Raman Spectroscopy Cell-based Biosensors

Abstract
:1. Introduction
2. Raman spectroscopy and instrumentation
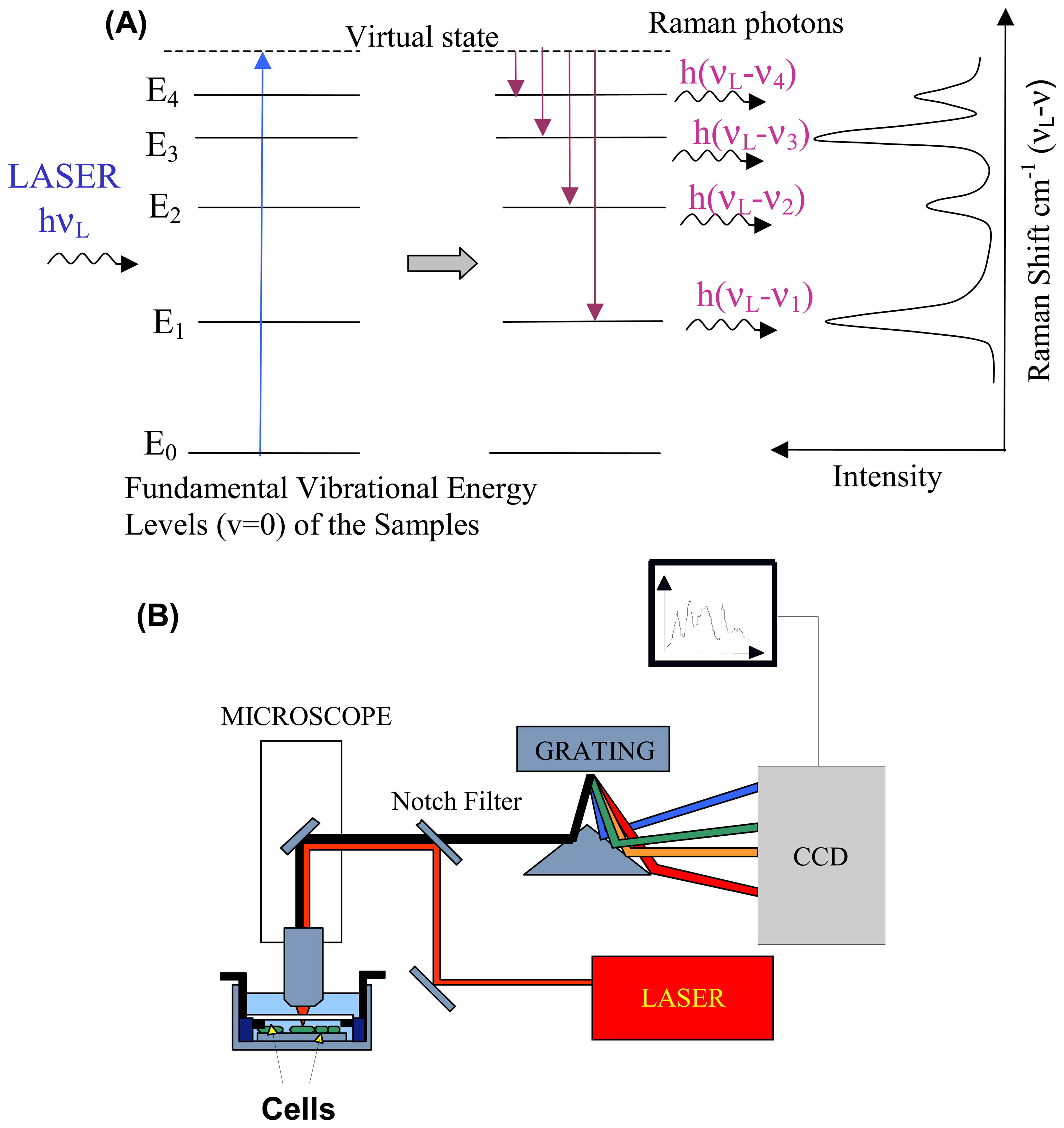
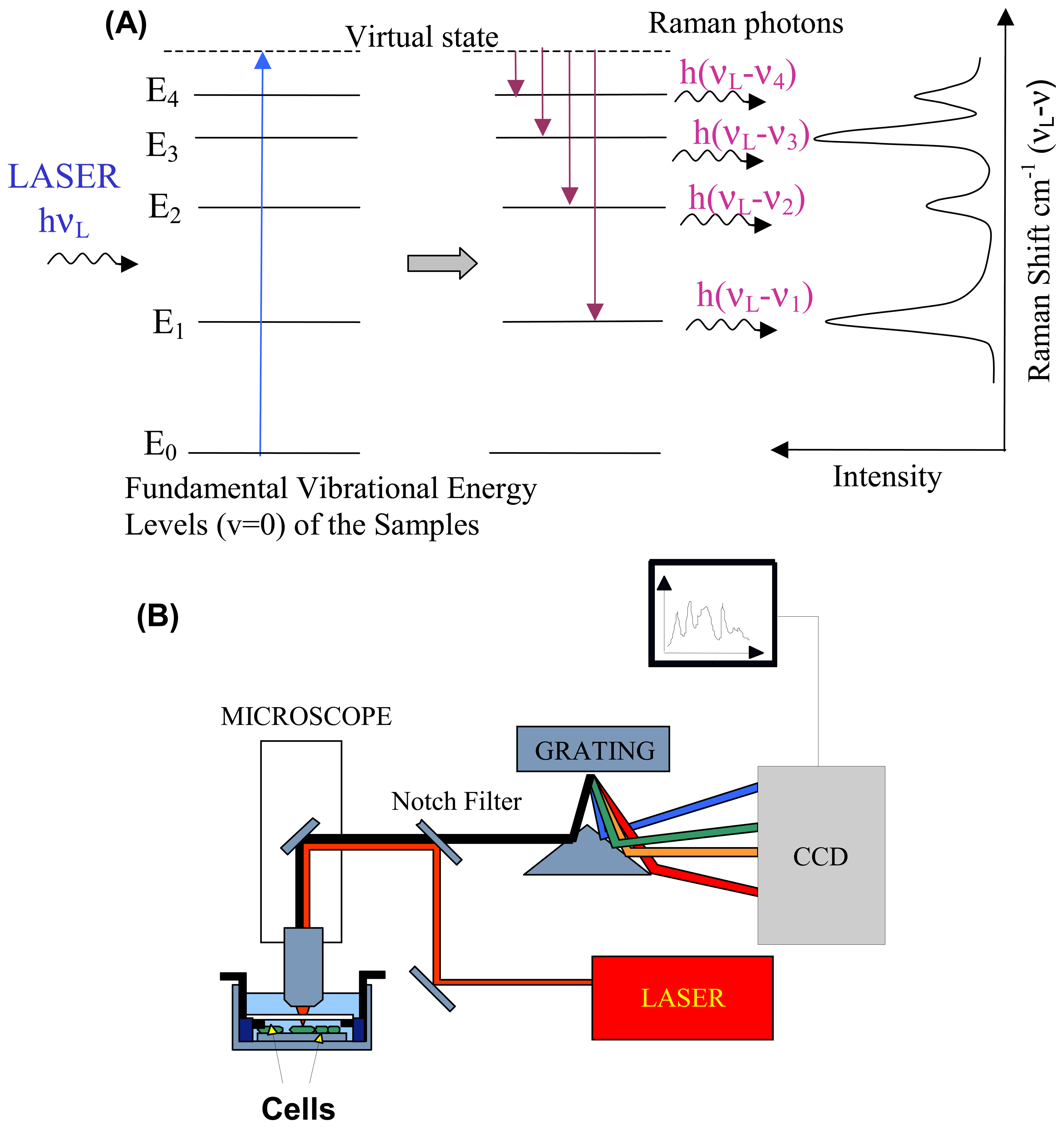
2.1. Raman scattering
2.2. Instrumentation
3. Applications
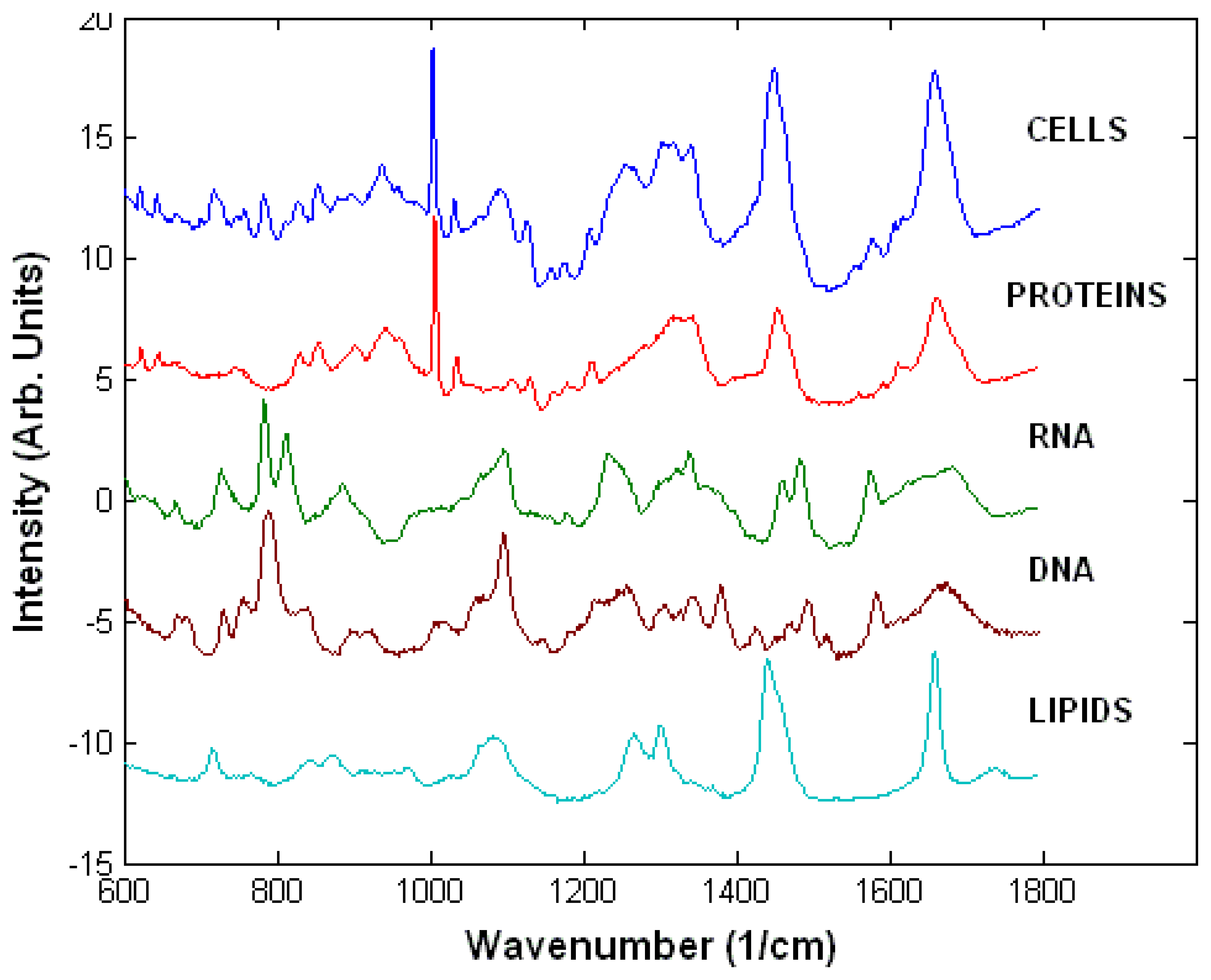
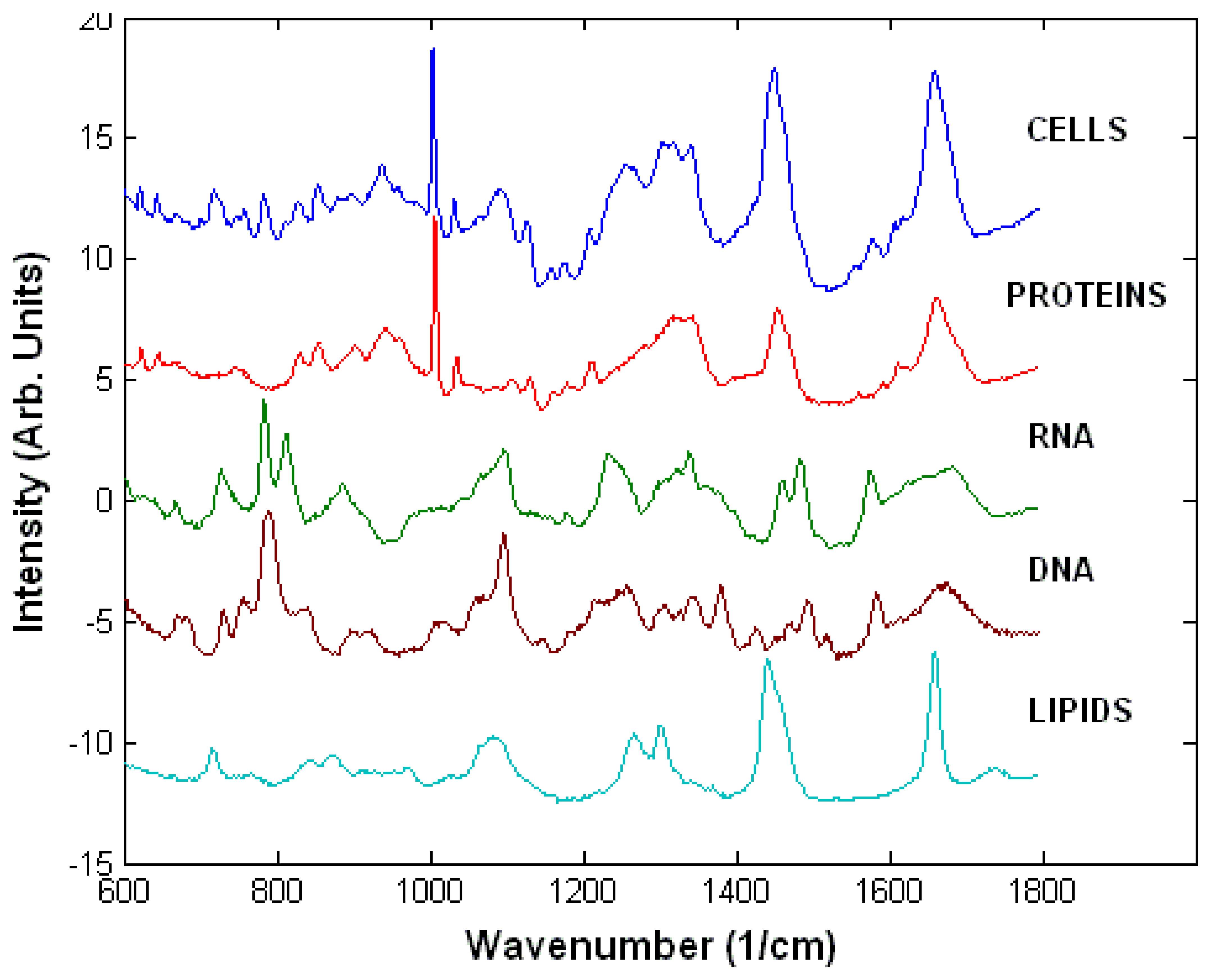
3.1. Raman spectra of live cells
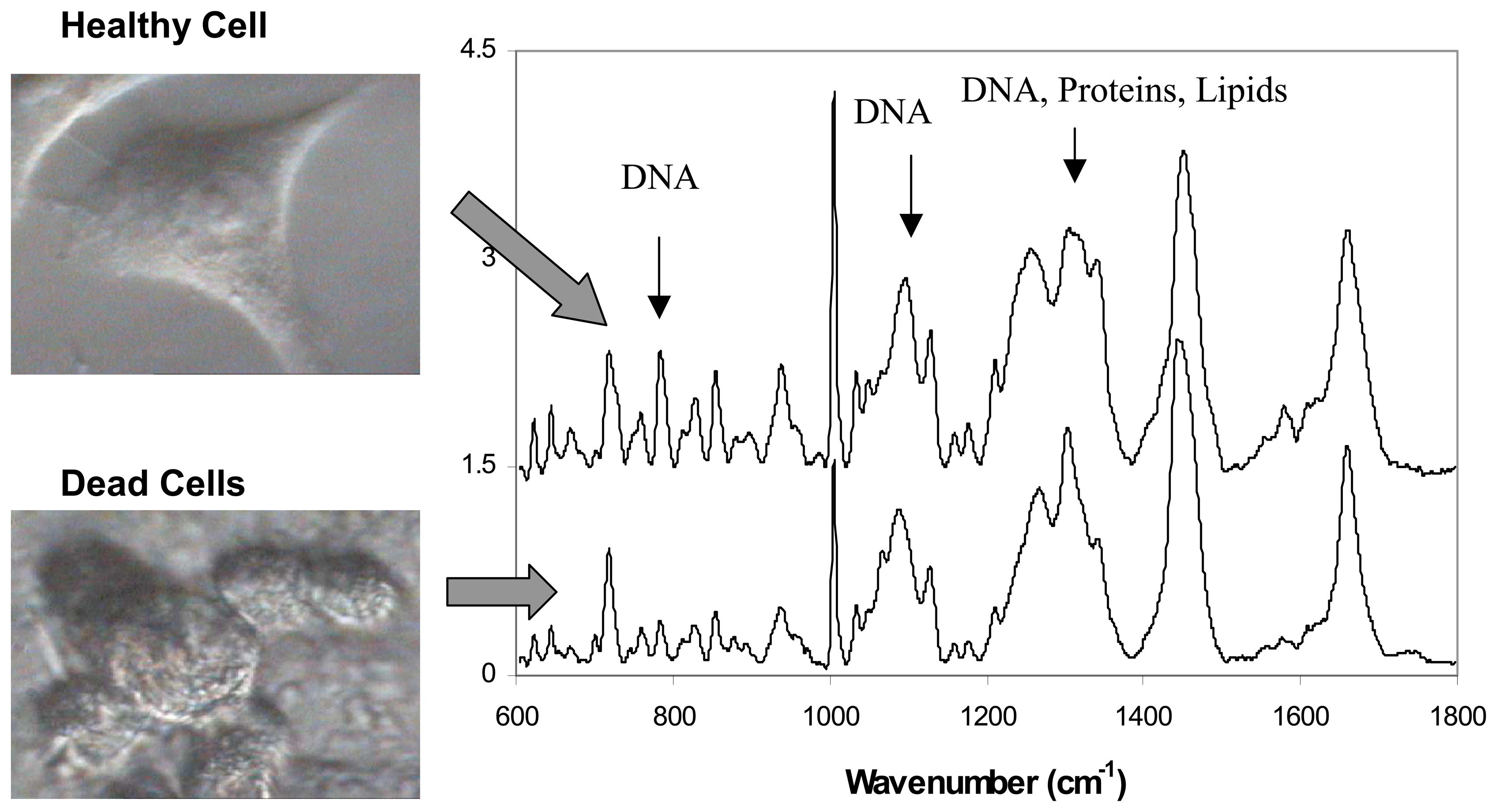
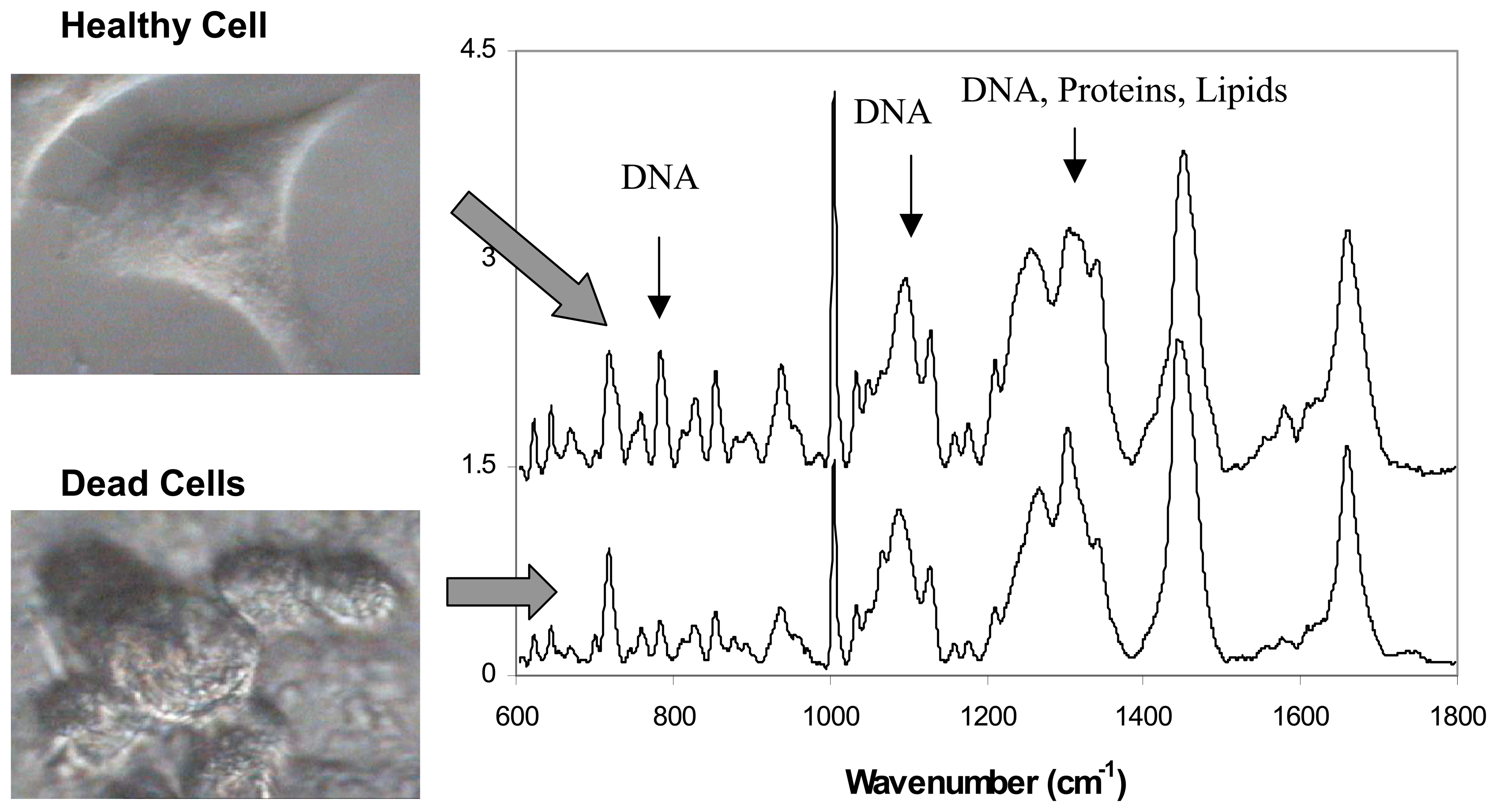
3.2. Live versus dead cells
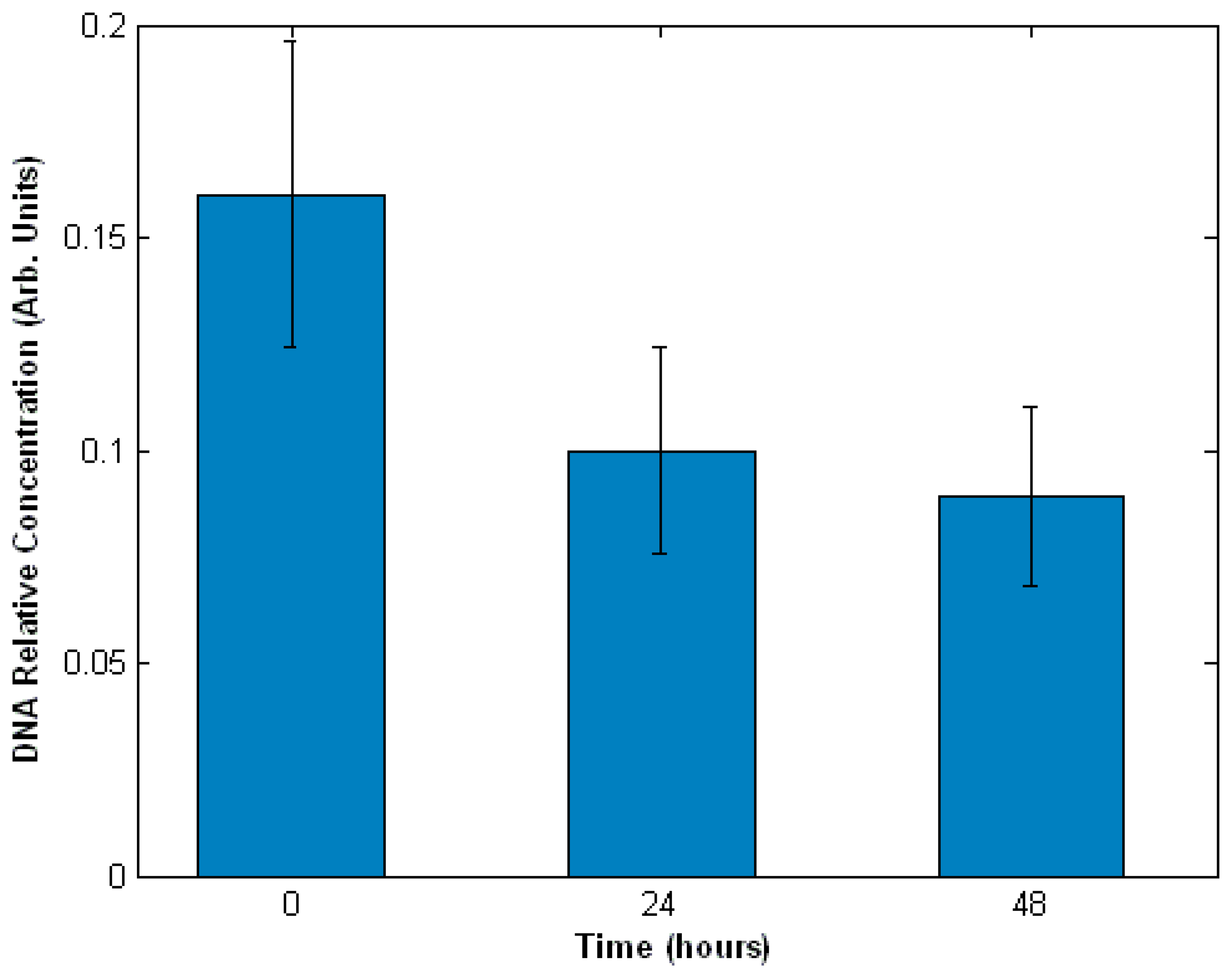
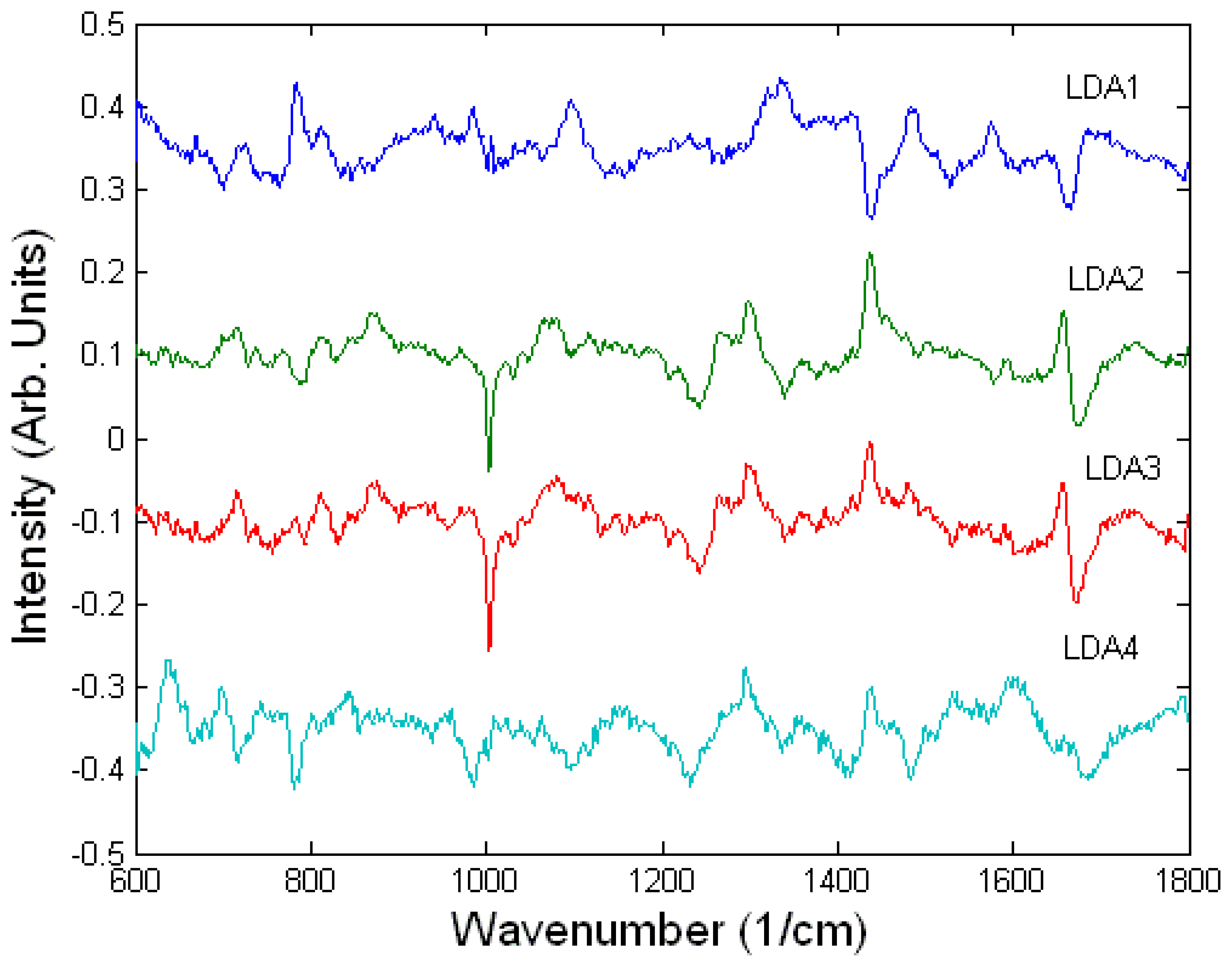
3.3. Interaction of cells with drugs
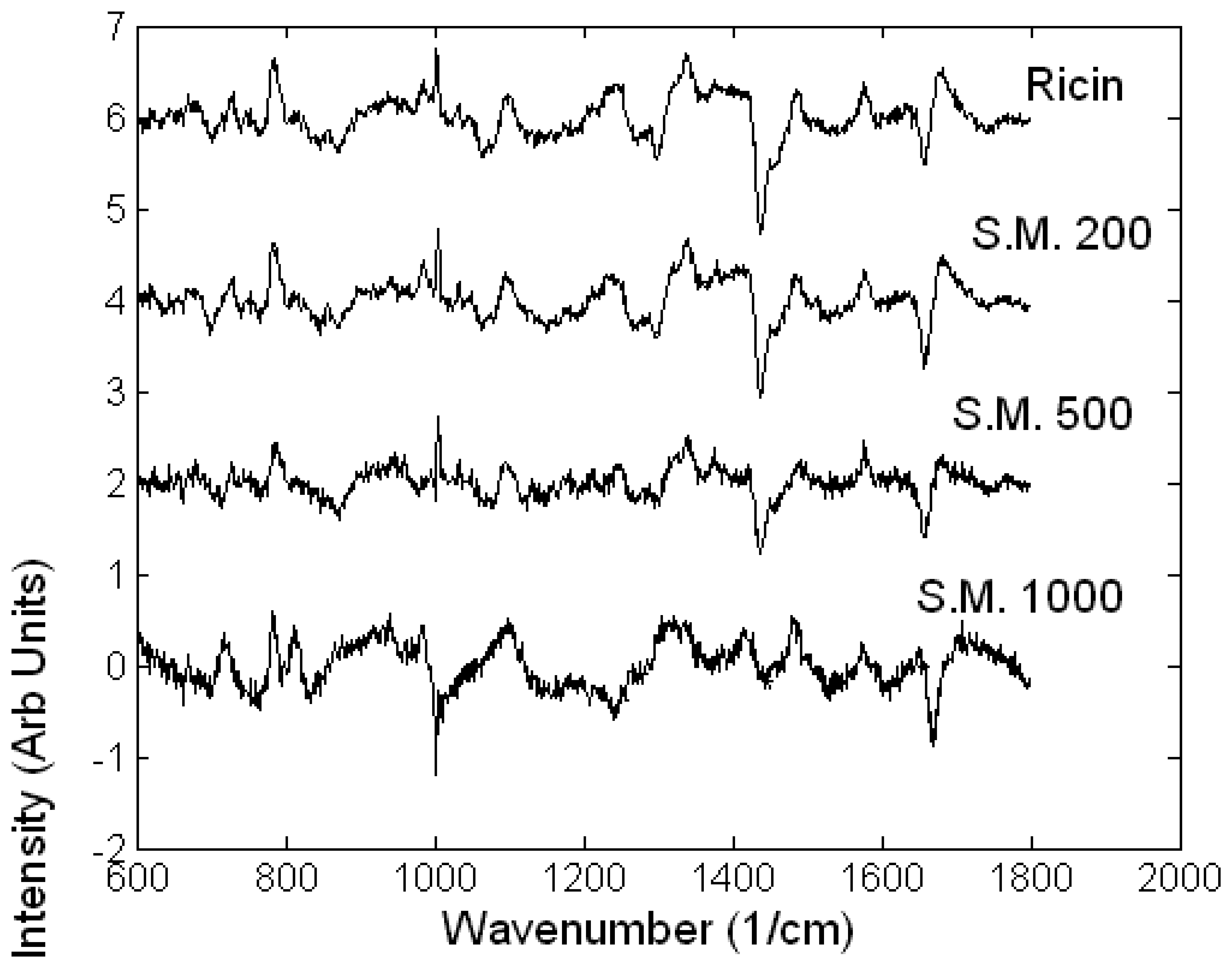
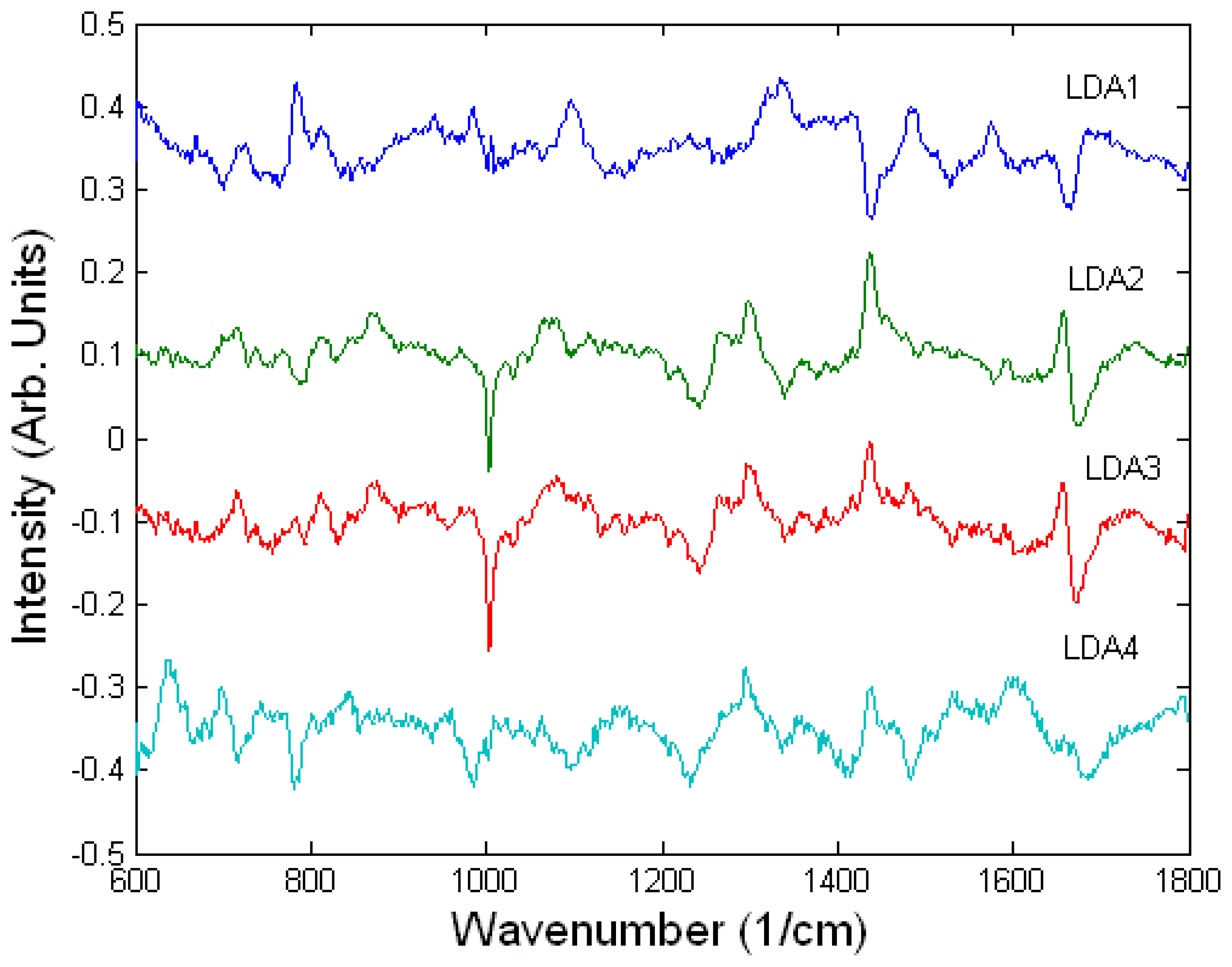
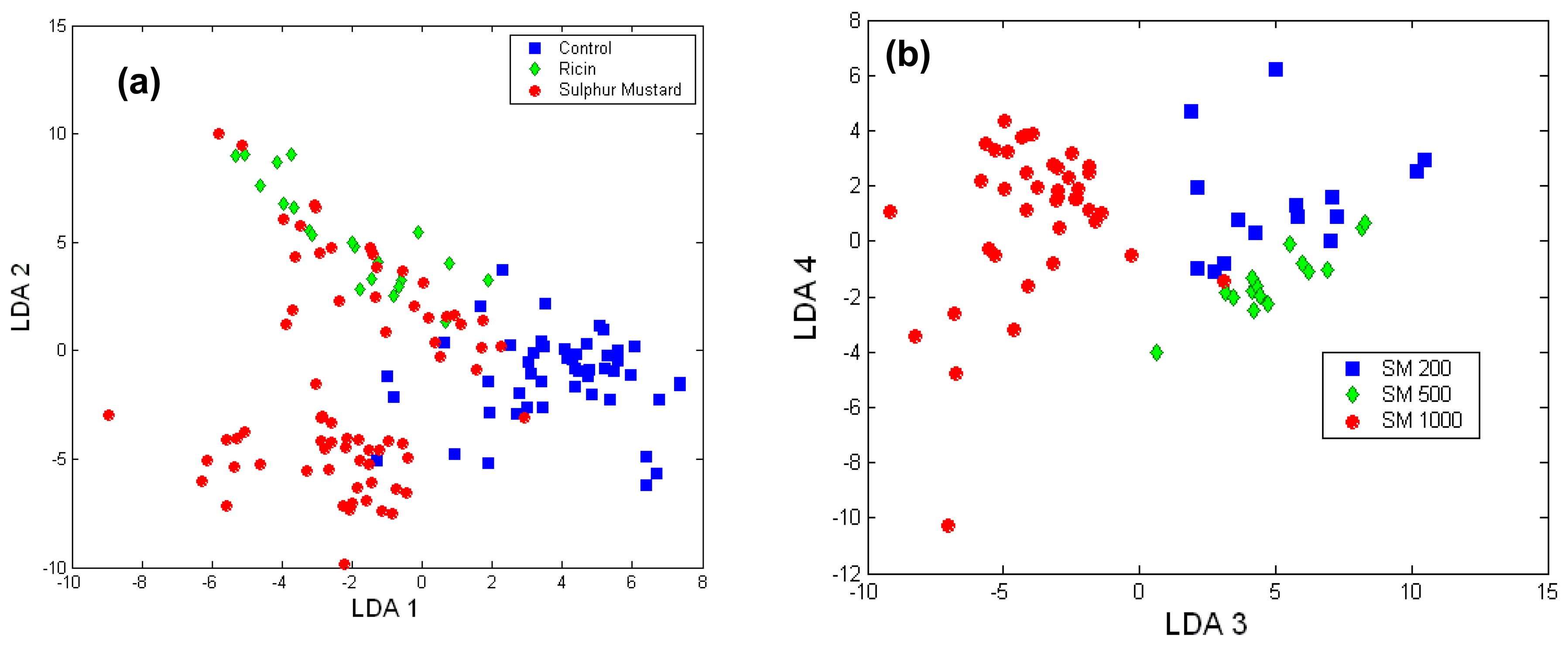
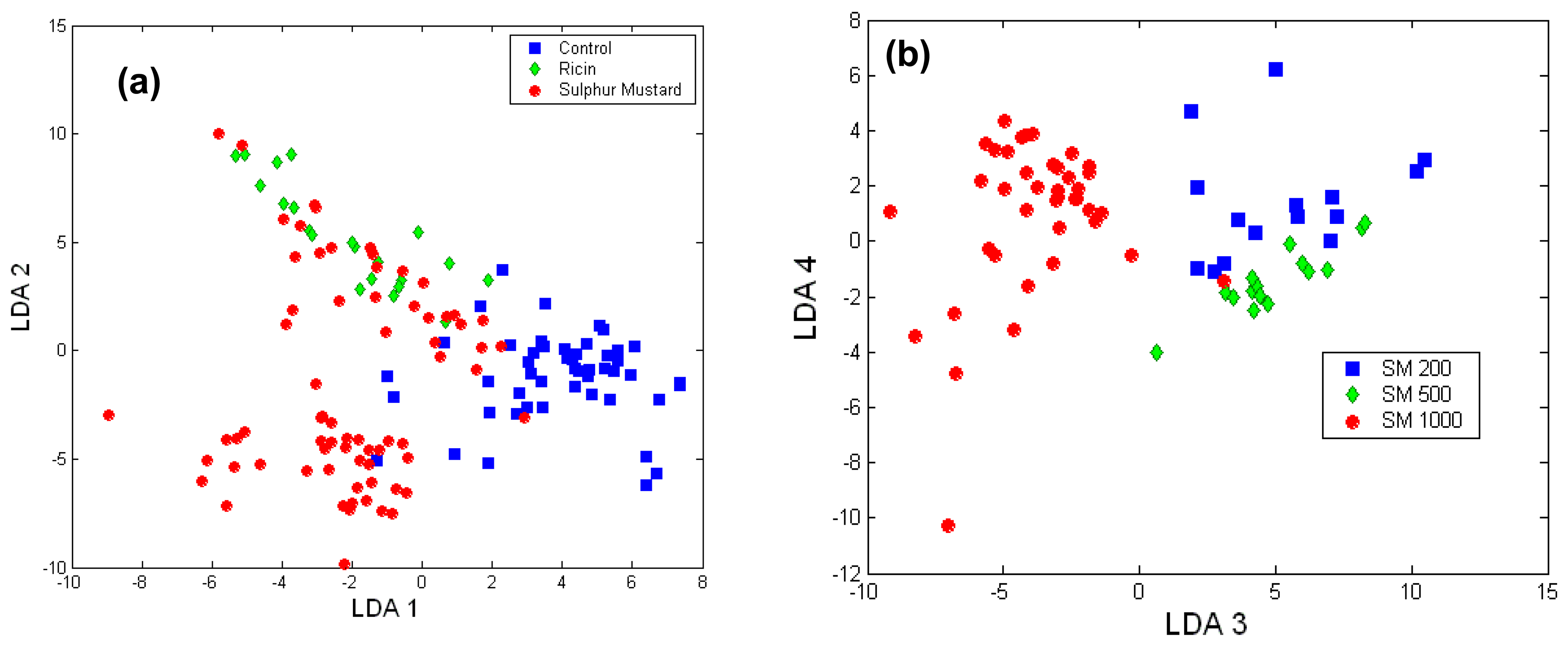
Toxicology of chemical and biological warfare
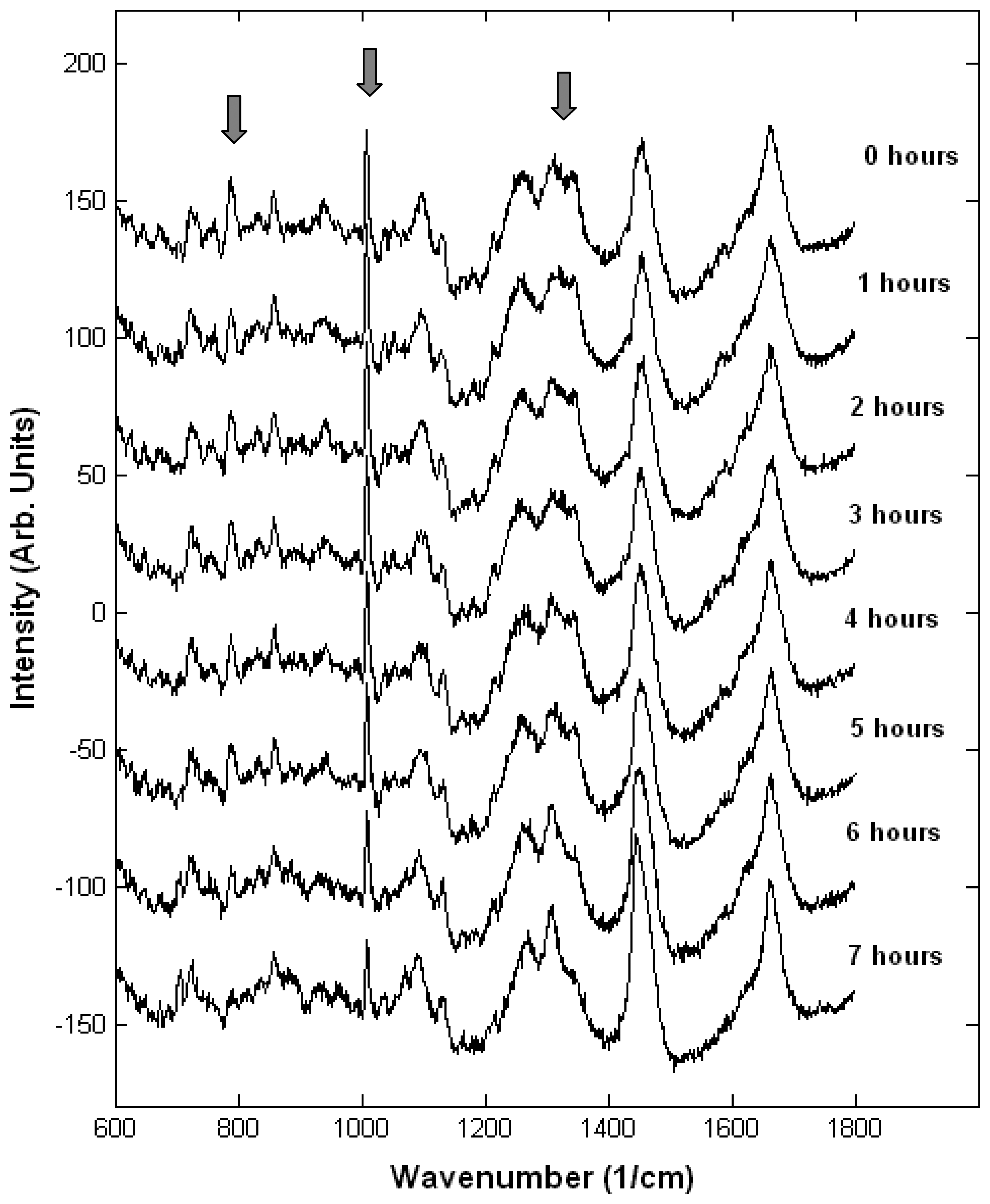
Monitoring single live cells
4. Conclusions
Acknowledgments
References
- Pancrazio, J.J.; Whelan, J.P.; Borkholder, D.A.; Ma, W.; Stenger, D.A. Development and applications of cell-base biosensors. Ann. Biomed. Eng. 1999, 27, 697–711. [Google Scholar]
- Ziegler, C. Cell-based biosensors. Fresenius J. Anal. Chem. 2000, 366, 552–559. [Google Scholar]
- Sørensen, S.J.; Burmølle, M.; Hansen, L.H. Making bio-sense of toxicity: new developments in whole-cell biosensors. Curr. Opin. Biotechnol. 2006, 17, 11–16. [Google Scholar]
- Lee, H.J.; Villaume, J.; Cullen, D.C.; Kim, B.C.; Gu, M.B. Monitoring and classification of PAH tocixity using an immobilised bioluminescent bacteria. Biosensensors Bioelectron. 2003, 18, 571–577. [Google Scholar]
- Kim, B.C.; Park, K.S.; Kim, S.D.; Gu, M.B. Evaluation of ahigh throughput toxicity biosensor and comparison with Daphnia magna bioassay. Biosensensors Bioelectron 2003, 18, 821–826. [Google Scholar]
- Fernandes, P.B. Technological advances in high-throughput screening. Curr. Opin. Chem. Biol. 1998, 2, 597–603. [Google Scholar]
- Gross, G.W.; Rhoades, B.K.; Jordan, R.J. Neural networks for biochemical sensing. Sensors Actuators 1992, 6, 1–8. [Google Scholar]
- Gross, G.W.; Azzazy, H.M.E.; Wu, M.C.; Rhodes, B.K. The use of neuronal networks on microelectrode arrays as biosensors. Biosensors Bioelectron 1995, 10, 553–567. [Google Scholar]
- Potter, S.; DeMarse, T.B. A new approach to neural cell culture for long-term studies. J. Neurosci. Methods 2001, 110, 17–24. [Google Scholar]
- Chiappalone, M.; Vato, A.; Tedesco, M.B.; Marcoli, M.; Davide, F.; Martinoia, S. Networks of neurons coupled to microelectrode arrays: a neuronal sensory system for pharmacological applications. Biosensensors Bioelectron 2003, 18, 627–634. [Google Scholar]
- Pancrazio, J.J.; Kulagina, N.V.; Shaffer, K.M.; Gray, S.A.; O'Shaughnessy, T.J. Sensitivity of the neuronal network biosensor to environmental threats. J. Toxicol. Env. Heal. A. 2004, 67, 809–818. [Google Scholar]
- Natarajan, A.; Molnar, P.; Sieverdes, K.; Jamshidi, A.; Hickman, J.J. Microelectrode array recordings of cardiac action potentials as a high throughput method to evaluate pesticide toxicity. Toxicol In Vitro. 2006, 20, 375–381. [Google Scholar]
- Liu, Q.; Cai, H.; Xu, Y.; Xiao, L.; Yang, M.; Wang, P. Detection of heavy metal toxicity using cardiac cell-based biosensor. Biosensors Bioelectron 2007, 22, 3224–3229. [Google Scholar]
- Yang, M.; Prasad, S.; Zhang, X.; Morgan, A.; Ozkan, M.; Ozkan, C.S. Cellular microarrays for chemical sensing. Sensor Mater 2003, 15, 313–333. [Google Scholar]
- Lorenzelli, L.; Margesin, B.; Martinoia, S.; Tedesco, M.T.; Valle, M. Bioelectrochemical signal monitoring og in vitro cultured cells by means of an automated microsystem based on solid state sensor. Biosensensors Bioelectron 2003, 18, 621–627. [Google Scholar]
- Rainina, E.I.; Efremenco, E.N.; Varfolomeyev, S.D.; Simonian, A.L.; Wild, J.R. The development of a new biosensor based on recombinant E. coli for the direct detection of organophosphorus neurotoxins. Biosens. Bioelectron 1996, 11, 991–1000. [Google Scholar]
- May, K.M.L.; Wang, Y.; Bachas, L.G.; Anderson, K.W. Development of a whole-cell-based biosensor for detecting histamine as a model toxin. Anal. Chem. 2004, 76, 5313–5318. [Google Scholar]
- Giaever, I.; Keese, C.R. A morphological biosensor for mammalian cells. Nature 1993, 366, 591–592. [Google Scholar]
- Tiruppathi, C.; Malik, A.B.; Del Vecchio, P.J.; Keese, C.R.; Giaever, I. Electrical method for detection of endothelial cell shape change in real time: assessment of endothelial barrier function. Proc. Natl. Acad. Sci. USA 1992, 89, 7919–7023. [Google Scholar]
- Mahadevan Jansen, A.; Richards Kortum, R. Raman Spectroscopy for the detection of cancers and precancers. J. Biomed. Optics. 1996, 1, 31–70. [Google Scholar]
- Gremlich, H.U.; Yan, B. Infrared and Raman Spectroscopy of Biological Materials; Marcel Dekker Inc.: New York, 2001. [Google Scholar]
- Thomas, G.J., Jr. Raman Spectroscopy of Protein and Nucleic Acid Assemblies. Annu. Rev. Biomol. Struct. 1999, 28, 1–27. [Google Scholar]
- Notingher, I.; Hench, L.L. Raman microspectroscopy: a non-invasive tool for studies of individual living cells in vitro. Expert. Rev. Med. Devices 2006, 3, 215–234. [Google Scholar]
- Wood, B.R.; McNaughton, D. Resonance Raman spectroscopy in malaria research. Expert Rev. Proteomics 2006, 3, 525–544. [Google Scholar]
- Kneipp, K.; Kneipp, H.; Kneipp, J. Surface-enhanced Raman scattering in local optical fields of silver and gold nanoaggregates-from single-molecule Raman spectroscopy to ultrasensitive probing in live cells. Acc. Chem. Res. 2006, 39, 443–450. [Google Scholar]
- Nan, X.; Potma, E.O.; Xie, X.S. Nonperturbative chemical imaging of organelle transport in living cells with coherent anti-stokes Raman scattering microscopy. Biophys. J. 2006, 91, 728–735. [Google Scholar]
- Zhang, X.; Yonzon, C.R.; Young, M.A.; Stuart, D.A.; Van Duyne, R.P. Surface-enhanced Raman spectroscopy biosensors: excitation spectroscopy for optimisation of substrates fabricated by nanosphere lithography. IEE. Proc. Nanobiotechnol 2005, 152, 195–206. [Google Scholar]
- Ferraro, J.R.; Nakamoto, K.; Brown, C.W. Introductory Raman Spectroscopy; Academic Press: San Diego, USA, 2003. [Google Scholar]
- Long, D.A. Raman spectroscopy; McGraw-Hill: New York, USA, 1977. [Google Scholar]
- Lewis, I.; Edwards, H. Handbook of Raman Spectroscopy; Marcel Dekker: New York, USA, 2001. [Google Scholar]
- Uzunbajakava, N.; Lenferink, A.; Kraan, Y.; Willekens, B.; Vrensen, G.; Greve, J.; Otto, C. Non resonant Raman imaging of protein distribution in single human cells. Biopolymers 2003, 72, 1–9. [Google Scholar]
- Neuman, K.C.; Chadd, E.H.; Liou, G.F.; Bergman, K.; Block, S.M. Characterization of photodamage to escherichia coli in optical traps. Biophys. J. 1999, 77, 2856–2863. [Google Scholar]
- Puppels, G.J.; Olminkhof, J.H.F.; Segers-Nolten, G.M.J.; Otto, C.; de Mul, F.F.; Greve, J. Laser irradiation and Raman spectroscopy of single living cells and chromosomes: sample degradation occurs with 514.5 nm but not with 660 nm laser light. Exp. Cell Res. 1991, 195, 361–367. [Google Scholar]
- Wood, B.R.; Caspers, P.; Puppels, G.J.; Pandiancherri, S.; McNaughton, D. Resonance Raman spectroscopy of red blood cells using near-infrared laser excitation. Anal. Bioanal. Chem. 2007, 387, 1691–1703. [Google Scholar]
- Notingher, I.; Verrier, S.; Haque, S.; Polak, J.M.; Hench, L.L. Spectroscopic study of human lung epithelial cells (A549) in culture: Living cells versus dead cells. Biopolymers 2003, 72, 230–240. [Google Scholar]
- Owen, C.A.; Selvakumaran, J.; Notingher, I.; Jell, G.; Hench, L.L.; Stevens, M.M. In vitro toxicology evaluation of pharmaceuticals using Raman micro-spectroscopy. J. Cell. Biochem. 2006, 99, 178–86. [Google Scholar]
- Notingher, I.; Green, C.; Dyer, C.; Perkins, E.; Hopkins, N.; Lindsay, C.; Hench, L.L. 2004a Discrimination between ricin and sulphur mustard toxicity in vitro using Raman spectroscopy. J. R. Soc.:Interface 2004, 1, 79–90. [Google Scholar]
- Notingher, I.; Selvakumaran, J.; Hench, L.L. New Detection System for Toxic Agents Based on Continuous Spectroscopic Monitoring of Living Cells. Biosens. Bioelectron 2004, 20, 780–789. [Google Scholar]
- Verrier, S.; Notingher, I.; Polak, J.M.; Hench, L.L. In situ monitoring of cell death using Raman microspectroscopy. Biopolymers 2004, 74, 57–62. [Google Scholar]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Peak (cm-1) | Assignment | |||
|---|---|---|---|---|
| Nucleic Acids | Proteins | Lipids | Carbohydrates | |
| 1736 | C=O ester | |||
| 1680-1655 | Amide I | C=C str. | ||
| 1617 | C=C Tyr, Trp | |||
| 1607 | C=C Phe, Tyr | |||
| 1578 | G, A | |||
| 1480-1420 | G, A, CH def | C-H | CH def | CH def |
| 1342 | A, G | C-H | CH def | |
| 1320 | G | C-H | ||
| 1301 | CH2 twist | |||
| 1284-1220 | T, A | Amide III | =CH bend | |
| 1209 | C-C6H5. Phe, Trp | |||
| 1176 | C-H bend Tyr | |||
| 1158 | C-C/C-N str. | |||
| 1128 | C-N str. | C-O str | ||
| 1095-1060 | PO2- str. | Chain C-C str. | C-O, C-C str | |
| 1033 | C-H in-plane Phe | |||
| 1005 | Sym. Ring br Phe | |||
| 980 | C-C BK str. b-sheet | =CH bend | ||
| 937 | C-C BK str. a-helix | C-O-C glycos. | ||
| 877 | C-C-N+ sym str | C-O-C ring | ||
| 854 | Ring br Tyr | |||
| 828 | O-P-O asym.str. | Ring br. Tyr | ||
| 811 | O-P-O str. RNA | |||
| 788 | O-P-O str. DNA | |||
| 782 | U,C,T ring br | |||
| 760 | Ring breath Trp | |||
| 729 | A | |||
| 717 | CN+(CH3)3 str | |||
| 667 | T, G | |||
| 645 | C-C twist Tyr | |||
| 621 | C-C twist Phe | |||
© 2007 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Notingher, I. Raman Spectroscopy Cell-based Biosensors. Sensors 2007, 7, 1343-1358. https://doi.org/10.3390/s7081343
Notingher I. Raman Spectroscopy Cell-based Biosensors. Sensors. 2007; 7(8):1343-1358. https://doi.org/10.3390/s7081343
Chicago/Turabian StyleNotingher, Ioan. 2007. "Raman Spectroscopy Cell-based Biosensors" Sensors 7, no. 8: 1343-1358. https://doi.org/10.3390/s7081343