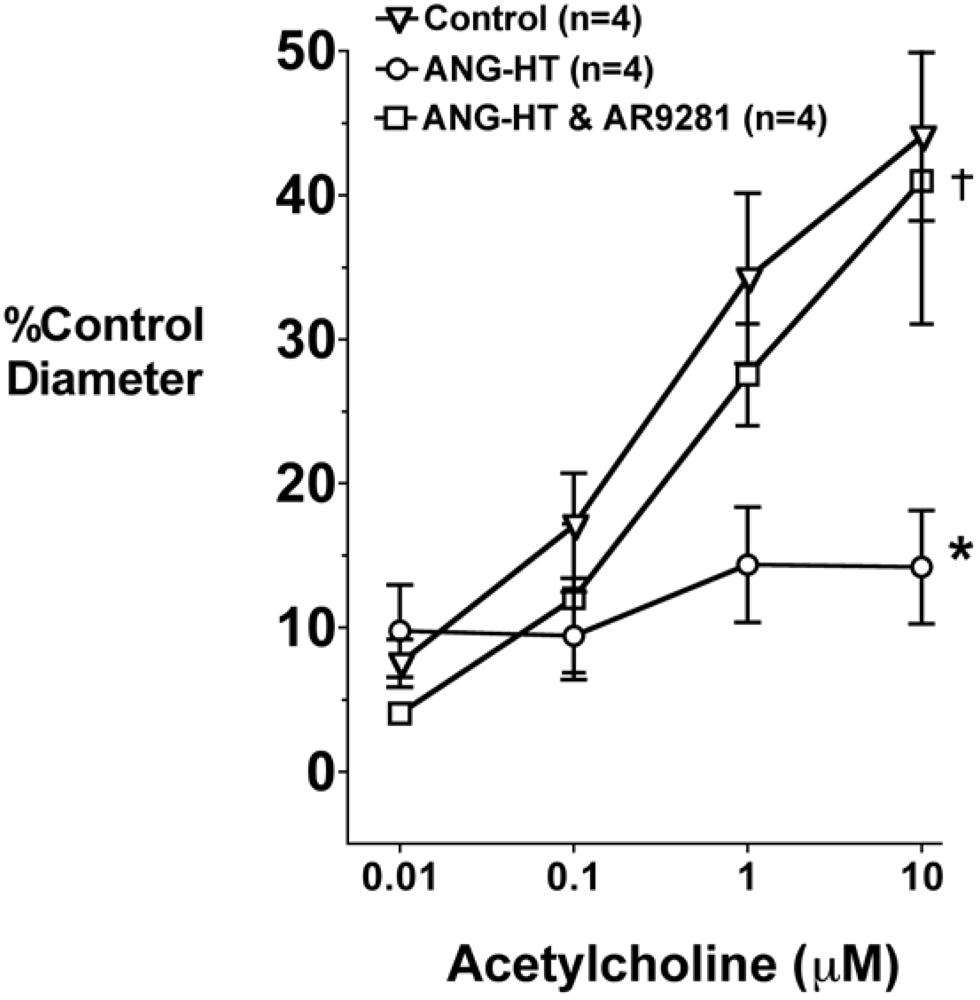
2.4. Renal and Mesenteric Vascular Function
3. Experimental Section
3.1. Animals
3.2. Evaluation of Glomerular Injury
3.3. Real-Time Polymerase Chain Reaction (PCR) Array Gene Expression Profiling
3.4. In Vitro Perfused Juxtamedullary Nephron Experiments
3.5. Mesenteric Resistance Artery Diameter responses
3.6. Statistics
4. Conclusions
Acknowledgements
References and Notes
- Spector, A.A. Arachidonic acid cytochrome P450 epoxygenase pathway. J. Lipid Res. 2009, 50, S52–S56. [Google Scholar]
- Newman, J.W.; Morisseau, C.; Hammock, B.D. Epoxide hydrolases: their roles and interactions with lipid metabolism. Prog. Lipid Res. 2005, 44, 1–51. [Google Scholar]
- Marino, J.P., Jr. Soluble epoxide hydrolase, a target with multiple opportunities for cardiovascular drug discovery. Curr. Top. Med. Chem. 2009, 9, 452–463. [Google Scholar]
- Imig, J.D. Cardiovascular therapeutic aspects of soluble epoxide hydrolase inhibitors. Cardiovasc. Drug Rev. 2006, 24, 169–188. [Google Scholar]
- Loch, D.; Hoey, A.; Morisseau, C.; Hammock, B.D.; Brown, L. Prevention of hypertension in DOCA-salt rats by an inhibitor of soluble epoxide hydrolase. Cell Biochem. Biophys. 2007, 47, 87–98. [Google Scholar]
- Jung, O.; Brandes, R.P.; Kim, I.H.; Schweda, F.; Schmidt, R.; Hammock, B.D.; Busse, R.; Fleming, I. Soluble epoxide hydrolase is a main effector of angiotensin II-induced hypertension. Hypertension 2005, 45, 759–765. [Google Scholar]
- Zhao, X.; Yamamoto, T.; Newman, J.W.; Kim, I.H.; Watanabe, T.; Hammock, B.D.; Stewart, J.; Pollock, J.S.; Pollock, D.M.; Imig, J.D. Soluble epoxide hydrolase inhibition protects the kidney from hypertension-induced damage. J. Am. Soc. Nephrol. 2004, 15, 1244–1253. [Google Scholar]
- Ghosh, S.; Chiang, P.C.; Wahlstrom, J.L.; Fujiwara, H.; Selbo, J.G.; Roberds, S.L. Oral delivery of 1,3-dicyclohexylurea nanosuspension enhances exposure and lowers blood pressure in hypertensive rats. Basic Clin. Pharmacol. Toxicol. 2008, 102, 453–458. [Google Scholar]
- Imig, J.D.; Zhao, X.; Zaharis, C.Z.; Olearczyk, J.J.; Pollock, D.M.; Newman, J.W.; Kim, I.H.; Watanabe, T.; Hammock, B.D. An orally active epoxide hydrolase inhibitor lowers blood pressure and provides renal protection in salt-sensitive hypertension. Hypertension 2005, 46, 975–981. [Google Scholar]
- Schmelzer, K.R.; Kubala, L.; Newman, J.W.; Kim, I.H.; Eiserich, J.P.; Hammock, B.D. Soluble epoxide hydrolase is a therapeutic target for acute inflammation. Proc. Natl. Acad. Sci. USA 2005, 102, 9772–9777. [Google Scholar]
