An Electrochemical Detection of Metallothioneins at the Zeptomole Level in Nanolitre Volumes

Abstract
:1. Introduction
2. Experimental
2.1 Chemicals, pH measurements and pipetting
2.2 Electrochemical measurements
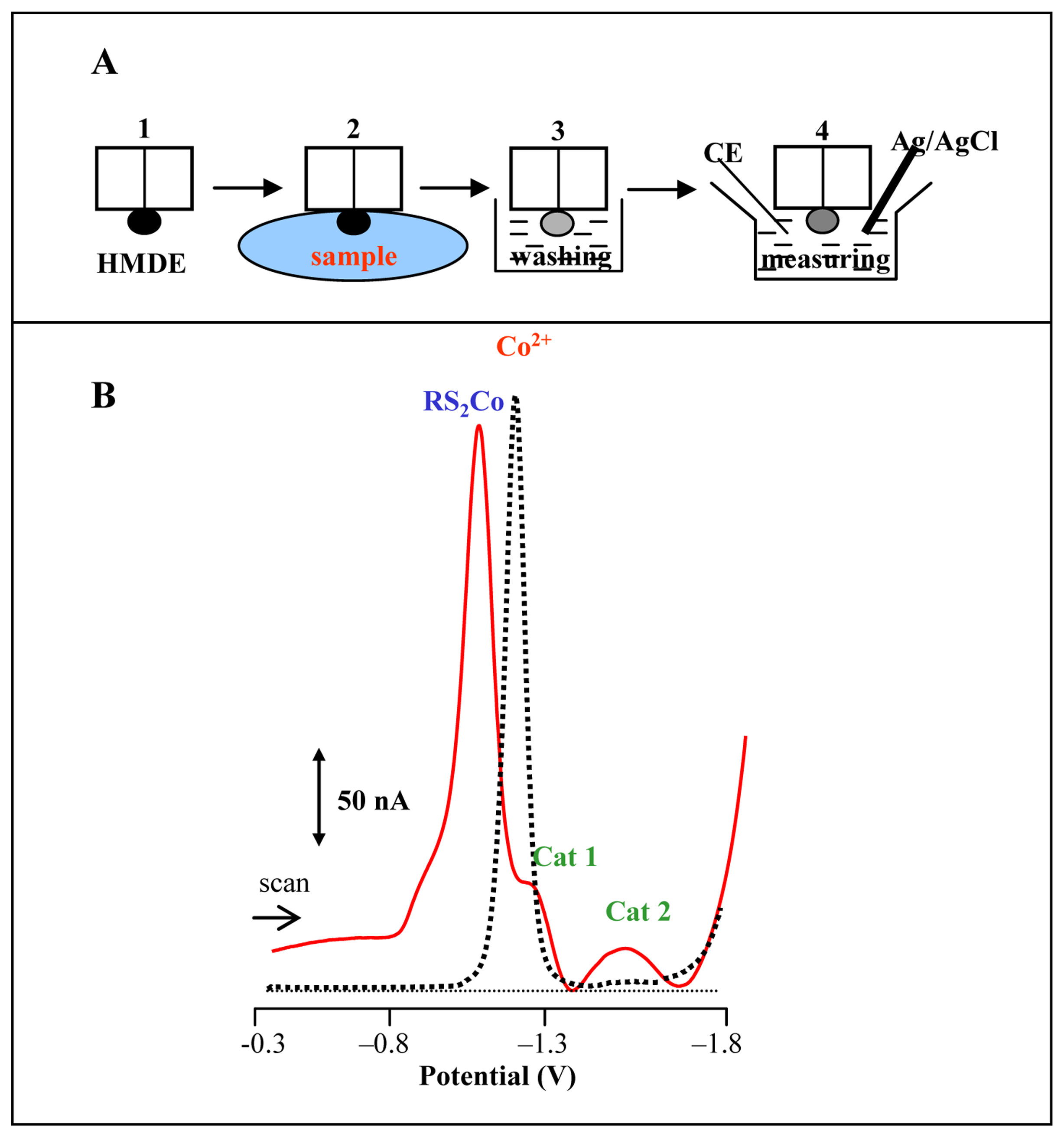
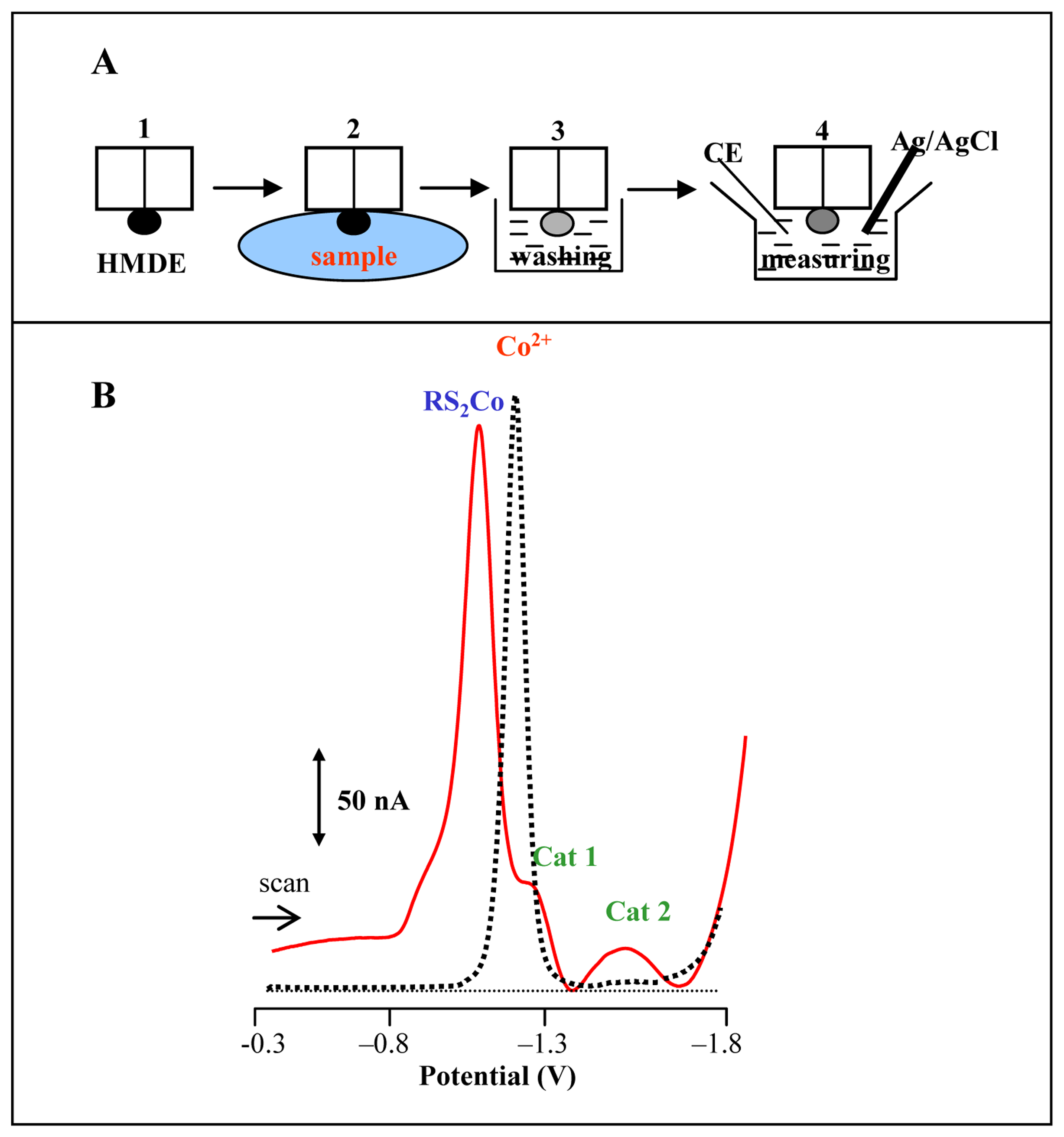
Adsorptive transfer stripping technique
Brdicka reaction of MT
2.3 Clinical material
2.4 Descriptive statistics
3. Results and Discussion
3.1 Utilizing of adsorptive transfer technique for analysis of MT
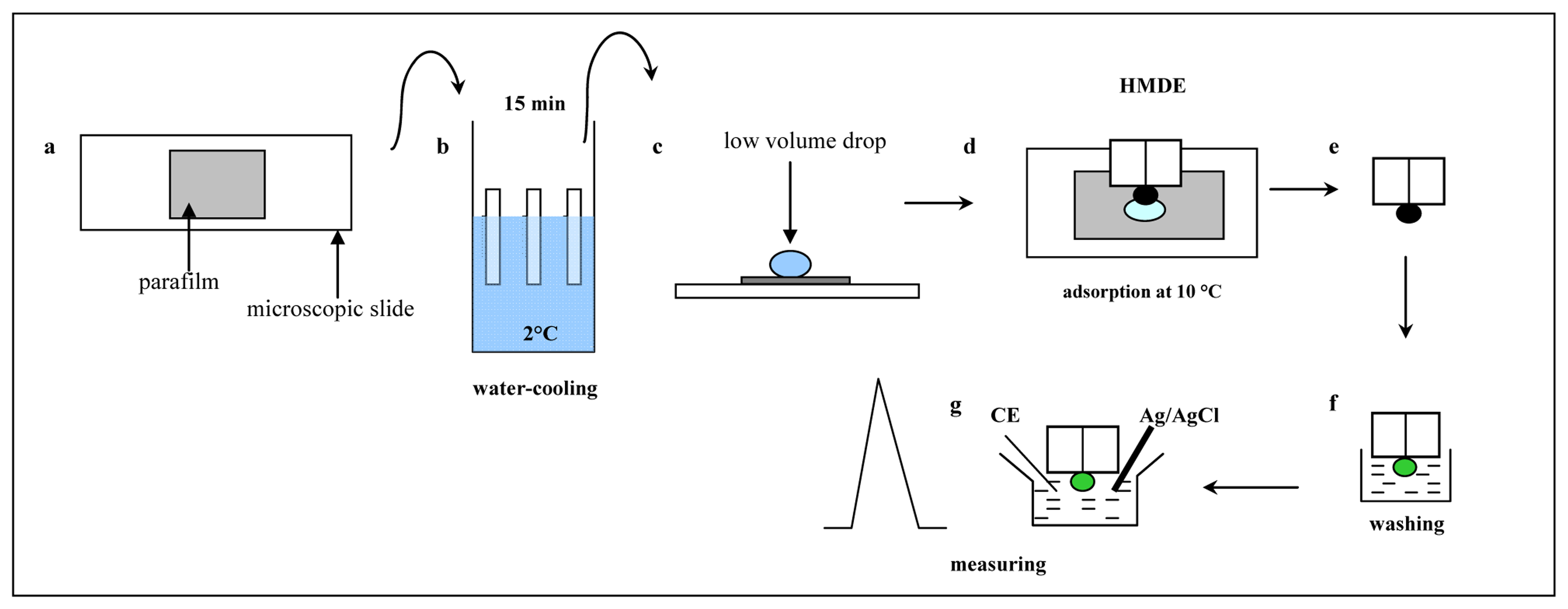
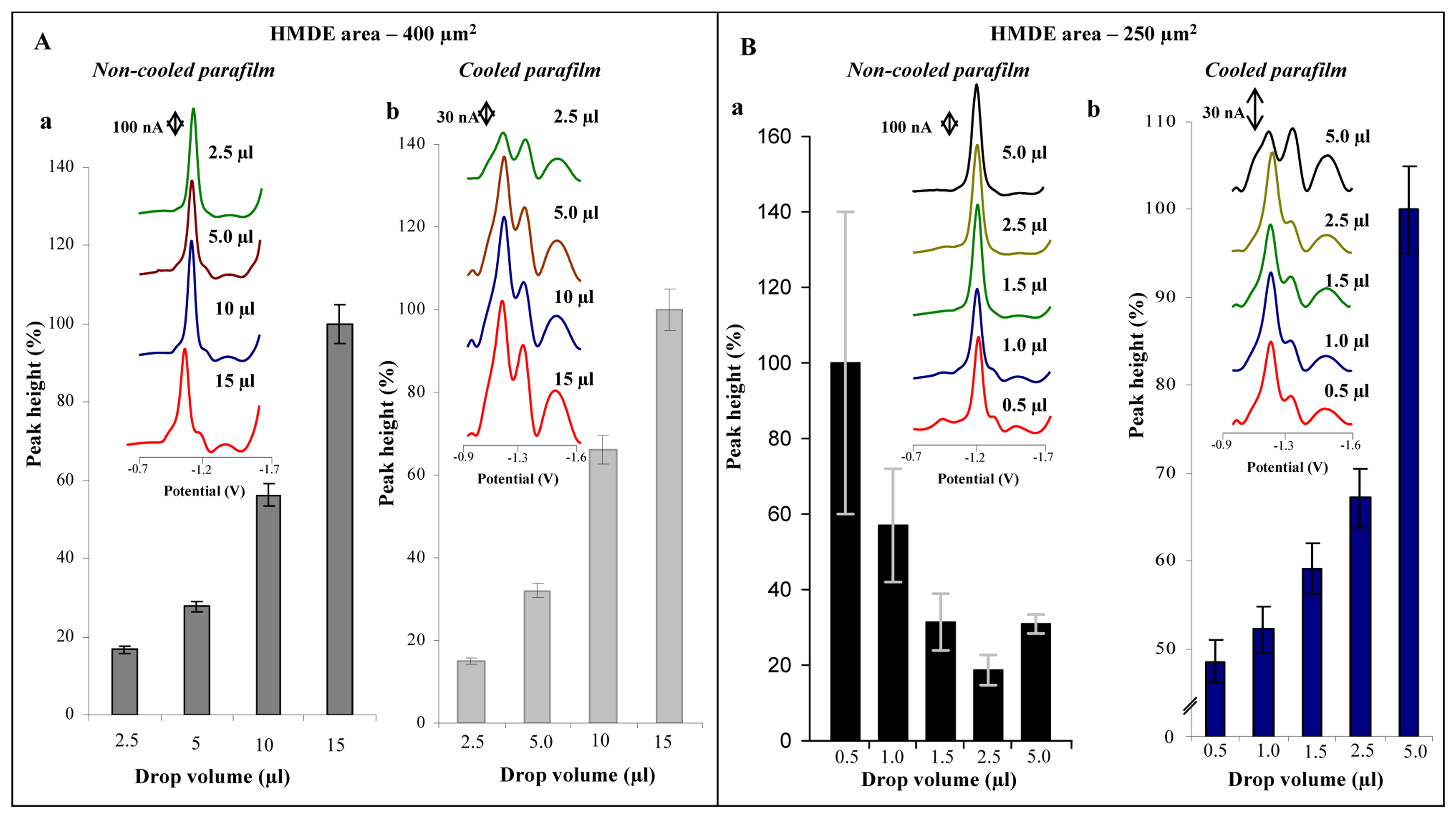
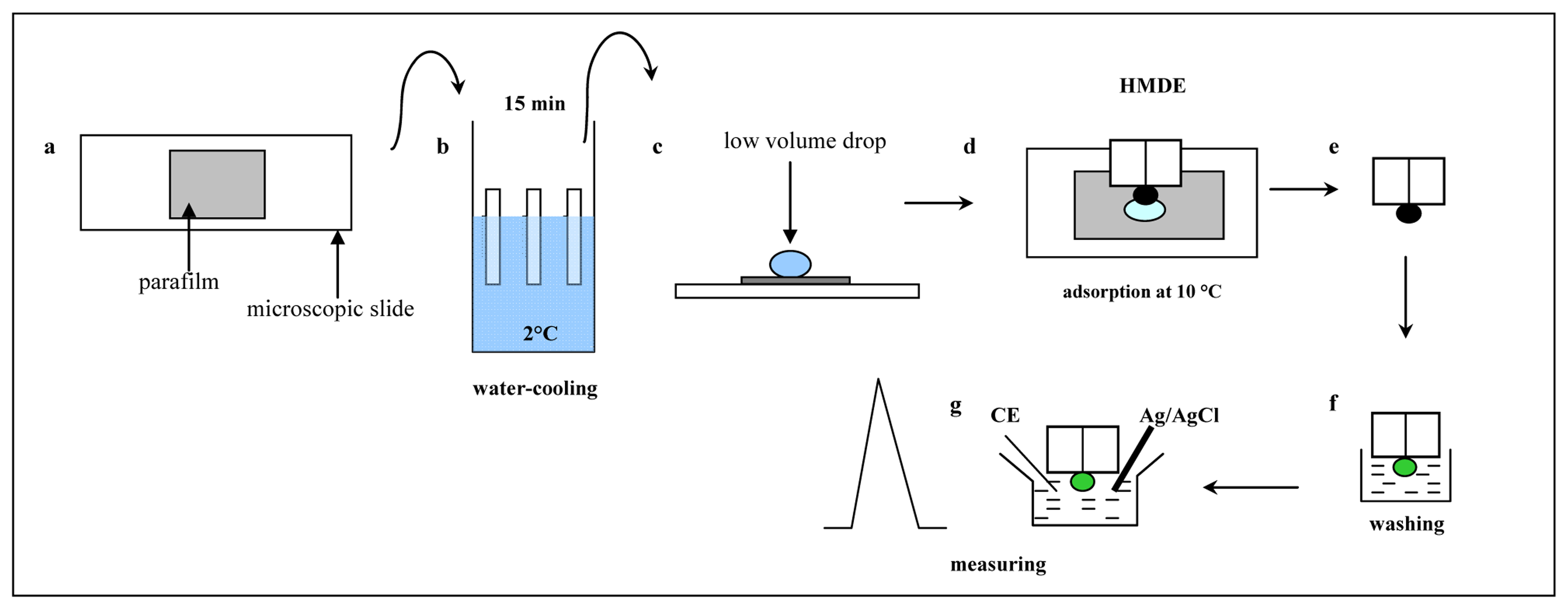
3.2 Improvement of the adsorptive transfer technique
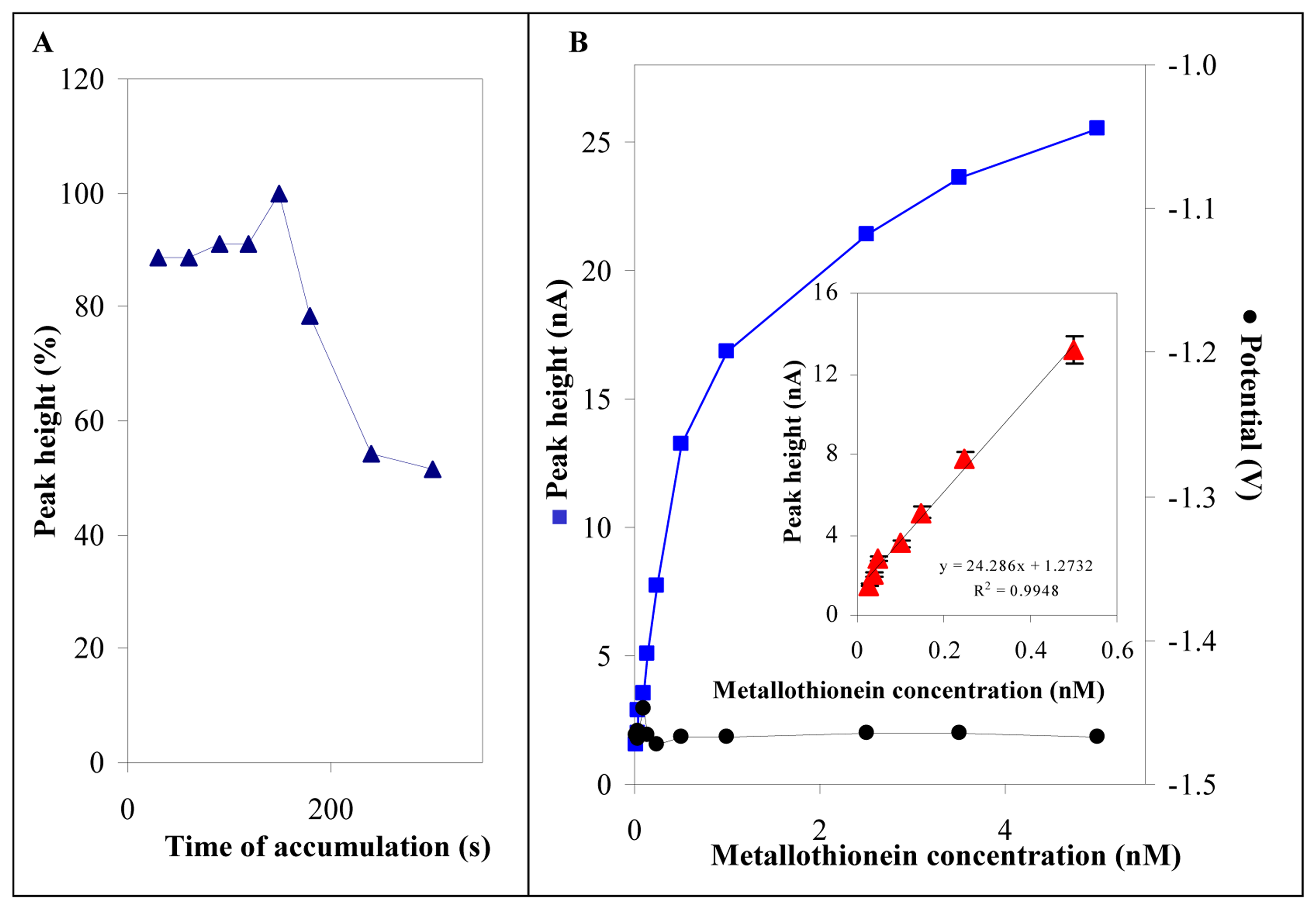
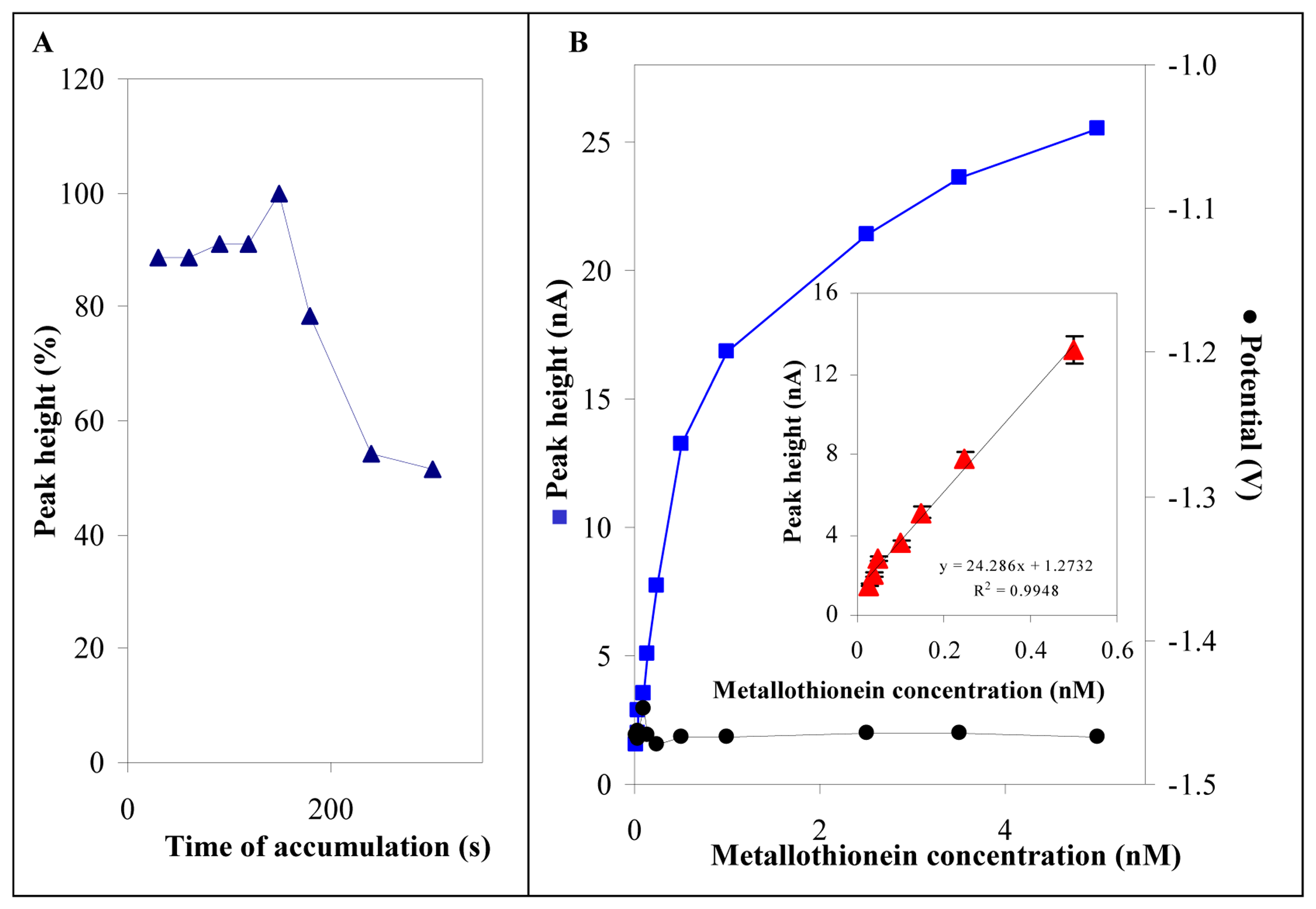
3.3 Electrochemical analysis of MT in low volume samples
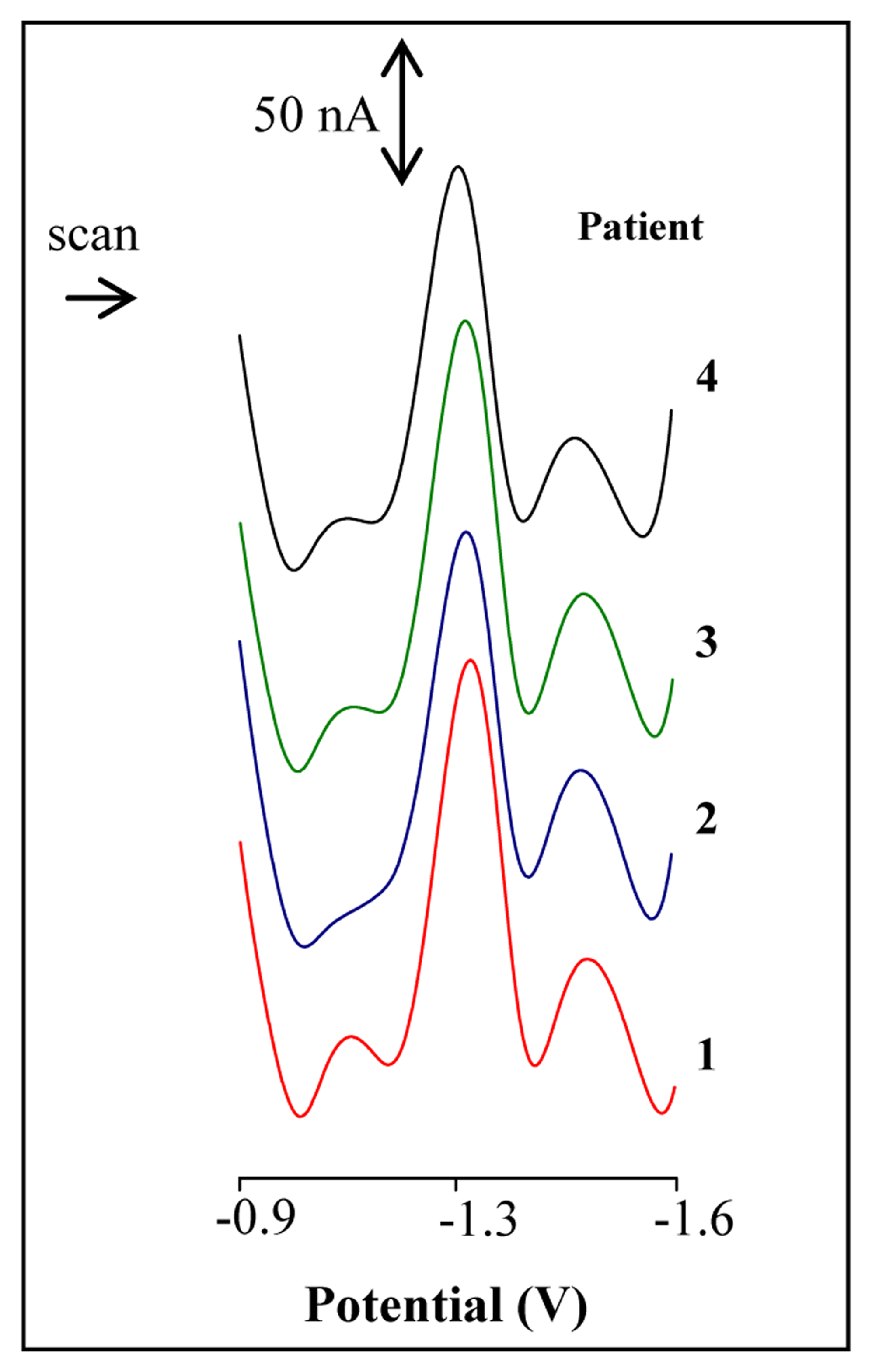
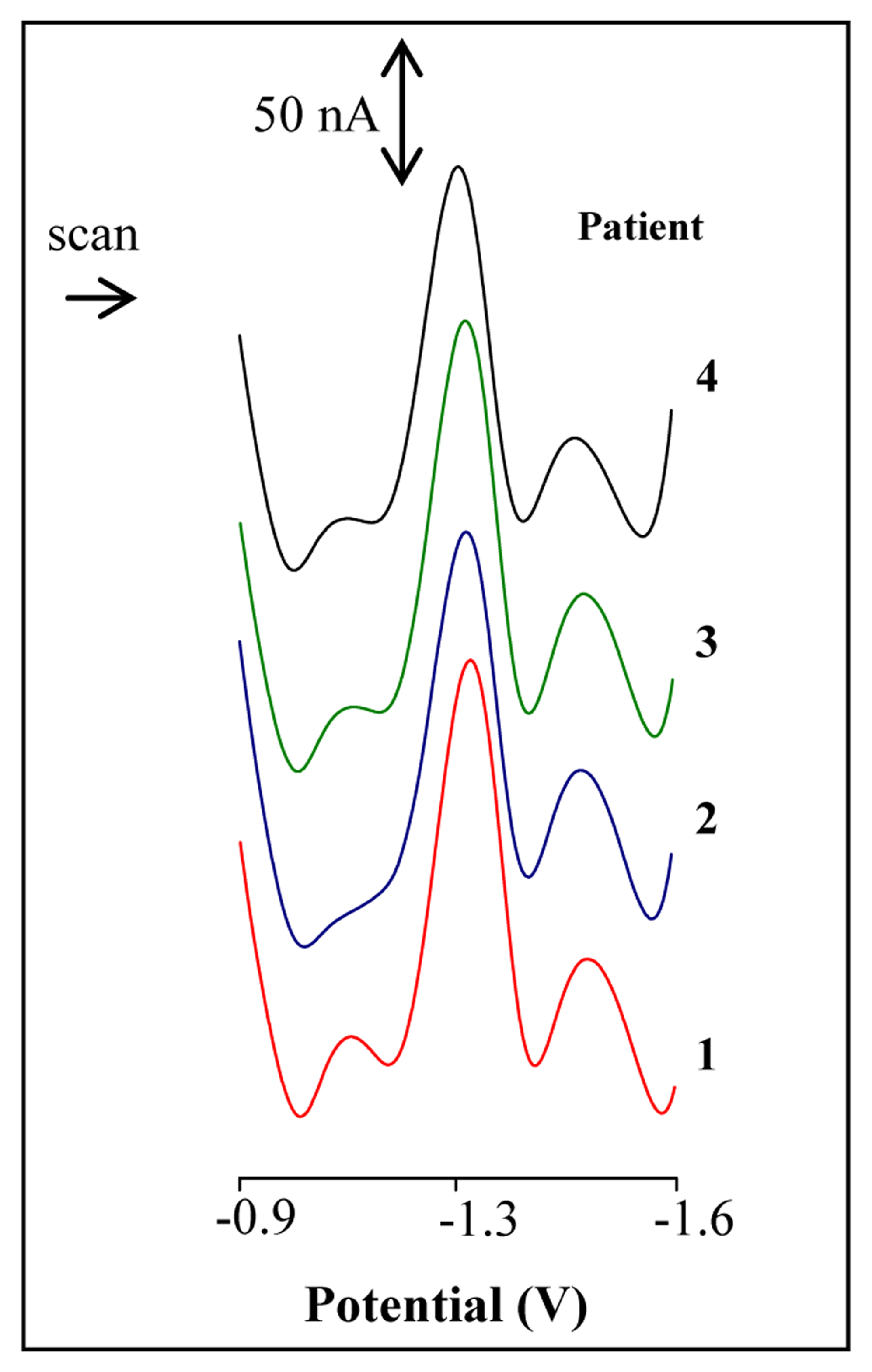
3.4 Analysis of blood serum from patients with a tumour disease
4. Conclusions
Acknowledgments
References
- Fredriksson, S.; Dixon, W.; Ji, H.; Koong, A.C.; Mindrinos, M.; Davis, R.W. Multiplexed protein detection by proximity ligation for cancer biomarker validation. Nat. Methods 2007, 4, 327–329. [Google Scholar]
- Bertolini, F.; Shaked, Y.; Mancuso, P.; Kerbel, R.S. The multifaceted circulating endothelial cell in cancer: towards marker and target identification. Nat. Rev. Cancer 2006, 6, 833–845. [Google Scholar]
- Kagi, J.H.R. Overview of Metallothionein. Method Enzymol. 1991, 205, 613–626. [Google Scholar]
- Kagi, J.H.R.; Schaffer, A. Biochemistry of Metallothionein. Biochemistry 1988, 27, 8509–8515. [Google Scholar]
- Hamer, D.H. Metallothionein. Annu. Rev. Biochem. 1986, 55, 913–951. [Google Scholar]
- Angel, P.; Karin, M. The Role of Jun, Fos and the Ap-1 Complex in Cell-Proliferation and Transformation. Biochim. Biophys. Acta 1991, 1072, 129–157. [Google Scholar]
- Lee, W.; Haslinger, A.; Karin, M.; Tjian, R. Activation of Transcription by 2 Factors That Bind Promoter and Enhancer Sequences of the Human Metallothionein Gene and Sv40. Nature 1987, 325, 368–372. [Google Scholar]
- Karin, M.; Haslinger, A.; Holtgreve, H.; Richards, R.I.; Krauter, P.; Westphal, H.M.; Beato, M. Characterization of DNA-Sequences through Which Cadmium and Glucocorticoid Hormones Induce Human Metallothionein-Iia Gene. Nature 1984, 308, 513–519. [Google Scholar]
- Palmiter, R.D.; Norstedt, G.; Gelinas, R.E.; Hammer, R.E.; Brinster, R.L. Metallothionein Human Gh Fusion Genes Stimulate Growth of Mice. Science 1983, 222, 809–814. [Google Scholar]
- Szpunar, J. Advances in analytical methodology for bioinorganic speciation analysis: metallomics, metalloproteomics and heteroatom-tagged proteomics and metabolomics. Analyst 2005, 130, 442–465. [Google Scholar]
- Wolf, C.; Schaumloffel, D.; Richarz, A.N.; Prange, A.; Bratter, P. CZE-ICP-MS separation of metallothioneins in human brain cytosols: comparability of electropherograms obtained from different sample matrices. Analyst 2003, 128, 576–580. [Google Scholar]
- Szpunar, J. Bio-inorganic speciation analysis by hyphenated techniques. Analyst 2000, 125, 963–988. [Google Scholar]
- Szpunar, J. Trace element speciation analysis of biomaterials by high-performance liquid chromatography with inductively coupled plasma mass spectrometric detection. Trac-Trends Anal. Chem. 2000, 19, 127–137. [Google Scholar]
- Ferrarello, C.N.; de la Campa, M.D.F.; Carrasco, J.F.; Sanz-Medel, A. Speciation of metallothionein-like proteins of the mussel Mytilus edulis at basal levels by chromatographic separations coupled to quadrupole and double-focusing magnetic sector ICPMS. Anal. Chem. 2000, 72, 5874–5880. [Google Scholar]
- Harrington, C.F.; Vidler, D.S.; Watts, M.J.; Hall, J.F. Potential for using isotopically altered metalloproteins in species-specific isotope dilution analysis of proteins by HPLC coupled to inductively coupled plasma mass spectrometry. Anal. Chem. 2005, 77, 4034–4041. [Google Scholar]
