5-aza-2′,2′-Difluoro Deoxycytidine (NUC013): A Novel Nucleoside DNA Methyl Transferase Inhibitor and Ribonucleotide Reductase Inhibitor for the Treatment of Cancer

Abstract
:1. Introduction
2. Results
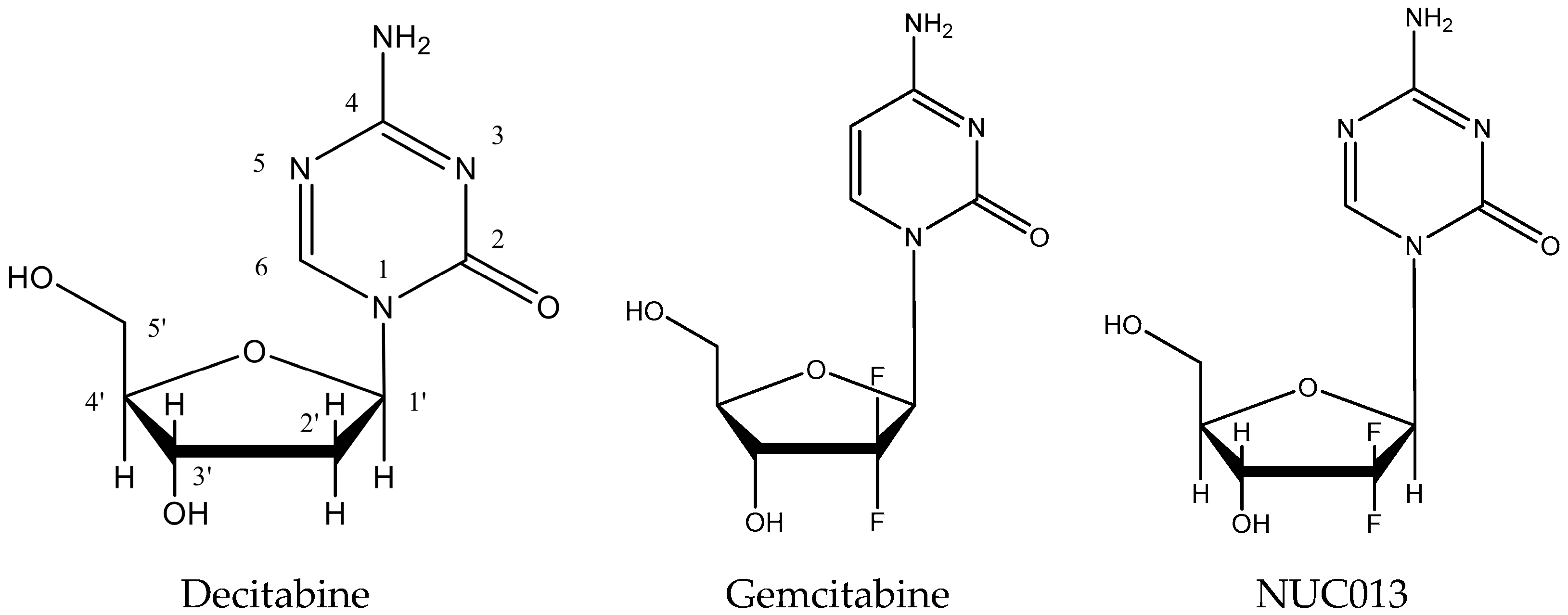
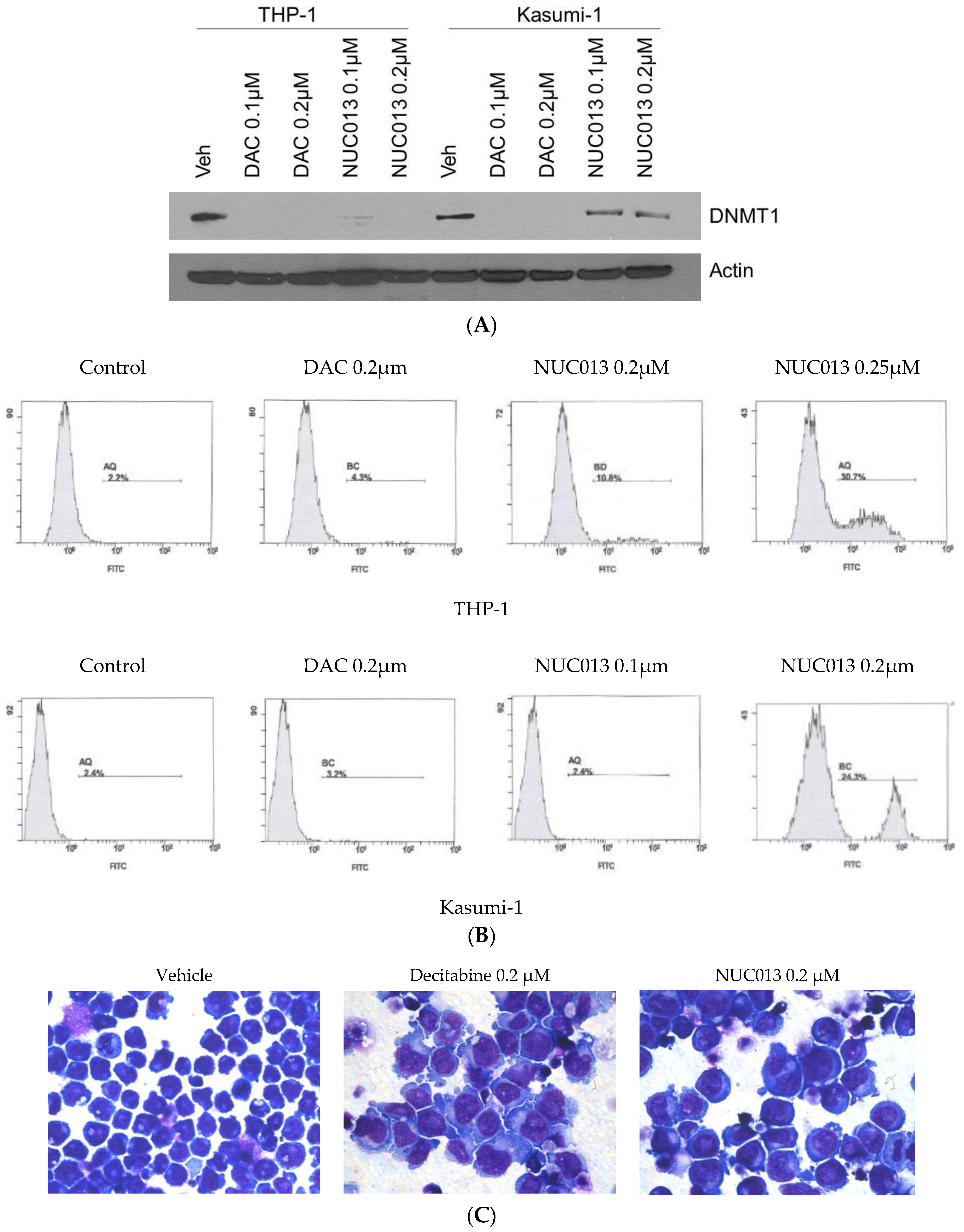
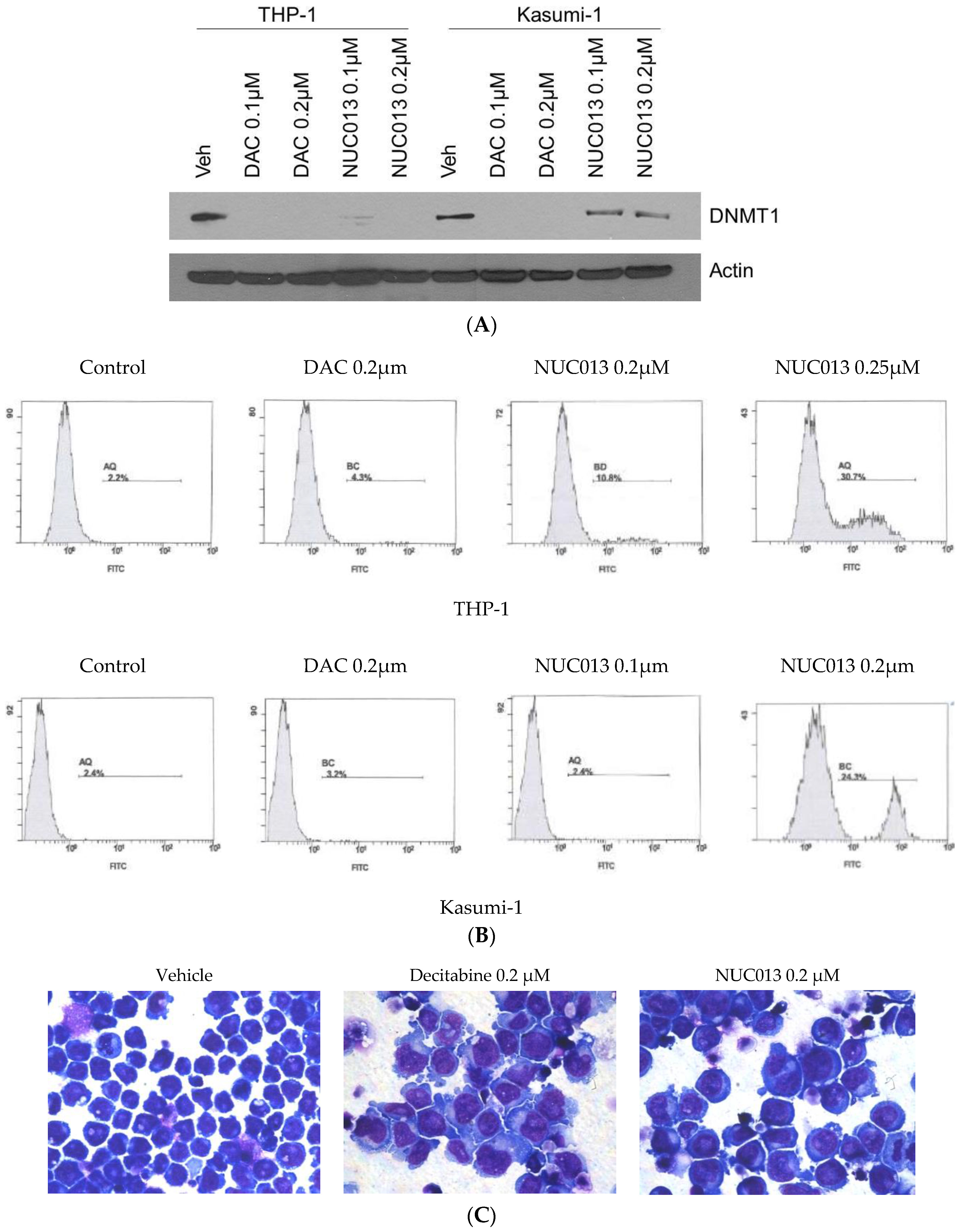
2.1. NUC013 DNMT Inhibition
2.2. NUC013 RNR Inhibition
2.3. NUC013 Cancer Cell Line Activity
2.4. NUC013 Pharmacology
2.5. NUC013 Tolerability
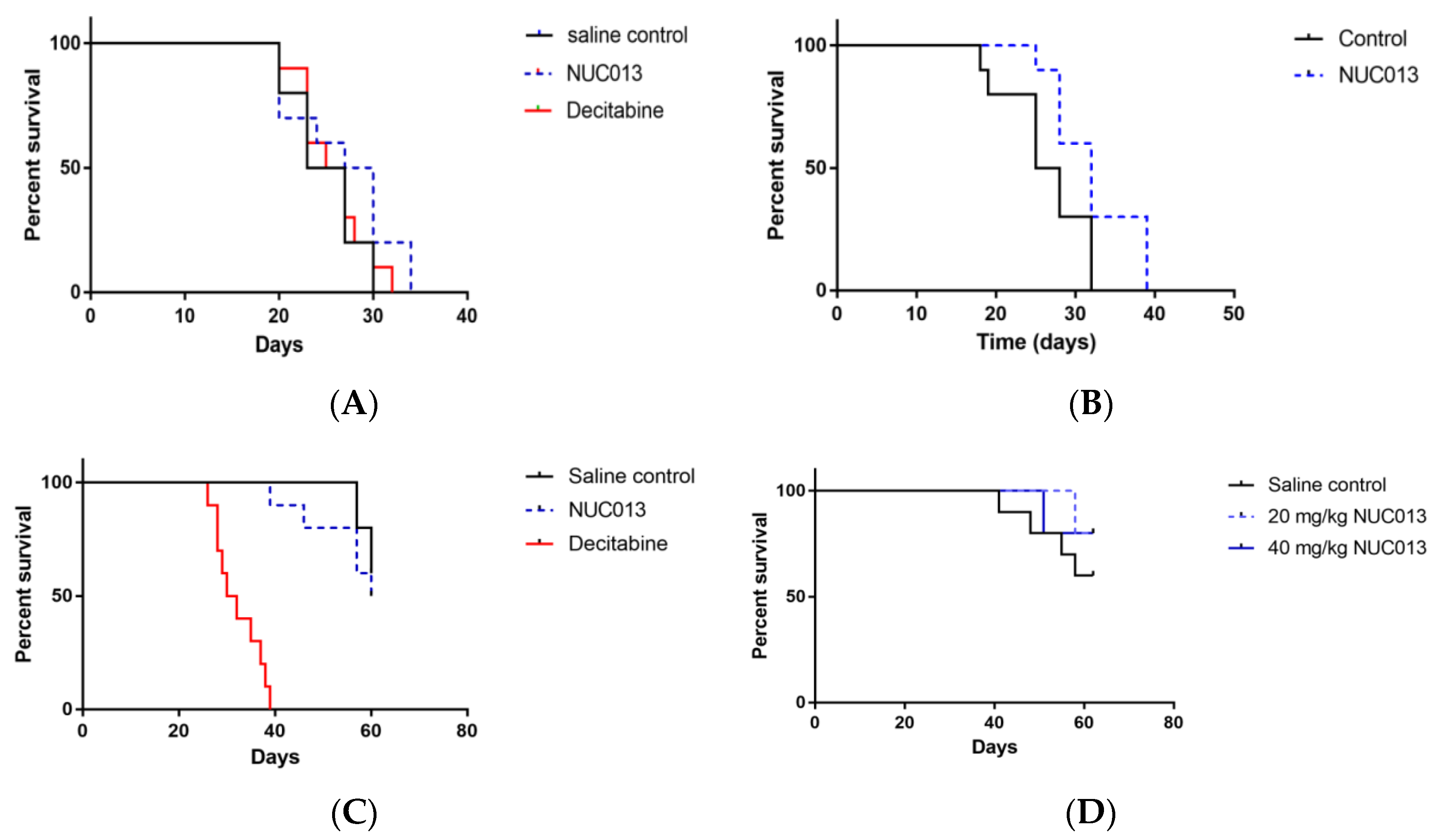
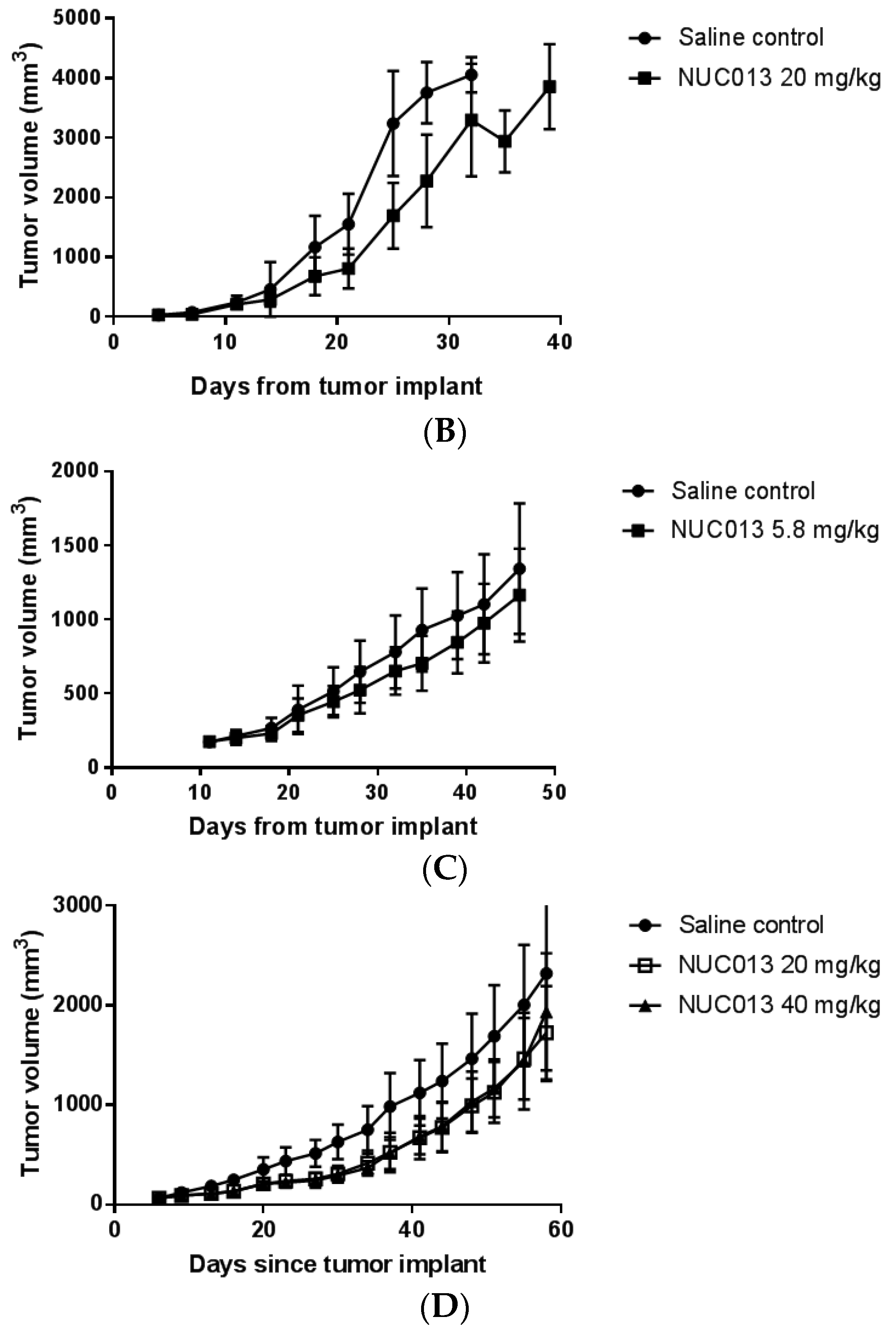
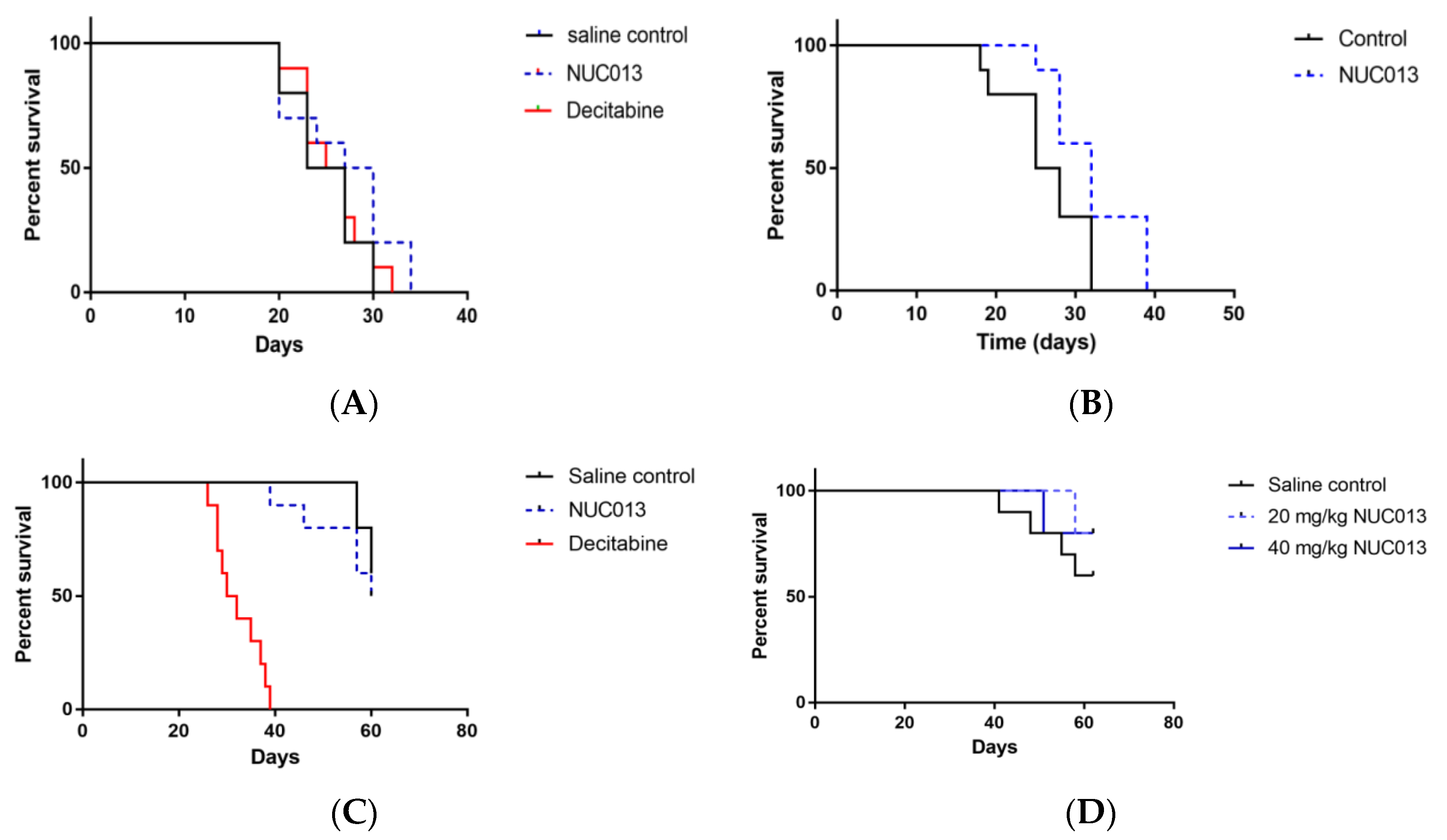
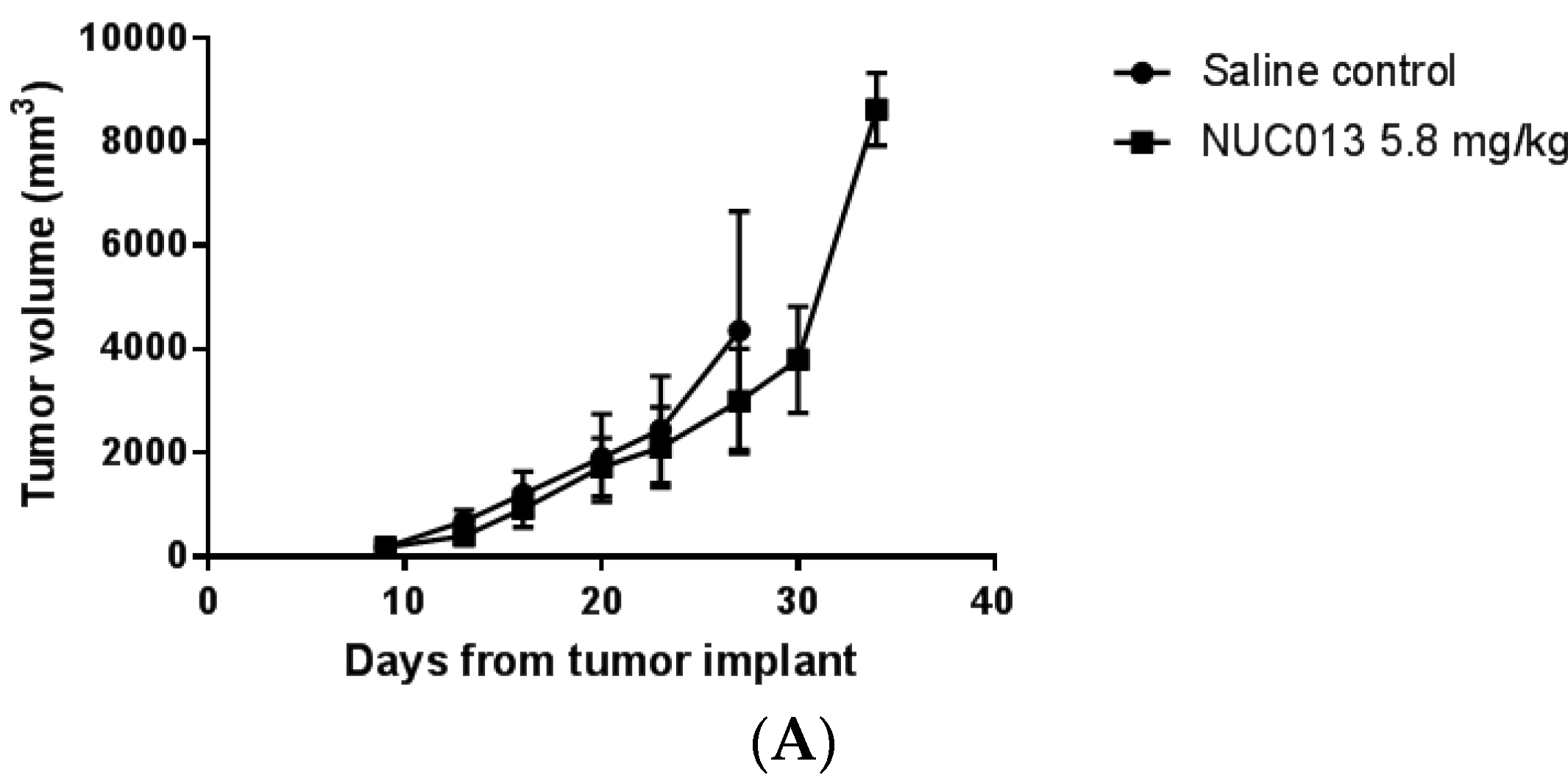
2.6. Xenograft Studies with NUC013
2.7. HL-60 Xenograft Model
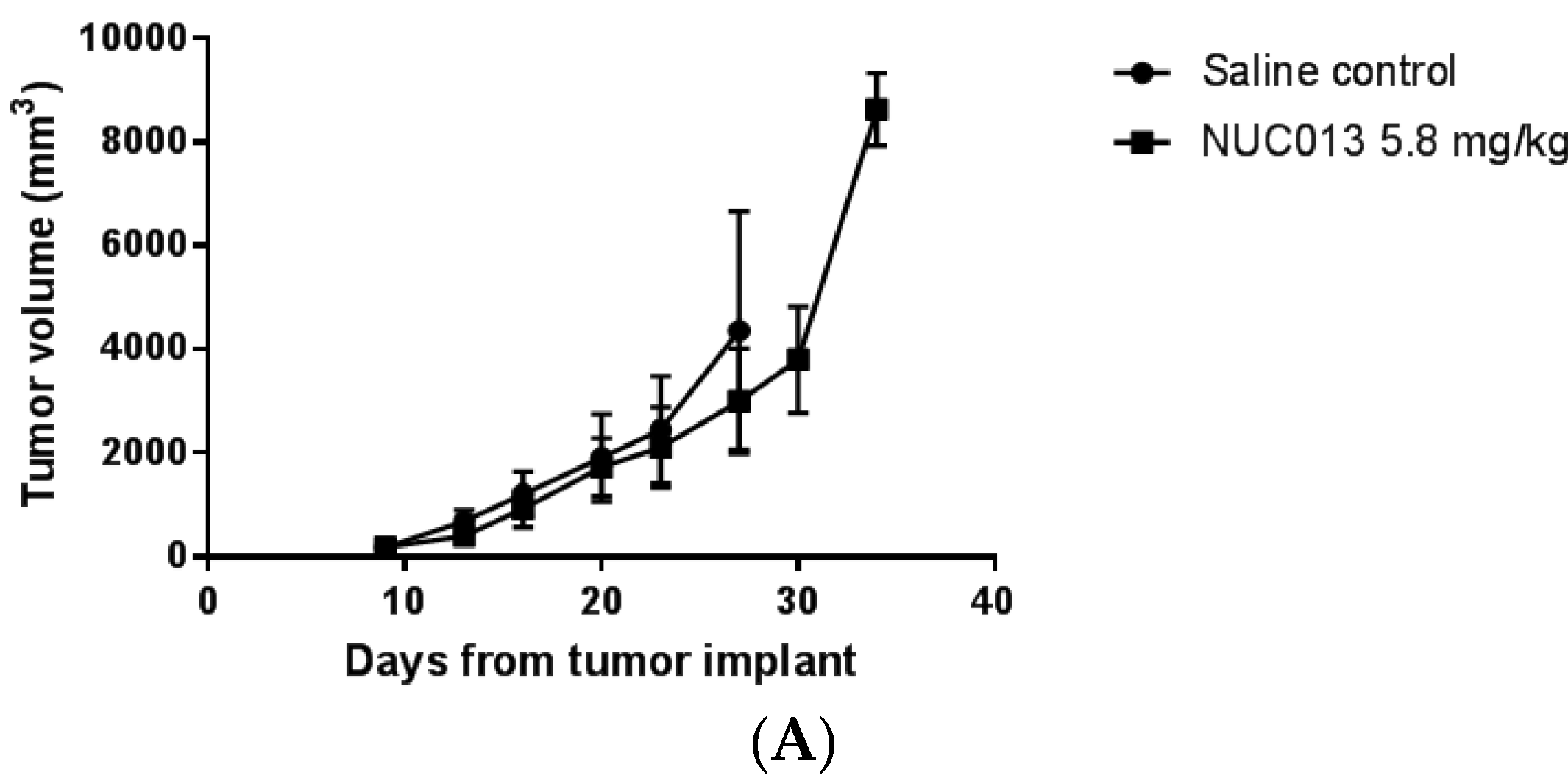
2.8. LoVo Xenograft Model
3. Discussion
4. Materials and Methods
4.1. NUC013
4.2. Total DNMT Inhibition
4.3. Western Blot for DNMT1
4.4. Apoptosis Assay
4.5. RNR Inhibition Assay
4.6. NCI 60 Cell Line Panel
4.7. In Vitro Activity
4.8. Pharmacokinetic Study
4.9. Tolerability Study
4.10. Tumor Xenograft Studies
- 100 to 250 mm3 (target group mean tumor volume of approximately 175 mm3) for the first set of studies.
- 14 to 63 mm3 (target group mean tumor weight of approximately 30 mm3) for the second set of studies.
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Kazanets, A.; Shorstova, T.; Hilmi, K.; Marques, M.; Witcher, M. Epigenetic silencing of tumor suppressor genes: Paradigms, puzzles, and potential. Biochim. Biophys. Acta 2016, 1865, 275–288. [Google Scholar] [PubMed]
- Momparler, R.L. Epigenetic therapy of cancer with 5-aza-2′-deoxycytidine (decitabine). Semin Oncol. 2005, 32, 443–451. [Google Scholar] [CrossRef] [PubMed]
- The European Medicines Agency. Dacogen (Decitabine). 2016. Available online: http://www.ema.europa.eu/ema/index.jsp?curl=pages/medicines/human/medicines/002221/human_med_001589.jsp&mid=WC0b01ac058001d124 (accessed on 16 July 2017).
- Sorm, F.; Pískala, A.; Cihák, A.; Veselý, J. 5-Azacytidine, a new, highly effective cancerostatic. Experientia 1964, 20, 202–203. [Google Scholar] [CrossRef] [PubMed]
- Pliml, J.; Sorm, F. Synthesis of 2′-deoxy-d-ribofuranosyl-5azacytosine. Collect. Czech. Chem. Commun. 1964, 29, 2576–2577. [Google Scholar] [CrossRef]
- Bouchard, J.; Momparler, R.L. Incorporation of 5-Aza-2′-deoxycytidine-5′-triphosphate into DNA. Interactions with mammalian DNA polymerase alpha and DNA methylase. Mol. Pharmacol. 1983, 24, 109–114. [Google Scholar] [PubMed]
- Cellgene Corporation. Highlights of Prescribing Information. Vidaza (Azacytidine for Injection). 2016. Available online: http://www.celgene.com/content/uploads/vidaza-pi.pdf (accessed on 16 July 2017).
- Otsuka America Pharmaceutical Inc. Highlights of Prescribing Information. Dacogen® (Decitabine) for Injection. 2014. Available online: http://www.dacogen.com/HCP/Index.aspx (accessed on 16 July 2017).
- Issa, J.-P. DNA methylation as a therapeutic target in cancer. Clin. Cancer Res. 2007, 13, 1634–1637. [Google Scholar] [CrossRef] [PubMed]
- Voytek, P.; Beisier, J.A.; Abbasi, M.M.; Wolpert-DeFilippes, M.K. Comparative studies of the cytostatic action and metabolism of 5-azacytidine and 5,6-dihydro-5-azacytidine. Cancer Res. 1977, 37, 1956–1961. [Google Scholar] [PubMed]
- Eli Lilly and Company. Highlights of Prescribing Information, Gemzar (Gemcitabine for Injection). Available online: http://pi.lilly.com/us/gemzar.pdf (accessed on 16 July 2017).
- Leroy, B.; Girard, L.; Hollestelle, A.; Minna, J.D.; Gazdar, A.F.; Soussi, T. Analysis of TP53 mutation status in human cancer cell lines: A reassessment. Hum. Mutat. 2014, 35, 756–765. [Google Scholar] [CrossRef] [PubMed]
- Christman, J.K. 5-azacytidine and 5-aza-2′-deoxycytidine as inhibitors of DNA methylation: Mechanistic studies and their implication for cancer therapy. Oncogene 2002, 21, 5483–5495. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Lohman, G.J.; Stubbe, J. Enhanced subunit interactions with gemcitabine-5′ diphosphate inhibit ribonucleotide reductases. Proc. Natl. Acad. Sci. USA 2007, 104, 14324–14329. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Lohman, G.J.S.; Stubbe, J.A. Mechanism of inactivation of human ribonucleotide reductase with p53R2 by gemcitabine 5′-diphosphate. Biochemistry 2009, 48, 11612–11621. [Google Scholar] [CrossRef] [PubMed]
- Zielinski, W.S.; Sprinzl, M. Chemical synthesis of 5-azacytidine nucleotides and preparation of tRNAs containing 5-azacytidine in its 3′-terminus. Nucleic Acids Res. 1984, 12, 5025–5036. [Google Scholar] [CrossRef] [PubMed]
- Piall, E.M.; Aherne, G.W.; Marks, V. The quantitative determination of 2′-deoxycytidine-5′ triphosphate in cell extracts by radioimmunoassay. Anal. Biochem. 1986, 154, 276–281. [Google Scholar] [CrossRef]
- Bianchi, V.; Pontis, E.; Reichard, P. Changes of deoxyribonucleoside triphosphate pools induced by hydroxyurea and their relation to DNA synthesis. J. Biol. Chem. 1986, 261, 16037–16042. [Google Scholar] [PubMed]
- Novocib. Cellular Assay for RNR Inhibitors. 2014. Available online: http://www.novocib.com/RNR_Inhibition_Whole_Cell_Screening.html (accessed on 16 July 2017).
- Chen, P.; Liu, Z.; Liu, S.; Xie, Z.; Aimiuwu, J.; Pang, J.; Klisovic, R.; Blum, W.; Grever, M.R.; Marcucci, G.; et al. A LC-MS/MS method for the analysis of intracellular nucleoside triphosphate levels. Pharm. Res. 2009, 26, 1504–1515. [Google Scholar] [CrossRef] [PubMed]
- Link, P.A.; Baer, M.R.; James, S.R.; Jones, D.A.; Karpf, A.R. p53-inducible ribonucleotide reductase (p53R2/RRM2B) is a DNA hypomethylation-independent decitabine gene target that correlates with clinical response in myelodysplastic syndrome/acute myelogenous leukemia. Cancer Res. 2008, 68, 9358–9366. [Google Scholar] [CrossRef] [PubMed]
- Yousefi, B.; Samadi, N.; Ahmadi, Y. Akt and p53R2, partners that dictate the progression and invasiveness of cancer. DNA Repair 2014, 22, 24–29. [Google Scholar] [CrossRef] [PubMed]
- Qi, J.J.; Liu, L.; Cao, J.X.; An, G.S.; Li, S.Y.; Li, G.; Jia, H.T.; Ni, J.H. E2F1 regulates p53R2 gene expression in p53-deficient cells. Mol. Cell. Biochem. 2015, 399, 179–188. [Google Scholar] [CrossRef] [PubMed]
- Matsushita, S.; Ikeda, R.; Fukushige, T.; Tajitsu, Y.; Gunshin, K.; Okumura, H.; Ushiyama, M.; Akiyama, S.; Kawai, K.; Takeda, Y.; et al. p53R2 is a prognostic factor of melanoma and regulates proliferation and chemosensitivity of melanoma cells. J. Dermatol. Sci. 2012, 68, 19–24. [Google Scholar] [CrossRef] [PubMed]
- Pontarin, G.; Ferraro, P.; Bee, L.; Reichard, P.; Bianchi, V. Mammalian ribonucleotide reductase subunit p53R2 is required for mitochondrial DNA replication and DNA repair in quiescent cells. Proc. Natl. Acad. Sci. USA 2012, 109, 13302–13307. [Google Scholar] [CrossRef] [PubMed]
- Momparler, R.L. Pharmacology of 5-Aza-2′-deoxycytidine (decitabine). Semin. Hematol. 2005, 42, S9–S16. [Google Scholar] [CrossRef] [PubMed]
- Al-Salihi, M.; Yu, M.; Burnett, D.M.; Alexander, A.; Samlowski, W.E.; Fitzpatrick, F.A. The depletion of DNA methyltransferase-1 and the epigenetic effects of 5-aza-2′deoxycytidine (decitabine) are differentially regulated by cell cycle progression. Epigenetics 2011, 6, 1021–1028. [Google Scholar] [CrossRef] [PubMed]
- Mini, E.; Nobili, S.; Caciagli, B.; Landini, I.; Mazzei, T. Cellular pharmacology of gemcitabine. Ann. Oncol. 2006, 17, v7–v12. [Google Scholar] [CrossRef] [PubMed]
- Zhu, W.G.; Hileman, T.; Ke, Y.; Wang, P.; Lu, S.; Duan, W.; Dai, Z.; Tong, T.; Villalona-Calero, M.A.; Plass, C.; et al. 5-aza-2′-deoxycytidine activates the p53/p21Waf1/Cip1 pathway to inhibit cell proliferation. J. Biol. Chem. 2004, 279, 15161–15166. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Zeng, S.X.; Lu, H. Targeting p53-MDM2-MDMX loop for cancer therapy. Subcell. Biochem. 2014, 85, 281–319. [Google Scholar] [PubMed]
- Nieto, M.; Samper, E.; Fraga, M.F.; González de Buitrago, G.; Esteller, M.; Serrano, M. The absence of p53 is critical for the induction of apoptosis by 5-aza-2′-deoxycytidine. Oncogene 2004, 23, 735–743. [Google Scholar] [CrossRef] [PubMed]