- Inceoglu, B.; Jinks, S.L.; Schmelzer, K.R.; Waite, T.; Kim, I.H.; Hammock, B.D. Inhibition of soluble epoxide hydrolase reduces LPS-induced thermal hyperalgesia and mechanical allodynia in a rat model of inflammatory pain. Life Sci. 2006, 79, 2311–2319. [Google Scholar]
- Olearczyk, J.J.; Quigley, J.E.; Mitchell, B.C.; Yamamoto, T.; Kim, I.H.; Newman, J.W.; Luria, A.; Hammock, B.D.; Imig, J.D. Administration of a substituted adamantyl urea inhibitor of soluble epoxide hydrolase protects the kidney from damage in hypertensive Goto-Kakizaki rats. Clin. Sci. (Lond) 2009, 116, 61–70. [Google Scholar] [PubMed]
- Simpkins, A.N.; Rudic, R.D.; Schreihofer, D.A.; Roy, S.; Manhiani, M.; Tsai, H.J.; Hammock, B.D.; Imig, J.D. Soluble epoxide inhibition is protective against cerebral ischemia via vascular and neural protection. Am. J. Pathol. 2009, 174, 2086–2095. [Google Scholar]
- Manhiani, M.; Quigley, J.E.; Knight, S.F.; Tasoobshirazi, S.; Moore, T.; Brands, M.W.; Hammock, B.D.; Imig, J.D. Soluble epoxide hydrolase gene deletion attenuates renal injury and inflammation with DOCA-salt hypertension. Am. J. Physiol. Renal Physiol. 2009, 297, F740–F748. [Google Scholar]
- Yu, Z.; Xu, F.; Huse, L.M.; Morisseau, C.; Draper, A.J.; Newman, J.W.; Parker, C.; Graham, L.; Engler, M.M.; Hammock, B.D.; Zeldin, D.C.; Kroetz, D.L. Soluble epoxide hydrolase regulates hydrolysis of vasoactive epoxyeicosatrienoic acids. Circ. Res. 2000, 87, 992–998. [Google Scholar]
- Hercule, H.C.; Schunck, W.H.; Gross, V.; Seringer, J.; Leung, F.P.; Weldon, S.M.; da Costa Goncalves, A.; Huang, Y.; Luft, F.C.; Gollasch, M. Interaction between P450 eicosanoids and nitric oxide in the control of arterial tone in mice. Arterioscler. Thromb. Vasc. Biol. 2009, 29, 54–60. [Google Scholar]
- Shen, H.C.; Ding, F.X.; Wang, S.; Xu, S.; Chen, H.S.; Tong, X.; Mitra, K.; Kumar, S.; Zhang, X.; Chen, Y.; Zhou, G.; Pai, L.Y.; Alonso-Galicia, M.; Chen, X.; Zhang, B.; Tata, J.R.; Berger, J.P.; Colletti, S.L. Discovery of spirocylic secondary amine-derived tertiary ureas as highly potent, selective and bioavailable soluble epoxide hydrolase inhibitors. Bioorg. Med. Chem. Lett. 2009, 19, 3398–3404. [Google Scholar] [PubMed]
- Mori, T.; Cowley, A.W., Jr. Role of pressure in angiotensin II-induced renal injury: chronic servo-control of renal perfusion pressure in rats. Hypertension 2004, 43, 752–759. [Google Scholar]
- Parrish, A.R.; Chen, G.; Burghardt, R.C.; Watanabe, T.; Morisseau, C.; Hammock, B.D. Attenuation of cisplatin nephrotoxicity by inhibition of soluble epoxide hydrolase. Cell Biol. Toxicol. 2008, 25, 217–225. [Google Scholar]
- Motoki, A.; Merkel, M.J.; Packwood, W.H.; Cao, Z.; Liu, L.; Iliff, J.; Alkayed, N.J.; Van Winkle, D.M. Soluble epoxide hydrolase inhibition and gene deletion are protective against myocardial ischemia-reperfusion injury in vivo. Am. J. Physiol. Heart Circ. Physiol. 2008, 295, H2128–H2134. [Google Scholar]
- Xu, D.; Li, N.; He, Y.; Timofeyev, V.; Lu, L.; Tsai, H.J.; Kim, I.H.; Tuteja, D.; Mateo, R.K.; Singapuri, A.; Davis, B.B.; Low, R.; Hammock, B.D.; Chiamvimonvat, N. Prevention and reversal of cardiac hypertrophy by soluble epoxide hydrolase inhibitors. Proc. Natl. Acad. Sci. USA 2006, 103, 18733–18738. [Google Scholar]
- Seubert, J.M.; Sinal, C.J.; Graves, J.; DeGraff, L.M.; Bradbury, J.A.; Lee, C.R.; Goralski, K.; Carey, M.A.; Luria, A.; Newman, J.W.; Hammock, B.D.; Falck, J.R.; Roberts, H.; Rockman, H.A.; Murphy, E.; Zeldin, D.C. Role of soluble epoxide hydrolase in postischemic recovery of heart contractile function. Circ. Res. 2006, 99, 442–450. [Google Scholar]
- Franco, M.; Martinez, F.; Rodriguez-Iturbe, B.; Johnson, R.J.; Santamaria, J.; Montoya, A.; Nepomuceno, T.; Bautista, R.; Tapia, E.; Herrera-Acosta, J. Angiotensin II, interstitial inflammation, and the pathogenesis of salt-sensitive hypertension. Am. J. Physiol. Renal Physiol. 2006, 291, F1281–F1287. [Google Scholar] [CrossRef] [PubMed]
- Zoccali, C.; Mallamaci, F.; Tripepi, G. Traditional and emerging cardiovascular risk factors in end-stage renal disease. Kidney Int. 2003, 63, S105–S110. [Google Scholar]
- Ruiz-Ortega, M.; Lorenzo, O.; Ruperez, M.; Esteban, V.; Mezzano, S.; Egido, J. Renin-angiotensin system and renal damage: emerging data on angiotensin II as a proinflammatory mediator. Contrib. Nephrol. 2001, 135, 123–137. [Google Scholar]
- Elmarakby, A.A.; Quigley, J.E.; Olearczyk, J.J.; Sridhar, A.; Cook, A.K.; Inscho, E.W.; Pollock, D.M.; Imig, J.D. Chemokine receptor 2b inhibition provides renal protection in angiotensin II - salt hypertension. Hypertension 2007, 50, 1069–1076. [Google Scholar]
- Muller, D.N.; Shagdarsuren, E.; Park, J.K.; Dechend, R.; Mervaala, E.; Hampich, F.; Fiebeler, A.; Ju, X.; Finckenberg, P.; Theuer, J.; Viedt, C.; Kreuzer, J.; Heidecke, H.; Haller, H.; Zenke, M.; Luft, F.C. Immunosuppressive treatment protects against angiotensin II-induced renal damage. Am. J. Pathol. 2002, 161, 1679–1693. [Google Scholar]
- Guzik, T.J.; Hoch, N.E.; Brown, K.A.; McCann, L.A.; Rahman, A.; Dikalov, S.; Goronzy, J.; Weyand, C.; Harrison, D.G. Role of the T cell in the genesis of angiotensin II induced hypertension and vascular dysfunction. J. Exp. Med. 2007, 204, 2449–2460. [Google Scholar]
- Hoch, N.E.; Guzik, T.J.; Chen, W.; Deans, T.; Maalouf, S.A.; Gratze, P.; Weyand, C.; Harrison, D.G. Regulation of T-cell function by endogenously produced angiotensin II. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2009, 296, R208–R216. [Google Scholar]
- Kang, K.T.; Sullivan, J.C.; Sasser, J.M.; Imig, J.D.; Pollock, J.S. Novel nitric oxide synthase--dependent mechanism of vasorelaxation in small arteries from hypertensive rats. Hypertension 2007, 49, 893–901. [Google Scholar]
- Diep, Q.N.; El Mabrouk, M.; Cohn, J.S.; Endemann, D.; Amiri, F.; Virdis, A.; Neves, M.F.; Schiffrin, E.L. Structure, endothelial function, cell growth, and inflammation in blood vessels of angiotensin II-infused rats: role of peroxisome proliferator-activated receptor-gamma. Circulation 2002, 105, 2296–2302. [Google Scholar] [PubMed]
- Schulman, I.H.; Zhou, M.S.; Raij, L. Nitric oxide, angiotensin II, and reactive oxygen species in hypertension and atherogenesis. Curr. Hypertens. Rep. 2005, 7, 61–67. [Google Scholar] [CrossRef] [PubMed]
- Goto, K.; Fujii, K.; Onaka, U.; Abe, I.; Fujishima, M. Renin-angiotensin system blockade improves endothelial dysfunction in hypertension. Hypertension 2000, 36, 575–580. [Google Scholar]
© 2009 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Imig, J.D.; Carpenter, M.A.; Shaw, S. The Soluble Epoxide Hydrolase Inhibitor AR9281 Decreases Blood Pressure, Ameliorates Renal Injury and Improves Vascular Function in Hypertension. Pharmaceuticals 2009, 2, 217-227. https://doi.org/10.3390/ph2030217
Imig JD, Carpenter MA, Shaw S. The Soluble Epoxide Hydrolase Inhibitor AR9281 Decreases Blood Pressure, Ameliorates Renal Injury and Improves Vascular Function in Hypertension. Pharmaceuticals. 2009; 2(3):217-227. https://doi.org/10.3390/ph2030217
Chicago/Turabian StyleImig, John D., Margaret A. Carpenter, and Sean Shaw. 2009. "The Soluble Epoxide Hydrolase Inhibitor AR9281 Decreases Blood Pressure, Ameliorates Renal Injury and Improves Vascular Function in Hypertension" Pharmaceuticals 2, no. 3: 217-227. https://doi.org/10.3390/ph2030217
APA StyleImig, J. D., Carpenter, M. A., & Shaw, S. (2009). The Soluble Epoxide Hydrolase Inhibitor AR9281 Decreases Blood Pressure, Ameliorates Renal Injury and Improves Vascular Function in Hypertension. Pharmaceuticals, 2(3), 217-227. https://doi.org/10.3390/ph2030217