- Wang, Z.X.; Prange, A. Use of surface-modified capillaries in the separation and characterization of metallothionein isoforms by capillary electrophoresis inductively coupled plasma mass spectrometry. Anal. Chem. 2002, 74, 626–631. [Google Scholar]
- Kizek, R.; Trnkova, L.; Palecek, E. Determination of metallothionein at the femtomole level by constant current stripping chronopotentiometry. Anal. Chem. 2001, 73, 4801–4807. [Google Scholar]
- Luo, D.B.; Lan, J.G.; Zhou, C.; Luo, C.X. Polarographic behavior of Co(II)-BSA or -HSA complex in the presence of a guanidine modifier. Anal. Chem. 2003, 75, 6346–6350. [Google Scholar]
- Erk, M.; Ivankovic, D.; Raspor, B.; Pavicic, J. Evaluation of different purification procedures for the electrochemical quantification of mussel metallothioneins. Talanta 2002, 57, 1211–1218. [Google Scholar]
- Long, G.L.; Winefordner, J.D. Limit of Detection. Anal. Chem. 1983, 55, A712–A724. [Google Scholar]
- Adam, V.; Hanustiak, P.; Krizkova, S.; Beklova, M.; Zehnalek, J.; Trnkova, L.; Horna, A.; Sures, B.; Kizek, R. Palladium biosensor. Electroanalysis 2007, 19, 1909–1914. [Google Scholar]
- Adam, V.; Krizkova, S.; Zitka, O.; Trnkova, L.; Petrlova, J.; Beklova, M.; Kizek, R. Determination of apo-metallothionein using adsorptive transfer stripping technique in connection with differential pulse voltammetry. Electroanalysis 2007, 19, 339–347. [Google Scholar]
- Hubalek, J.; Hradecky, J.; Adam, V.; Krystofova, O.; Huska, D.; Masarik, M.; Trnkova, L.; Horna, A.; Klosova, K.; Adamek, M.; Zehnalek, J.; Kizek, R. Spectrometric and voltammetric analysis of urease - nickel nanoelectrode as an electrochemical sensor. Sensors 2007, 7, 1238–1255. [Google Scholar]
- Huska, D.; Zitka, O.; Adam, V.; Beklova, M.; Krizkova, S.; Zeman, L.; Horna, A.; Havel, L.; Zehnalek, J.; Kizek, R. A sensor for investigating the interaction between biologically important heavy metals and glutathione. Czech J. Anim. Sci. 2007, 52, 37–43. [Google Scholar]
- Krizkova, S.; Adam, V.; Petrlova, J.; Zitka, O.; Stejskal, K.; Zehnalek, J.; Sures, B.; Trnkova, L.; Beklova, M.; Kizek, R. A suggestion of electrochemical biosensor for study of platinum(II)-DNA interactions. Electroanalysis 2007, 19, 331–338. [Google Scholar]
- Petrek, J.; Havel, L.; Petrlova, J.; Adam, V.; Potesil, D.; Babula, P.; Kizek, R. Analysis of salicylic acid in willow barks and branches by an electrochemical method. Russ. J. Plant Physiol. 2007, 54, 553–558. [Google Scholar]
- Petrlova, J.; Krizkova, S.; Zitka, O.; Hubalek, J.; Prusa, R.; Adam, V.; Wang, J.; Beklova, M.; Sures, B.; Kizek, R. Utilizing a chronopotentiometric sensor technique for metallothionein determination in fish tissues and their host parasites. Sens. Actuator B-Chem. 2007, 127, 112–119. [Google Scholar]
- Petrlova, J.; Masarik, M.; Potesil, D.; Adam, V.; Trnkova, L.; Kizek, R. Zeptomole detection of streptavidin using carbon paste electrode and square-wave voltammetry. Electroanalysis 2007, 19, 1177–1182. [Google Scholar]
- Supalkova, V.; Petrek, J.; Havel, L.; Krizkova, S.; Petrlova, J.; Adam, V.; Potesil, D.; Babula, P.; Beklova, M.; Horna, A.; Kizek, R. Electrochemical sensors for detection of acetylsalicylic acid. Sensors 2006, 6, 1483–1497. [Google Scholar]
- Vitecek, J.; Petrlova, J.; Petrek, J.; Adam, V.; Potesil, D.; Havel, L.; Mikelova, R.; Trnkova, L.; Kizek, R. Electrochemical study of S-nitrosoglutathione and nitric oxide by carbon fibre NO sensor and cyclic voltammetry - possible way of monitoring of nitric oxide. Electrochim. Acta 2006, 51, 5087–5094. [Google Scholar]
- Adam, V.; Petrlova, J.; Potesil, D.; Lubal, P.; Zehnalek, J.; Sures, B.; Kizek, R. New electrochemical biosensor to determine platinum cytostatics to DNA structure. Chem. Listy 2005, 99, 353–393. [Google Scholar]
- Adam, V.; Petrlova, J.; Potesil, D.; Zehnalek, J.; Sures, B.; Trnkova, L.; Jelen, F.; Kizek, R. Study of metallothionein modified electrode surface behavior in the presence of heavy metal ions-biosensor. Electroanalysis 2005, 17, 1649–1657. [Google Scholar]
- Adam, V.; Zehnalek, J.; Petrlova, J.; Potesil, D.; Sures, B.; Trnkova, L.; Jelen, F.; Vitecek, J.; Kizek, R. Phytochelatin modified electrode surface as a sensitive heavy-metal ion biosensor. Sensors 2005, 5, 70–84. [Google Scholar]
- Petrlova, J.; Potesil, D.; Zehnalek, J.; Sures, B.; Adam, V.; Trnkova, L.; Kizek, R. Cisplatin electrochemical biosensor. Electrochim. Acta 2006, 51, 5169–5173. [Google Scholar]
- Potesil, D.; Mikelova, R.; Adam, V.; Kizek, R.; Prusa, R. Change of the protein p53 electrochemical signal according to its structural form - Quick and sensitive distinguishing of native, denatured, and aggregated form of the “guardian of the genome”. Protein J. 2006, 25, 23–32. [Google Scholar]
- Trnkova, L.; Jelen, F.; Petrlova, J.; Adam, V.; Potesil, D.; Kizek, R. Elimination voltammetry with linear scan as a new detection method for DNA sensors. Sensors 2005, 5, 448–464. [Google Scholar]
- Li, L.; Boedicker, J.Q.; Ismagilov, R.F. Using a multijunction microfluidic device to inject substrate into an array of preformed plugs without cross-contamination: Comparing theory and experiments. Anal. Chem. 2007, 79, 2756–2761. [Google Scholar]
- Henares, T.G.; Takaishi, M.; Yoshida, N.; Terabe, S.; Mizutani, F.; Sekizawa, R.; Hisamoto, H. Integration of multianalyte sensing functions on a capillary-assembled microchip: Simultaneous determination of ion concentrations and enzymatic activities by a “drop-and-sip” technique. Anal. Chem. 2007, 79, 908–915. [Google Scholar]
- Du, W.B.; Fang, Q.; Fang, Z.L. Microfluidic sequential injection analysis in a short capillary. Anal. Chem. 2006, 78, 6404–6410. [Google Scholar]
- Lorenz, R.M.; Edgar, J.S.; Jeffries, G.D.M.; Chiu, D.T. Microfluidic and optical systems for the on-demand generation and manipulation of single femtoliter-volume aqueous droplets. Anal. Chem. 2006, 78, 6433–6439. [Google Scholar]
- Legendre, L.A.; Bienvenue, J.M.; Roper, M.G.; Ferrance, J.P.; Landers, J.P. A simple, valveless microfluidic sample preparation device for extraction and amplification of DNA from nanoliter-volume samples. Anal. Chem. 2006, 78, 1444–1451. [Google Scholar]
- Adam, V.; Zitka, O.; Dolezal, P.; Zeman, L.; Horna, A.; Hubalek, J.; Sileny, J.; Krizkova, S.; Trnkova, L.; Kizek, R. Lactoferrin isolation using monolithic column coupled with spectrometric or micro-amperometric detector. Sensors 2008, 8, 464–487. [Google Scholar]
- Babula, P.; Huska, D.; Hanustiak, P.; Baloun, J.; Krizkova, S.; Adam, V.; Hubalek, J.; Havel, L.; Zemlicka, M.; Horna, A.; Beklova, M.; Kizek, R. Flow injection analysis coupled with carbon electrodes as the tool for analysis of naphthoquinones with respect to their content and functions in biological samples. Sensors 2006, 6, 1466–1482. [Google Scholar]
- Prasek, J.; Adamek, M.; Hubalek, J.; Adam, V.; Trnkova, L.; Kizek, R. New hydrodynamic electrochemical arrangement for cadmium ions detection using thick-film chemical sensor electrodes. Sensors 2006, 6, 1498–1512. [Google Scholar]
- Zitka, O.; Horna, A.; Stejskal, K.; Zehnalek, J.; Adam, V.; Havel, L.; Zeman, L.; Kizek, R. Study of structural changes of lactoferrin using flow injection analysis with electrochemical detection on glassy carbon electrode. Acta Chim. Slov. 2007, 54, 68–73. [Google Scholar]
- Zitka, O.; Huska, D.; Krizkova, S.; Adam, V.; Chavis, G.J.; Trnkova, L.; Horna, A.; Hubalek, J.; Kizek, R. An investigation of glutathione-platinum(II) interactions by means of the flow injection analysis using glassy carbon electrode. Sensors 2007, 7, 1256–1270. [Google Scholar]
- Fojta, M.; Havran, L.; Billova, S.; Kostecka, P.; Masarik, M.; Kizek, R. Two-surface strategy in electrochemical DNA hybridization assays: Detection of osmium-labeled target DNA at carbon electrodes. Electroanalysis 2003, 15, 431–440. [Google Scholar]
- Fojta, M.; Havran, L.; Kizek, R.; Billova, S.; Palecek, E. Multiply osmium-labeled reporter probes for electrochemical DNA hybridization assays: detection of trinucleotide repeats. Biosens. Bioelectron. 2004, 20, 985–994. [Google Scholar]
- Kizek, R.; Masarik, M.; Kramer, K.J.; Potesil, D.; Bailey, M.; Howard, J.A.; Klejdus, B.; Mikelova, R.; Adam, V.; Trnkova, L.; Jelen, F. An analysis of avidin, biotin and their interaction at attomole levels by voltammetric and chromatographic techniques. Anal. Bioanal. Chem. 2005, 381, 1167–1178. [Google Scholar]
- Masarik, M.; Kizek, R.; Kramer, K.J.; Billova, S.; Brazdova, M.; Vacek, J.; Bailey, M.; Jelen, F.; Howard, J.A. Application of avidin-biotin technology and adsorptive transfer stripping square-wave voltammetry for detection of DNA hybridization and avidin in transgenic avidin maize. Anal. Chem. 2003, 75, 2663–2669. [Google Scholar]
- Palecek, E.; Kizek, R.; Havran, L.; Billova, S.; Fojta, M. Electrochemical enzyme-linked immunoassay in a DNA hybridization sensor. Anal. Chim. Acta 2002, 469, 73–83. [Google Scholar]
- Palecek, E.; Masarik, M.; Kizek, R.; Kuhlmeier, D.; Hassmann, J.; Schulein, J. Sensitive electrochemical determination of unlabeled MutS protein and detection of point mutations in DNA. Anal. Chem. 2004, 76, 5930–5936. [Google Scholar]
- Jelen, F.; Kourilova, A.; Pecinka, P.; Palecek, E. Microanalysis of DNA by stripping transfer voltammetry. Bioelectrochemistry 2004, 63, 249–252. [Google Scholar]
- Palecek, E. Surface-attached molecular beacons light the way for DNA sequencing. Trends Biotechnol. 2004, 22, 55–58. [Google Scholar]
- Palecek, E.; Jelen, F. Electrochemistry of nucleic acids and development of DNA sensors. Crit. Rev. Anal. Chem. 2002, 32, 261–270. [Google Scholar]
- Palecek, E.; Fojta, M. Detecting DNA hybridization and damage. Anal. Chem. 2001, 73, 74A–83A. [Google Scholar]
- Palecek, E. From polarography of DNA to microanalysis with nucleic acid-modified electrodes. Electroanalysis 1996, 8, 7–14. [Google Scholar]
- Palecek, E.; Postbieglova, I. Adsorptive Stripping Voltammetry of Biomacromolecules with Transfer of the Adsorbed Layer. J. Electroanal. Chem. 1986, 214, 359–371. [Google Scholar]
- Goulding, H.; Jasani, B.; Pereira, H.; Reid, A.; Galea, M.; Bell, J.A.; Elston, C.W.; Robertson, J.F.; Blamey, R.W.; Nicholson, R.A.; Schmid, K.W.; Ellis, I.O. Metallothionein Expression in Human Breast-Cancer. Br. J. Cancer 1995, 72, 968–972. [Google Scholar]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
© 2008 by MDPI (http://www.mdpi.org). Reproduction is permitted for noncommercial purposes.
Share and Cite
Adam, V.; Baloun, J.; Fabrik, I.; Trnkova, L.; Kizek, R. An Electrochemical Detection of Metallothioneins at the Zeptomole Level in Nanolitre Volumes. Sensors 2008, 8, 2293-2305. https://doi.org/10.3390/s8042293
Adam V, Baloun J, Fabrik I, Trnkova L, Kizek R. An Electrochemical Detection of Metallothioneins at the Zeptomole Level in Nanolitre Volumes. Sensors. 2008; 8(4):2293-2305. https://doi.org/10.3390/s8042293
Chicago/Turabian StyleAdam, Vojtech, Jiri Baloun, Ivo Fabrik, Libuse Trnkova, and Rene Kizek. 2008. "An Electrochemical Detection of Metallothioneins at the Zeptomole Level in Nanolitre Volumes" Sensors 8, no. 4: 2293-2305. https://doi.org/10.3390/s8042293
APA StyleAdam, V., Baloun, J., Fabrik, I., Trnkova, L., & Kizek, R. (2008). An Electrochemical Detection of Metallothioneins at the Zeptomole Level in Nanolitre Volumes. Sensors, 8(4), 2293-2305. https://doi.org/10.3390/s8042293