- Leonova, K.I.; Brodsky, L.; Lipchick, B.; Pal, M.; Novototskaya, L.; Chenchik, A.A.; Sen, G.C.; Komarova, E.A.; Gudkov, A.V.; et al. p53 cooperates with DNA methylation and a suicidal interferon response to maintain epigenetic silencing of repeats and noncoding RNAs. Proc. Natl. Acad. Sci. USA 2013, 110, E89–E98. [Google Scholar] [CrossRef] [PubMed]
- Yi, L.; Sun, Y.; Levine, A. Selected drugs that inhibit DNA methylation can preferentially kill p53 deficient cells. Oncotarget 2014, 5, 8924–8936. [Google Scholar] [CrossRef] [PubMed]
- Fandy, T.E.; Jiemjit, A.; Thakar, M.; Rhoden, P.; Suarez, L.; Gore, S.D. Decitabine induces delayed reactive oxygen species (ROS) accumulation in leukemia cells and induces the expression of ROS generating enzymes. Clin. Cancer Res. 2014, 20, 1249–1258. [Google Scholar] [CrossRef] [PubMed]
- Van Camp, J.A. Decitabine (Dacogen), A DNA methyltransferase inhibitor for cancer. In Modern Drug Synthesis; Li, J.J., Johnson, D.S., Eds.; Wiley: Hoboken, NJ, USA, 2010. [Google Scholar]
- Mahfouz, R.Z.; Jankowska, A.; Ebrahem, Q.; Gu, X.; Visconte, V.; Tabarroki, A.; Terse, P.; Covey, J.; Chan, K.; Ling, Y.; et al. Increased CDA expression/activity in males contributes to decreased cytidine analog half-life and likely contributes to worse outcomes with 5-azacytidine or decitabine therapy. Clin. Cancer Res. 2013, 19, 938–948. [Google Scholar] [CrossRef] [PubMed]
- Serdjebi, C.; Seitz, J.F.; Ciccolini, J.; Duluc, M.; Norguet, E.; Fina, F.; Lacarelle, B.; Ouafik, L.; Dahan, L. Rapid deaminator status is associated with poor clinical outcome in pancreatic cancer patients treated with a gemcitabine-based regimen. Pharmacogenomics 2013, 14, 1047–1051. [Google Scholar] [CrossRef] [PubMed]
- Epigentek Group Inc. EpiQuik™ DNA Methyltransferase Activity/Inhibition Assay Kit. 2014. Available online: https://www.epigentek.com/docs/P-3001.pdf (accessed on 16 July 2017).
- Novocib. RNR Inhibition—Whole Cell Assay. Available online: http://www.novocib.com/images/NovoCIB%20WCS-Nov6.pdf (accessed on 16 July 2017).
- National Cancer Institute. Developmental Therapeutics Program. NCI 60 Screening Methodology. 2015. Available online: https://dtp.cancer.gov/discovery_development/nci-60/methodology.htm (accessed on 16 July 2017).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Nucleotide | Control, T1 | Control, T2 | Gemcitabine (50 µM) | NUC013 (50 µM) |
|---|---|---|---|---|
| dATP, peak area, AU | 15.8 | 15.3 | 0 | 10.2 |
| dGDP, peak area, AU | 11.3 | 10.3 | 7.95 | 5.6 |
| dTDP, peak area, AU | 20.6 | 18.2 | 43.4 | 18.65 |
| dUTP, peak area, AU | 16.8 | 35.2 | 5.3 | 8.8 |
| ADP | 14,052 | 14,188 | 15,639 | 13,845 |
| ATP | 22,163 | 17,489 | 26,296 | 21,335 |
| GTP | 2003 | 1151 | 2036 | 2006 |
| CTP | 1209 | 848 | 1297 | 1279 |
| UTP | 4011 | 4146 | 4801 | 4070 |
| Growth Inhibition at 10 µM | Number of Cell Lines | |
|---|---|---|
| Decitabine | NUC013 | |
| >50% | 6 | 24 |
| ≤50% | 49 | 31 |
| Growth Inhibition at 10 µM | Decitabine | |
|---|---|---|
| TP53 (Null/Mutant) | TP53 (WT) | |
| >50% | 4 | 2 |
| ≤50% | 36 | 13 |
| Growth Inhibition at 10 µM | NUC013 | |
|---|---|---|
| TP53 (Null/Mutant) | TP53 (WT) | |
| >50% | 13 | 11 |
| ≤50% | 27 | 4 |
| Animal | t1/2 (h) | C0 (ng/mL) | AUClast (h·ng/mL) | AUCInf (h·ng/mL) | AUC Extr (%) | Vss (L/kg) | CL (mL/min/kg) | MRT (h) | Last time point for AUClast (h) |
|---|---|---|---|---|---|---|---|---|---|
| IV-Mouse | 0.335 | 36,941 | 11,082 | 11,086 | 0.04 | 0.26 | 9.10 | 0.292 | 4 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Daifuku, R.; Hu, Z.; Saunthararajah, Y. 5-aza-2′,2′-Difluoro Deoxycytidine (NUC013): A Novel Nucleoside DNA Methyl Transferase Inhibitor and Ribonucleotide Reductase Inhibitor for the Treatment of Cancer. Pharmaceuticals 2017, 10, 65. https://doi.org/10.3390/ph10030065
Daifuku R, Hu Z, Saunthararajah Y. 5-aza-2′,2′-Difluoro Deoxycytidine (NUC013): A Novel Nucleoside DNA Methyl Transferase Inhibitor and Ribonucleotide Reductase Inhibitor for the Treatment of Cancer. Pharmaceuticals. 2017; 10(3):65. https://doi.org/10.3390/ph10030065
Chicago/Turabian StyleDaifuku, Richard, Zhenbo Hu, and Yogen Saunthararajah. 2017. "5-aza-2′,2′-Difluoro Deoxycytidine (NUC013): A Novel Nucleoside DNA Methyl Transferase Inhibitor and Ribonucleotide Reductase Inhibitor for the Treatment of Cancer" Pharmaceuticals 10, no. 3: 65. https://doi.org/10.3390/ph10030065