
A Microchip for Integrated Single-Cell Gene Expression Profiling and Genotoxicity Detection

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Principle, Materials, and Experimental Setup
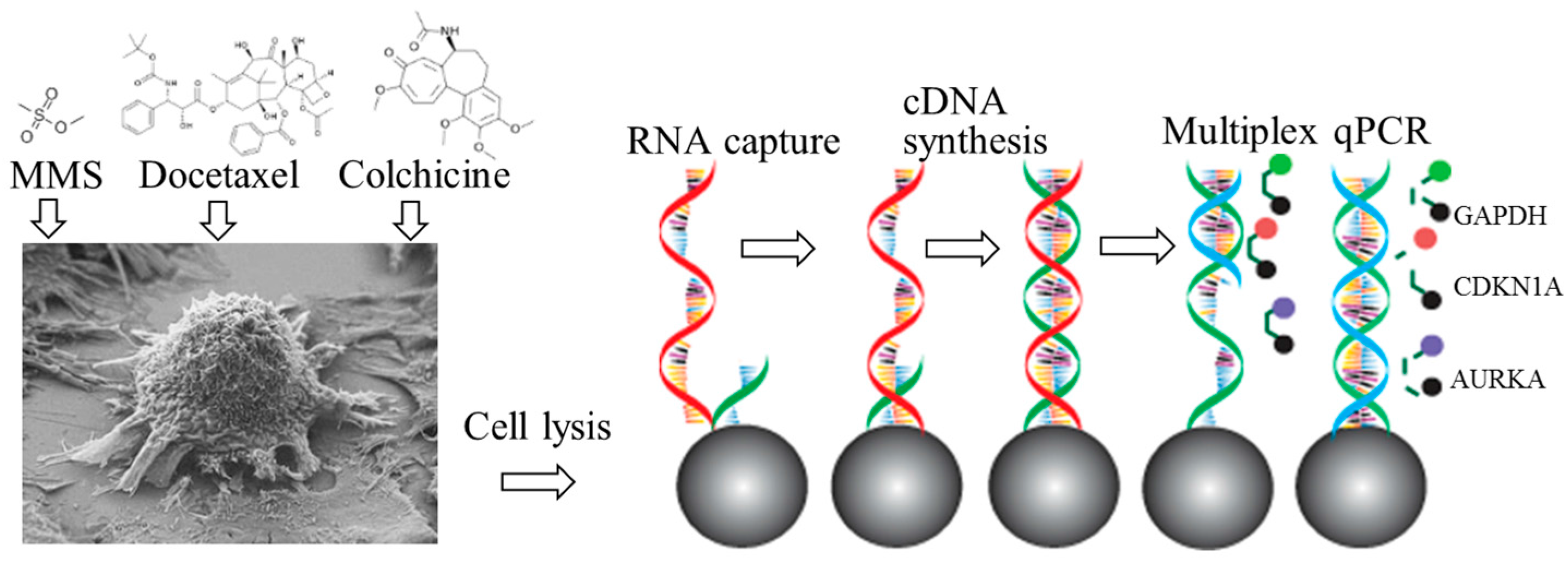
2.1. Principle
2.2. Materials and Experimental Set-up
3. Chip Design, Simulation, and Fabrication
3.1. Design
3.2. Cell Trapping Simulation
3.3. Device Fabrication
4. Results and Discussion
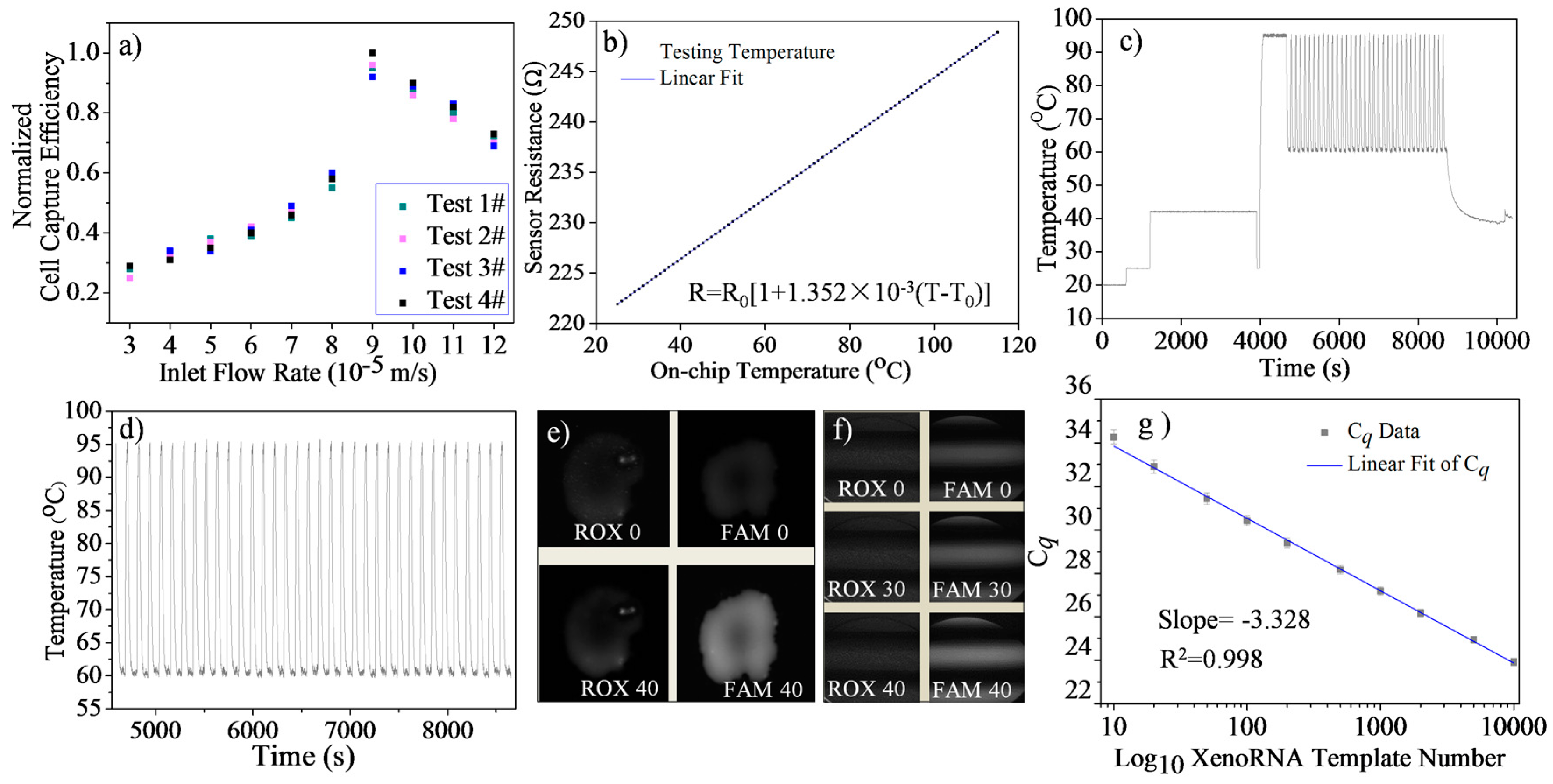
4.1. Single Cell Trapping Test
4.2. Temperature Sensor Calibration
4.3. Temperature Control Evaluation
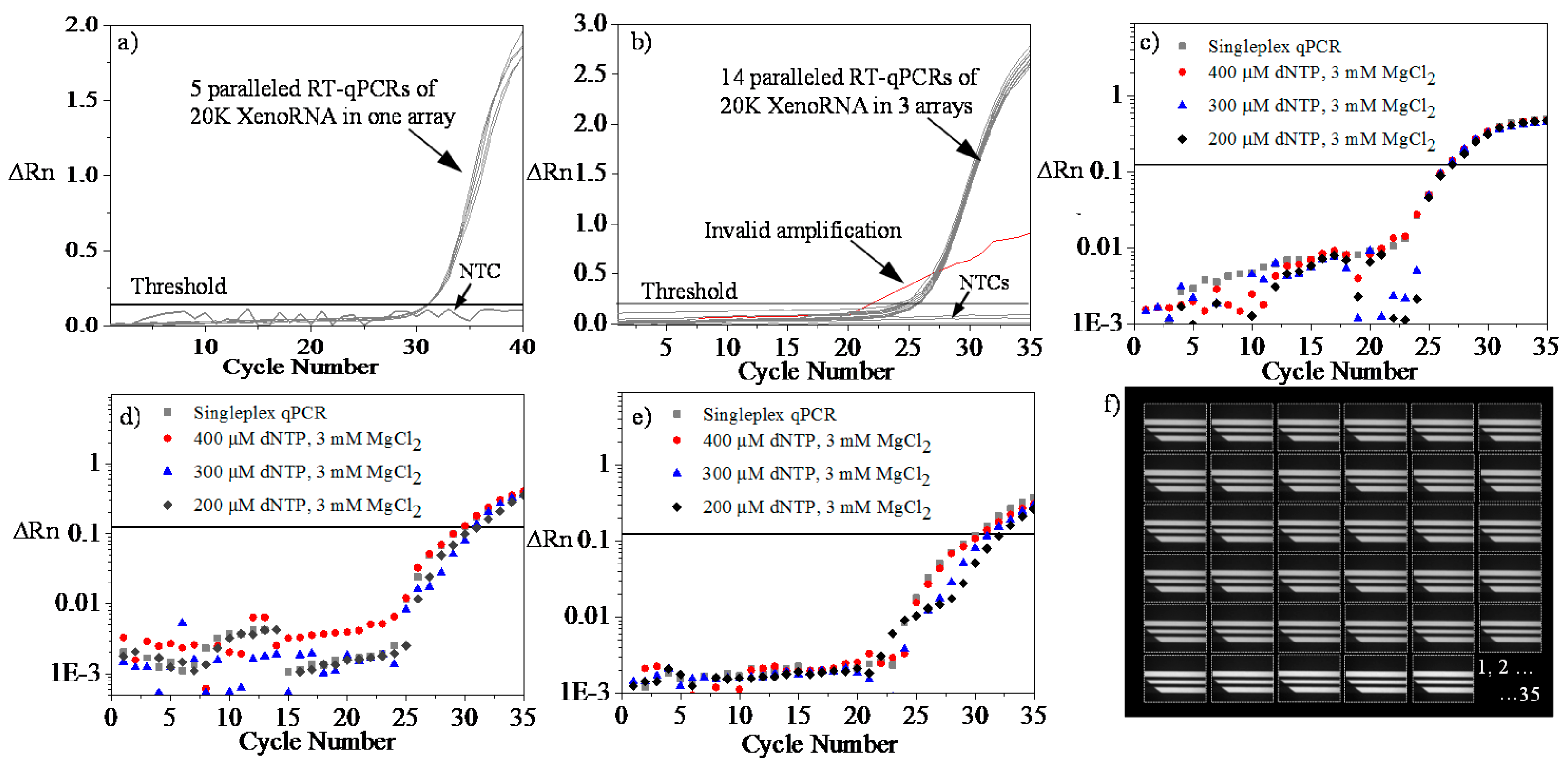
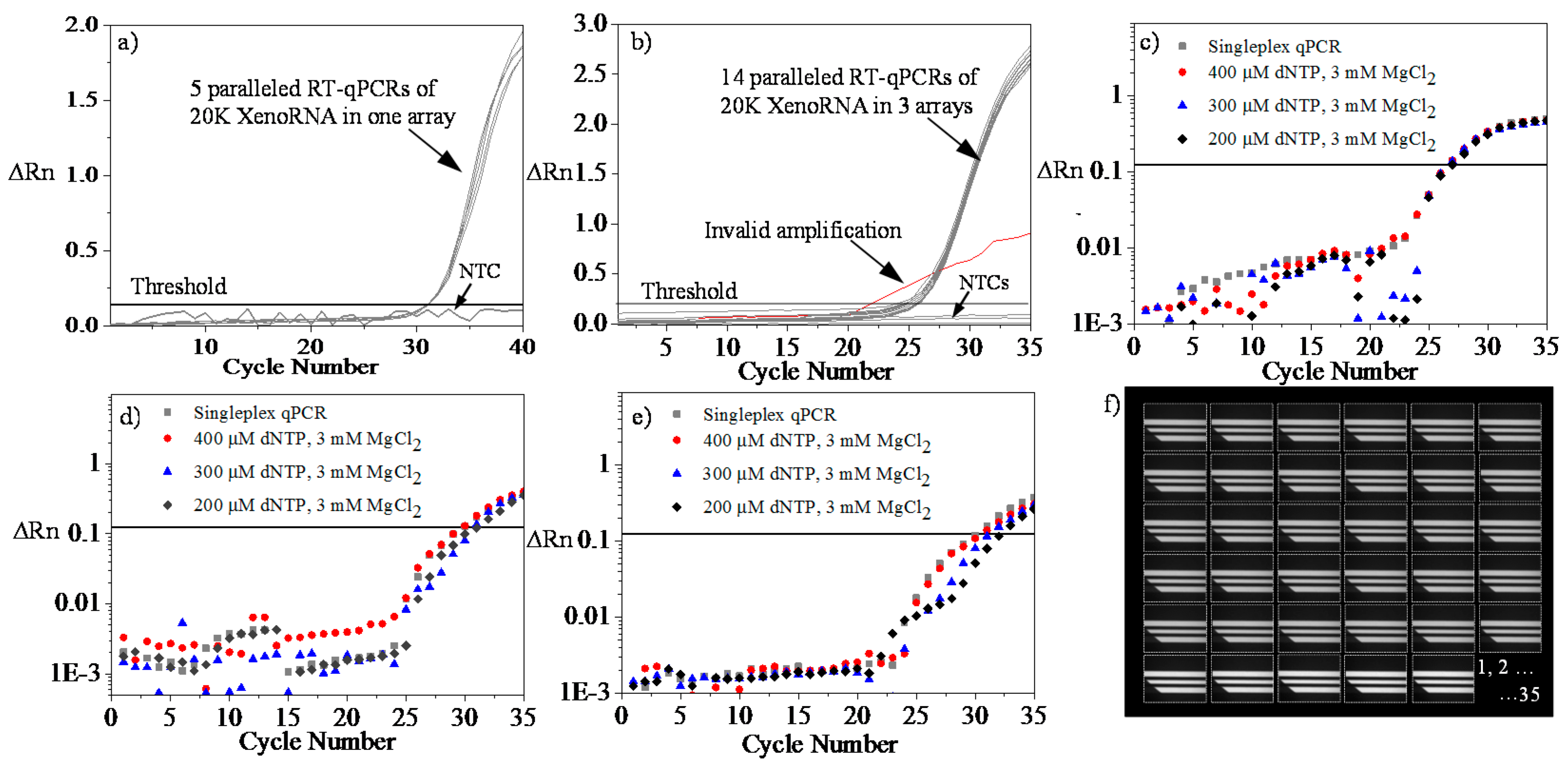
4.4. Microfluidic RT-qPCR Validation using XenoRNA
4.5. Repeatability and Reproducibility Test Using XenoRNA
4.6. Multiplex Amplification
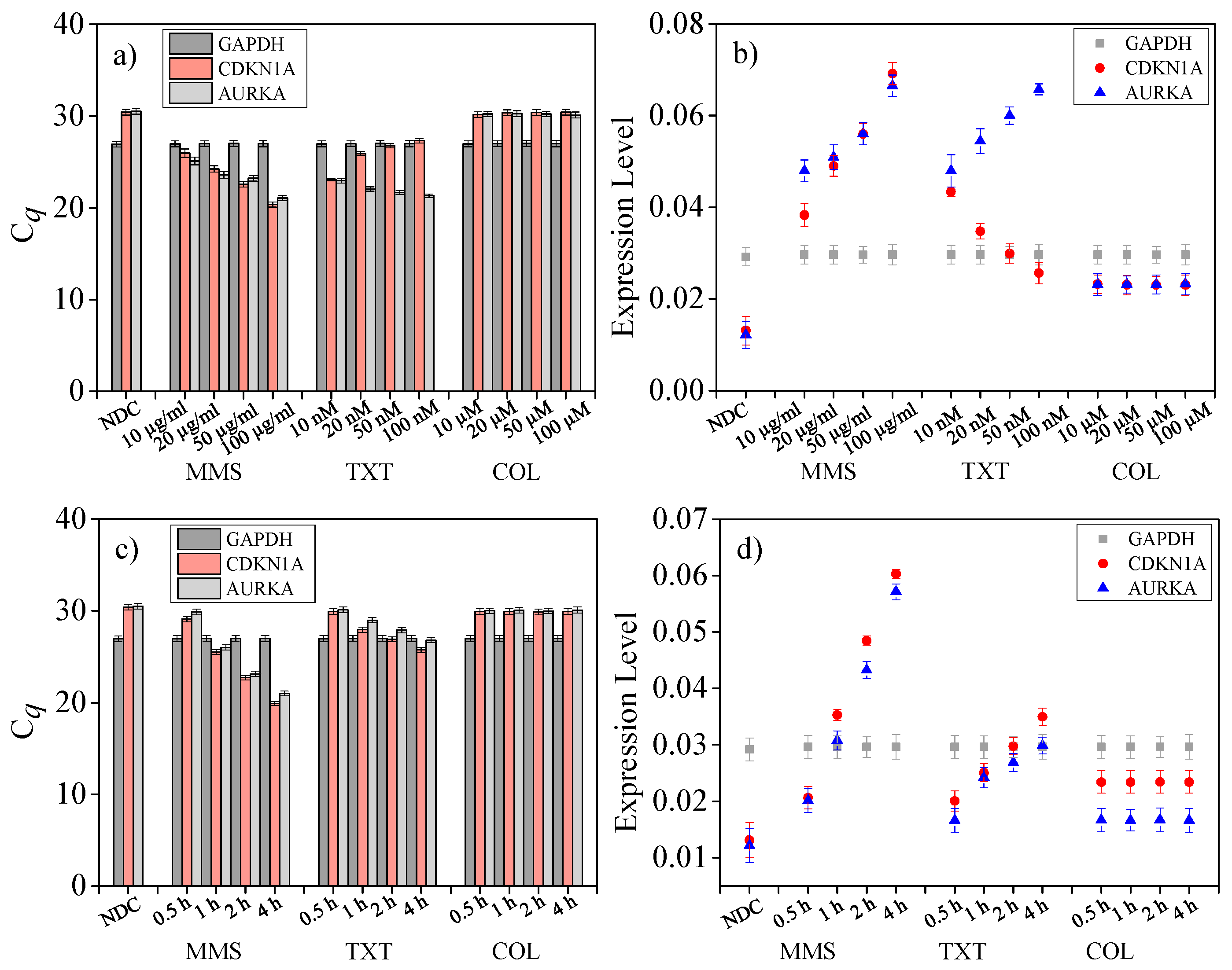
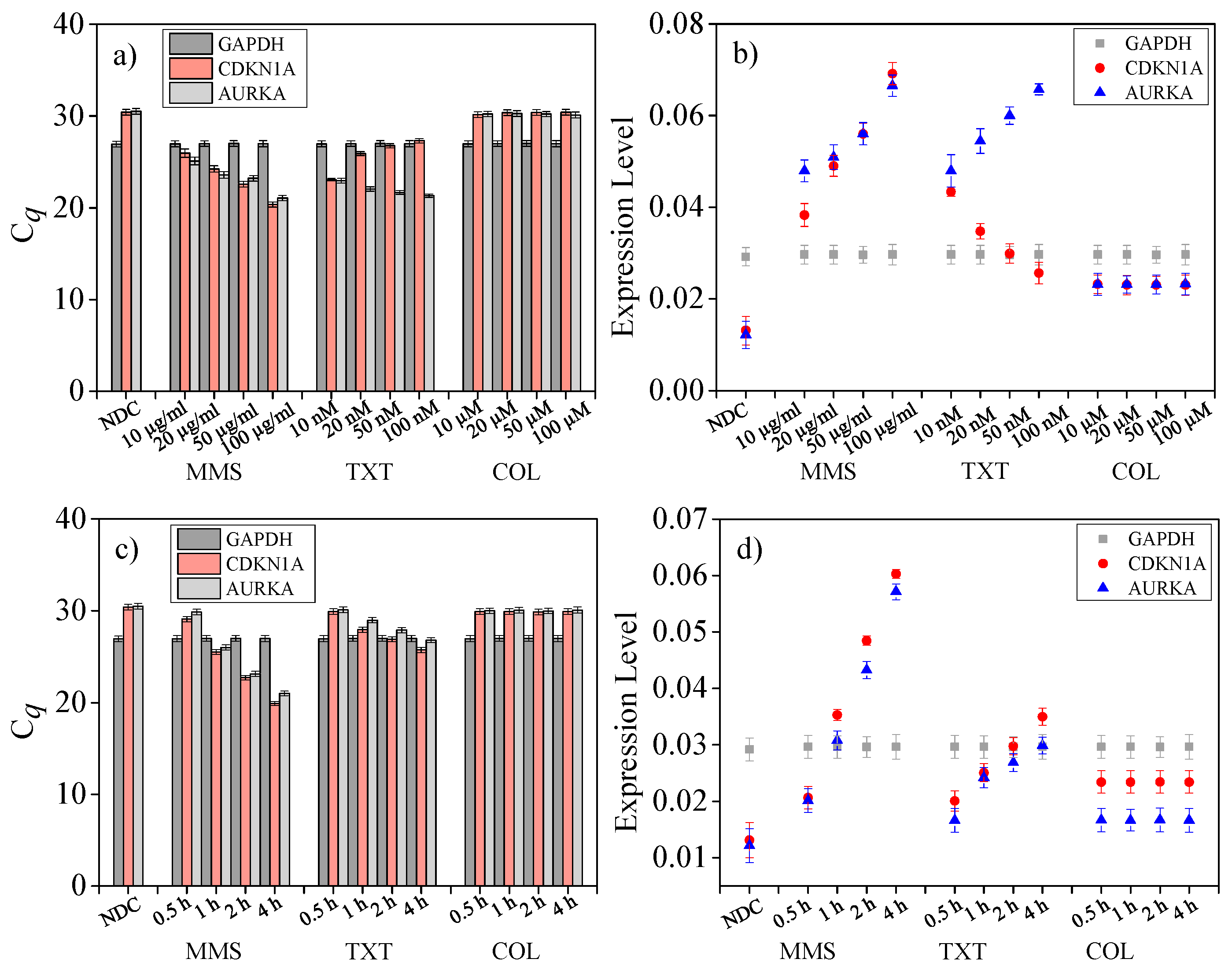
4.7. Drug Dose Assay on Single Cells
4.8. Drug Treat Time Length Assay on Single Cells
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Collins, F.S.; Varmus, H. A new initiative on precision medicine. N. Engl. J. Med. 2015, 372, 793–795. [Google Scholar] [CrossRef] [PubMed]
- Navin, N.E. Cancer genomics: One cell at a time. Genome Biol. 2014, 15. [Google Scholar] [CrossRef] [PubMed]
- Shapiro, E.; Biezuner, T.; Linnarsson, S. Single-cell sequencing-based technologies will revolutionize whole-organism science. Nat. Rev. Genet. 2013, 14, 618–630. [Google Scholar] [CrossRef] [PubMed]
- Gawad, C.; Koh, W.; Quake, S.R. Single-cell genome sequencing: Current state of the science. Nat. Rev. Genet. 2016, 17, 175–188. [Google Scholar] [CrossRef] [PubMed]
- Wills, Q.F.; Livak, K.J.; Tipping, A.J.; Enver, T.; Goldson, A.J.; Sexton, D.W.; Holmes, C. Single-cell gene expression analysis reveals genetic associations masked in whole-tissue experiments. Nat. Biotechnol. 2013, 31, 748–752. [Google Scholar] [CrossRef] [PubMed]
- Ennen, M.; Keime, C.; Kobi, D.; Mengus, G.; Lipsker, D.; Thibault-Carpentier, C.; Davidson, I. Single-cell gene expression signatures reveal melanoma cell heterogeneity. Oncogene 2015, 34, 3251–3263. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.T.; Lee, H.W.; Lee, H.O.; Kim, S.C.; Seo, Y.J.; Chung, W.; Eum, H.H.; Nam, D.H.; Kim, J.; Joo, K.M.; et al. Single-cell mRNA sequencing identifies subclonal heterogeneity in anti-cancer drug responses of lung adenocarcinoma cells. Genome Biol. 2015, 16. [Google Scholar] [CrossRef] [PubMed]
- Powell, A.A.; Talasaz, A.H.; Zhang, H.; Coram, M.A.; Reddy, A.; Deng, G.; Telli, M.L.; Advani, R.H.; Carlson, R.W.; Mollick, J.A.; et al. Single cell profiling of circulating tumor cells: Transcriptional heterogeneity and diversity from breast cancer cell lines. PLoS ONE 2012, 7, e33788. [Google Scholar] [CrossRef] [PubMed]
- Sandler, O.; Mizrahi, S.P.; Weiss, N.; Agam, O.; Simon, I.; Balaban, N.Q. Lineage correlations of single cell division time as a probe of cell-cycle dynamics. Nature 2015, 519, 468–471. [Google Scholar] [CrossRef] [PubMed]
- Almendro, V.; Marusyk, A.; Polyak, K. Cellular heterogeneity and molecular evolution in cancer. Annu. Rev. Pathol. Mech. Dis. 2013, 8, 277–302. [Google Scholar] [CrossRef] [PubMed]
- Janiszewska, M.; Liu, L.; Almendro, V.; Kuang, Y.; Paweletz, C.; Sakr, R.A.; Weigelt, B.; Hanker, A.B.; Chandarlapaty, S.; King, T.A.; et al. In situ single-cell analysis identifies heterogeneity for PIK3CA mutation and HER2 amplification in HER2-positive breast cancer. Nat. Genet. 2015, 47, 1212–1219. [Google Scholar] [CrossRef] [PubMed]
- Ramsköld, D.; Luo, S.; Wang, Y.C.; Li, R.; Deng, Q.; Faridani, O.R.; Daniels, G.A.; Khrebtukova, I.; Loring, J.F.; Laurent, L.C.; et al. Full-length mRNA-Seq from single-cell levels of RNA and individual circulating tumor cells. Nat. Biotechnol. 2012, 30, 777–782. [Google Scholar] [CrossRef] [PubMed]
- Carter, M.G.; Sharov, A.A.; VanBuren, V.; Dudekula, D.B.; Carmack, C.E.; Nelson, C.; Ko, M.S. Transcript copy number estimation using a mouse whole-genome oligonucleotide microarray. Genome Biol. 2005, 6. [Google Scholar] [CrossRef]
- Elvira, K.S.; Solvas, X.S.; Wootton, R.C.R. The past, present and potential for microfluidic reactor technology in chemical synthesis. Nat. Chem. 2013, 5, 905–915. [Google Scholar] [CrossRef] [PubMed]
- Qian, W.; Zhang, Y.; Chen, W. Capturing cancer: Emerging microfluidic technologies for the capture and characterization of circulating tumor cells. Small 2015, 11, 3850–3872. [Google Scholar] [CrossRef] [PubMed]
- Lin, M.; Chen, J.F.; Lu, Y.T.; Zhang, Y.; Song, J.; Hou, S.; Ke, Z.; