Electrochemical DNA Sensors for Detection of DNA Damage

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Overview
2. DNA-electrochemical biosensors
3. DNA oxidative biomarker
4. Electrochemical biosensors for detection of DNA damage
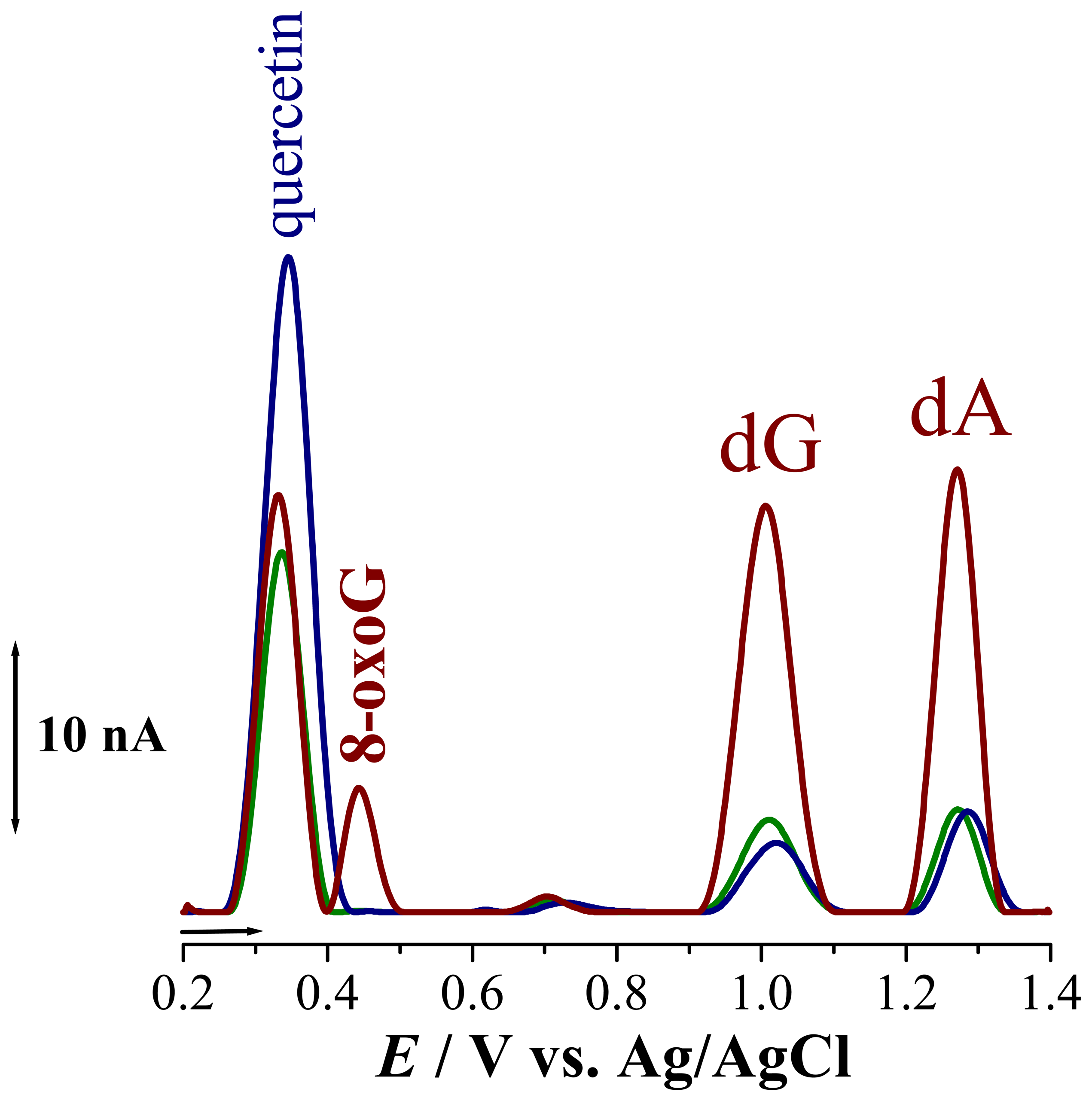
 . It is known that oxidation of quercetin is a complex process during which superoxide radicals are formed. These radicals readily attack the dsDNA disrupting the helix, causing contact between the electrode surface and the DNA nucleotides guanosine, Epa = + 1.02 V, and adenosine, Epa= +1.28 V, such that their oxidation peaks increase, Fig. 5
. It is known that oxidation of quercetin is a complex process during which superoxide radicals are formed. These radicals readily attack the dsDNA disrupting the helix, causing contact between the electrode surface and the DNA nucleotides guanosine, Epa = + 1.02 V, and adenosine, Epa= +1.28 V, such that their oxidation peaks increase, Fig. 5  . On the other hand, the radicals formed during the oxidation of quercetin incorporated into dsDNA lead to the formation of 8-oxoguanine, with peak at Epa = + 0.45 V. In order to prove the involvement of the oxygen radicals in DNA damage during oxidation of intercalated quercetin, experiments were also carried out in solutions saturated with N2, Fig. 5
. On the other hand, the radicals formed during the oxidation of quercetin incorporated into dsDNA lead to the formation of 8-oxoguanine, with peak at Epa = + 0.45 V. In order to prove the involvement of the oxygen radicals in DNA damage during oxidation of intercalated quercetin, experiments were also carried out in solutions saturated with N2, Fig. 5  . The differential pulse voltammogram obtained showed only small guanosine and adenosine oxidation peaks, demonstrating that no DNA damage had occurred. Also, no additional peak, at
. The differential pulse voltammogram obtained showed only small guanosine and adenosine oxidation peaks, demonstrating that no DNA damage had occurred. Also, no additional peak, at5. Conclusions
Acknowledgments
References
- Thevenot, D. R.; Toth, K.; Durst, R. A.; Wilson, G. S. Electrochemical biosensors: Recommended definitions and classification. Pure Appl. Chem. 1999, 71, 2333–2348. [Google Scholar]
- Oliveira Brett, A. M.; Serrano, S. H. P.; Piedade, J. A. P. Electrochemistry of DNA, Comprehensive Chemical Kinetics, Applications of Kinetic Modeling; Compton, R. G., Hancock, G., Eds.; Elsevier: Oxford, UK, 1999; vol. 37, chapter 3; pp. 91–119. [Google Scholar]
- Oliveira Brett, A. M. DNA-based biosensors, Comprehensive Analytical Chemistry. Biosensors and Modern Specific Analytical Techniques. 2005, vol. 44, 179–208. [Google Scholar]
- Palecek, E. Past, present and future of nucleic acids electrochemistry. Talanta 2002, 56, 809–819. [Google Scholar]
- Palecek, E.; Fojta, M.; Tomschik, M.; Wang, J. Electrochemical biosensors for DNA hybridization and DNA damage. Biosens. Bioelectron. 1998, 13, 621–628. [Google Scholar]
- Mascini, M.; Palchetti, I.; Marrazza, G. DNA electrochemical biosensors. Fresenius J. Anal. Chem. 2001, 369, 15–22. [Google Scholar]
- Oliveira-Brett, A. M.; Vivan, M.; Fernandes, I. R.; Piedade, J. A. P. Electrochemical detection of in situ adriamycin oxidative damage to DNA. Talanta 2002, 56, 959–970. [Google Scholar]
- Oliveira Brett, A. M.; Matysik, F. M. Voltammetric and sonovoltammetric studies on the oxidation of thymine and cytosine at a glassy carbon electrode. J. Electroanal. Chem. 1997, 429, 95–99. [Google Scholar]
- Oliveira Brett, A. M.; Matysik, F. M. Sonoelectrochemical studies of guanine and guanosine. Bioelectrochem. Bioenerg. 1997, 42, 111–116. [Google Scholar]
- Oliveira-Brett, A. M; Silva, L. A.; Brett, C. M. A. Adsorption of guanine, guanosine, and adenine at electrodes studied by differential pulse voltammetry and electrochemical impedance. Langmuir 2002, 18, 2326–2330. [Google Scholar]
- Oliveira Brett, A. M.; Diculescu, V.; Piedade, J. A. P. Electrochemical oxidation mechanism of guanine and adenine using a glassy carbon microelectrode. Bioelectrochem 2002, 55, 61–62. [Google Scholar]
- Oliveira Brett, A. M.; Piedade, J. A.; Silva, L. A.; Diculescu, V. C. Voltammetric determination of all DNA nucleotides. Anal. Biochem. 2004, 332, 321–329. [Google Scholar]
- Pividori, M. I.; Merkoçi, A.; Alegret, S. Electrochemical genosensor design: immobilization of oligonucleotides onto transducer surfaces and detection methods. Biosens. Bioelectron. 2000, 15, 291–303. [Google Scholar]
- Oliveira-Brett, A. M.; Chiorcea, A.-M. Effect of pH and applied potential on the adsorption of DNA on highly oriented pyrolytic graphite electrodes. Atomic force microscopy surface characterisation. Electrochem. Commun. 2003, 5, 178–183. [Google Scholar]
- Oliveira-Brett, A. M.; Chiorcea, A.-M. Atomic force microscopy of DNA immobilized onto a highly oriented pyrolytic graphite electrode surface. Langmuir 2003, 19, 3830–3839. [Google Scholar]
- Oliveira-Brett, A. M.; Chiorcea, A.-M. DNA imaged on a HOPG electrode surface by AFM with controlled potential. Bioelectrochem. 2004, in press. [Google Scholar]
- Chiorcea, A.-M.; Oliveira-Brett, A. M. Atomic force microscopy characterisation of an electrochemical DNA–biosensor. Bioelectrochem. 2003, 63, 229–232. [Google Scholar]
- Halliwell, B.; Gutteridge, J. M. C. Biologically relevant metal ion-dependent hydroxyl radical generation. FEBS Letters 1992, 307, 108–112. [Google Scholar]
- Halliwell, B.; Gutteridge, J. M. C. Free Radicals in Biology and Medicine, 3rd ed.; Oxford University Press: UK, 1999. [Google Scholar]
- Shibutani, S.; Takeshita, M.; Grollman, A. P. Insertion of specific bases during DNA synthesis past the oxidation-damaged base 8-oxodG. Nature 1991, 349, 431–434. [Google Scholar]
- Cysewski, P. An ab initio study of the tautomeric and coding of 8-oxo-guanine. J. Chem. Soc., Faraday. Trans. 1998, 94, 3117–3125. [Google Scholar]
- Kasai, H. Analysis of a form of oxidative DNA damage, 8-hydroxy-2′-deoxyguanosine, as a marker of cellular oxidative stress during carcinogenesis. Mutat. Res. 1997, 387, 147–163. [Google Scholar]
- Halliwell, B. Oxygen and nitrogen are pro-carcinogens. Damage to DNA by reactive oxygen, chlorine and nitrogen species: measurement, mechanism and the effects of nutrition. Mutat. Res. 1999, 443, 37–52. [Google Scholar]
- Oliveira Brett, A. M.; Piedade, J. A.; Serrano, S. H. P. Electrochemical oxidation of 8-oxoguanine. Electroanal 2000, 12, 969–973. [Google Scholar]
- Floyd, R. A.; Watson, J. J.; Harris, J.; West, M.; Wong, P. K. Formation of 8-hydroxydeoxyguanosine, hydroxyl free radical adduct of DNA in granulocytes exposed to the tumor promoter, tetradeconylphorbolacetate. Biochem. Biophys. Res. Comm. 1987, 137, 841–846. [Google Scholar]
- Rebelo, I. A.; Piedade, J. A. P.; Oliveira-Brett, A. M. Development of an HPLC method with electrochemical detection of femtomoles of 8-oxo-7,8-dihydroguanine and 8-oxo-7,8-dihydro-2′-deoxyguanosine in the presence of uric acid. Talanta 2004, 63, 323–331. [Google Scholar]
- Rebelo, I. A.; Piedade, J. A. P.; Oliveira-Brett, A. M. Electrochemical determination of 8-oxoguanine in presence of uric acid. Bioelectrochem. 2004, 63, 267–270. [Google Scholar]
- Fojta, M. Electrochemical sensors for DNA interactions and damage. Electroanal 2002, 14, 1449–1463. [Google Scholar]
- Larsen, I. K. A Textbook of drug design and development; Krosgaard-Larse, P., Bundgaard, H., Eds.; Harwood Academic Publisher: UK, 1991; p. p. 192. [Google Scholar]
- Saenger, W. (Ed.) Principles of Nucleic Acids Structure; Springer-Verlag: New York, 1984; p. p. 201.
- Eichorn, G. L. Metal Ions in Genetic Information Transfer; Eichorn, G. L., Marzilli, L. G., Eds.; Elsevier: New York, 1981; p. p1. [Google Scholar]
- Oliveira Brett, A. M.; Serrano, S. H. P.; Macedo, T. A.; Raimundo, D.; Marques, M. H.; LaScalea, M. A. Electrochemical determination of carboplatin in serum using DNA-modified glassy carbon electrode. Electroanal. 1996, 8, 992–997. [Google Scholar]
- Rice-Evans, C. A.; Miller, N. J.; Paganga, G. Structure-antioxidant activity relationships of flavonoids and phenolic acids. Free Rad. Biol. Med. 1996, 20, 933–956. [Google Scholar]
- Johnson, M. K.; Loo, G. Effects of epigallocatechin gallate and quercetin on oxidative damage to cellular DNA. Mut. Res./ DNA Repair. 2000, 459, 211–218. [Google Scholar]
- Ohshima, H.; Yoshie, Y.; Auriol, S.; Gilibert, I. Antioxidant and pro-oxidant actions of flavonoids: effects on DNA damage induced by nitric oxide, peroxynitrite and nitroxyl anion. Free Rad. Biol. Med. 1998, 25, 1057–1065. [Google Scholar]
- Labuda, J.; Bučková, M.; Heilerová, L.; Šilhár, S.; Štepánek, I. Evaluation of the redox properties and anti/pro-oxidant effects of selected flavonoids by means of a DNA-based electrochemical biosensor. Anal Bioanal Chem 2003, 376, 168–173. [Google Scholar]
- Korbut, O.; Buckova, M.; Labuda, J.; Grundler, P. Voltammetric detection of anti-oxidative properties of flavonoids using electrically heated DNA modified carbon paste electrodes. Sensors 2003, 3, 1–10. [Google Scholar]
- Sahu, S. C.; Washington, M. C. Effects of antioxidants on quercetin-induced nuclear DNA damage and lipid peroxidation. Cancer Lett. 1991, 60, 259–264. [Google Scholar]
- Duthie, S. J.; Johnson, W.; Dobson, V. L. The effect of dietary flavonoids on DNA damage (strand breaks and oxidized pyrimdines (sic)) and growth in human cells. Mutat. Res. 1997, 390, 141–151. [Google Scholar]
- Oliveira Brett, A. M.; Diculescu, V. C. Electrochemical study of quercetin-DNA interaction: Part I. Analysis in incubated solutions. Bioelectrochem. 2004, 64, 133–141. [Google Scholar]
- Oliveira Brett, A. M.; Diculescu, V. C. Electrochemical study of quercetin-DNA interaction: Part II. In situ sensing with DNA biosensor. Bioelectrochem. 2004, 64, 143–150. [Google Scholar]
- Fojta, M.; Kubicarova, T.; Palecek., E. Electrode potential-modulated cleavage of surface-confined DNA by hydroxyl radicals detected by an electrochemical biosensor. Biosens. Bioelectron. 2000, 15, 107–115. [Google Scholar]
- Seqúaris, J. M.; Valenta, P. AC voltammetry: a control method for the damage to DNA caused in vitro by alkylating mutagens. J. Electroanal. Chem. 1987, 227, 11–20. [Google Scholar]
- Oliveira Brett, A. M.; Macedo, T. R. A.; Raimundo, D.; Marques, M. H.; Serrano, S. H. P. Voltammetric behaviour of mitoxantrone at a DNA-biosensor. Biosens. Bioelectron. 1998, 13, 861–867. [Google Scholar]
- Oliveira Brett, A. M.; Serrano, S. H. P.; Gutz, I.; La-Scalea, M. A. Electrochemical reduction of metronidazole at a DNA-modified glassy carbon electrode. Bioelectrochem. Bioenerg. 1997, 42, 175–178. [Google Scholar]
- LaScalea, M. A.; Serrano, S. H. P.; Ferreira, E. I.; Oliveira Brett, A. M. Voltammetric behavior of benznidazole at a DNA electrochemical biosensor. J. Pharm. Biomed Anal. 2002, 29, 561–568. [Google Scholar]
- The Chemotherapy Source Book; Perry, M.C. (Ed.) Williams and Wilkins: Baltimore, USA, 1991.
- Nucleic Acids in Chemistry and BiologyBlackburn, C.M.; Gait, J. (Eds.) , 2nd Edition; OUP: UK, 1996.
- Rusling, J. F. Sensors for toxicity of chemicals and oxidative stress based on electrochemical catalytic DNA oxidation. Biosens. Bioelectroni. 2004, 20, 1022–1028. [Google Scholar]
- Oliveira Brett, A. M.; da Silva, L. A.; Fujii, H.; Mataka, S.; Thiemann, T. Detection of the damage caused to DNA by a thiophene-S-oxide using an electrochemical DNA-biosensor. J. Electroanal. Chem. 2003, 549, 91–99. [Google Scholar]
- Abreu, F. C.; Goulart, M. O. F.; Oliveira Brett, A. M. Detection of the damage caused to DNA by niclosamide using an electrochemical DNA-biosensor. Biosens. Bioelectron. 2002, 17, 913–919. [Google Scholar]
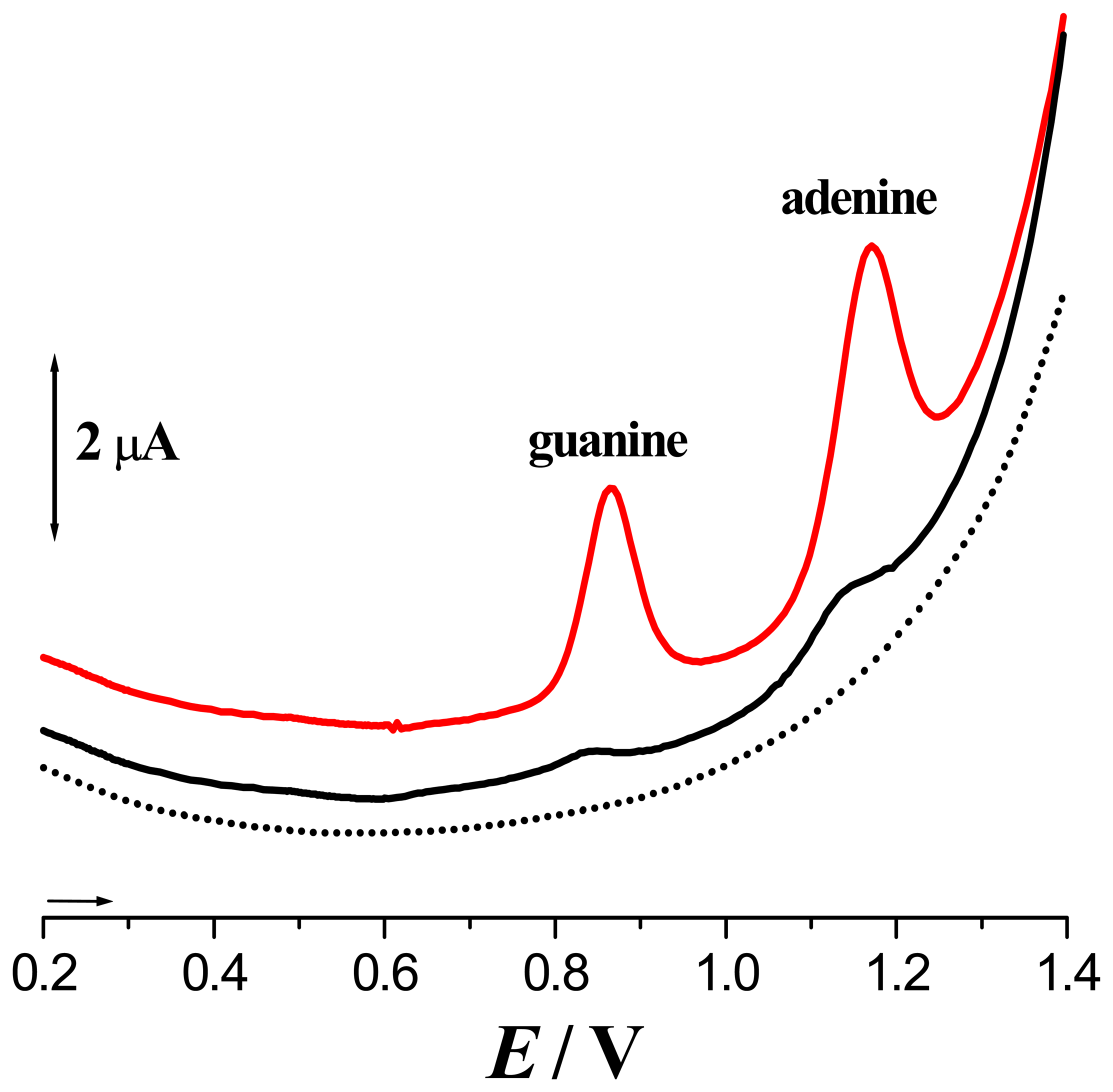
 ) 1st voltammogram and dsDNA - (▪▪▪▪) 1st and (
) 1st voltammogram and dsDNA - (▪▪▪▪) 1st and (
 ) 40th voltammogram [From Ref [7] with permission].
) 1st voltammogram and dsDNA - (▪▪▪▪) 1st and (
) 40th voltammogram [From Ref [7] with permission].
) 40th voltammogram [From Ref [7] with permission].
) 1st voltammogram and dsDNA - (▪▪▪▪) 1st and (
) 40th voltammogram [From Ref [7] with permission].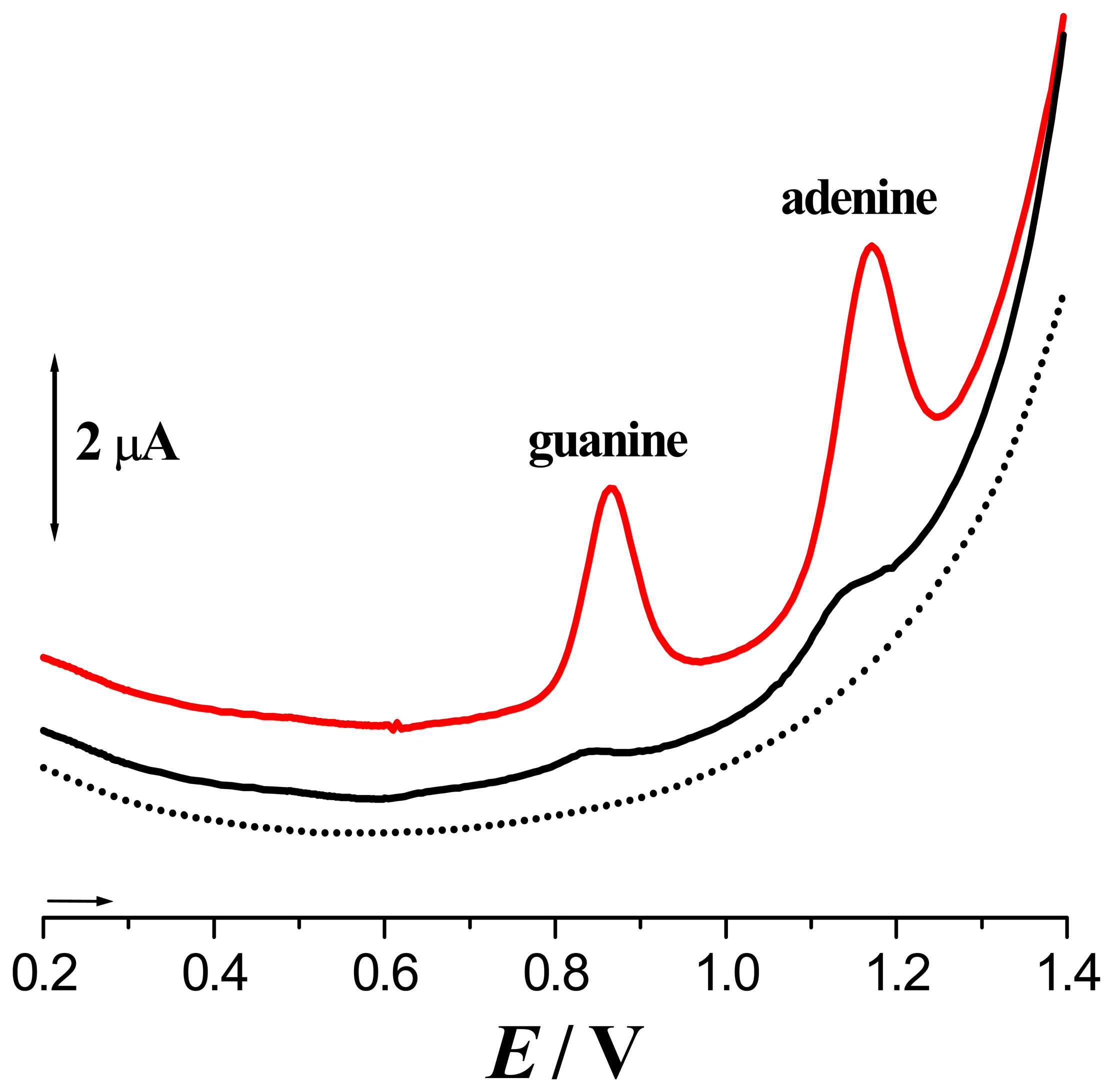


 ) before and after applying +0.400 V during 300 s in (
) before and after applying +0.400 V during 300 s in (
 ) normal atmosphere and in (
) normal atmosphere and in (
 ) N2 saturated buffer solution. [From Ref [40] with permission]
) before and after applying +0.400 V during 300 s in (
) normal atmosphere and in (
) N2 saturated buffer solution. [From Ref [40] with permission]
) N2 saturated buffer solution. [From Ref [40] with permission]
) before and after applying +0.400 V during 300 s in (
) normal atmosphere and in (
) N2 saturated buffer solution. [From Ref [40] with permission]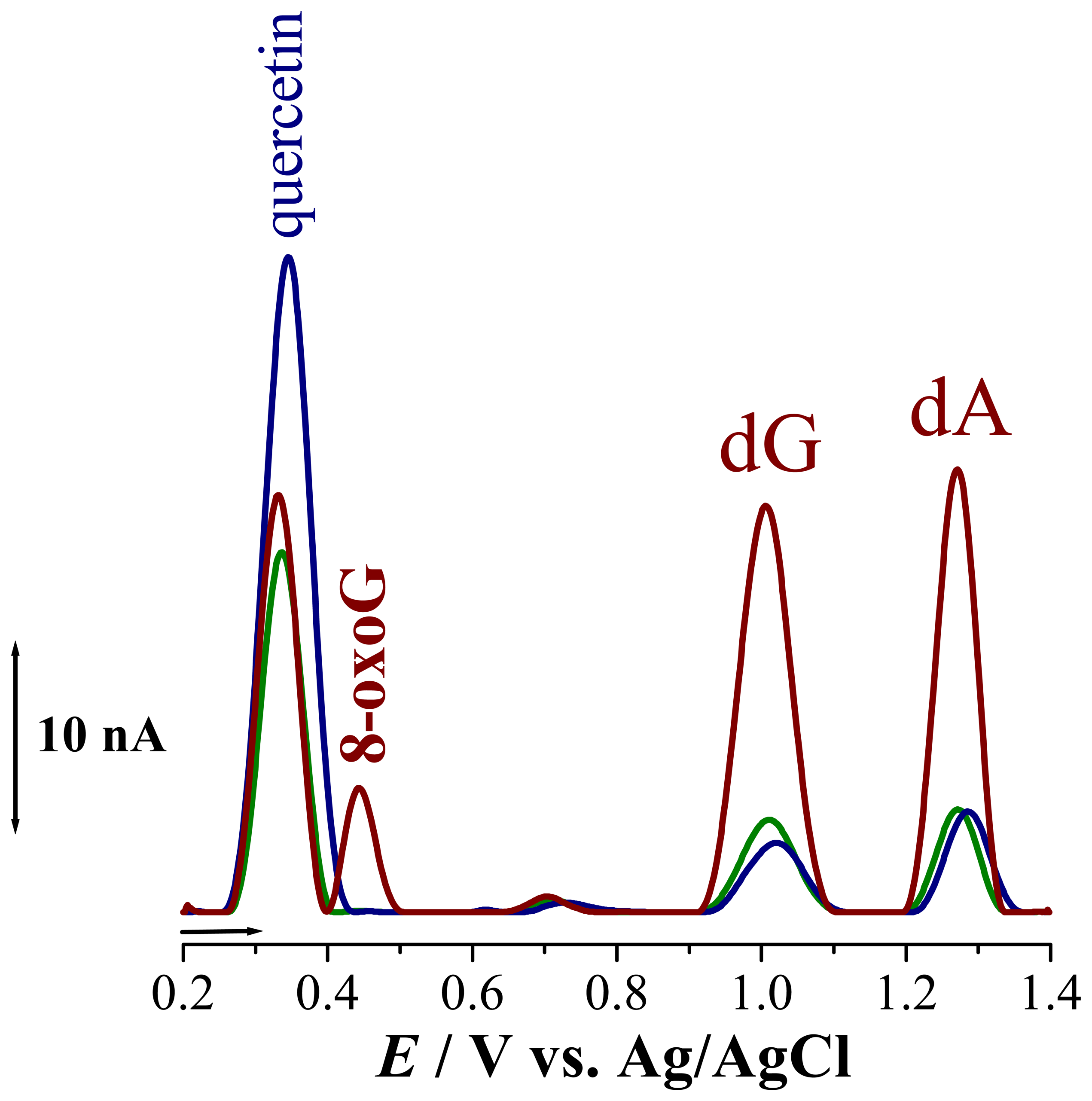
© 2005 by MDPI ( http://www.mdpi.org). Reproduction is permitted for noncommercial purposes.
Share and Cite
Diculescu, V.C.; Paquim, A.-M.C.; Brett, A.M.O. Electrochemical DNA Sensors for Detection of DNA Damage. Sensors 2005, 5, 377-393. https://doi.org/10.3390/s5060377
Diculescu VC, Paquim A-MC, Brett AMO. Electrochemical DNA Sensors for Detection of DNA Damage. Sensors. 2005; 5(6):377-393. https://doi.org/10.3390/s5060377
Chicago/Turabian StyleDiculescu, Victor Constantin, Ana-Maria Chiorcea Paquim, and Ana Maria Oliveira Brett. 2005. "Electrochemical DNA Sensors for Detection of DNA Damage" Sensors 5, no. 6: 377-393. https://doi.org/10.3390/s5060377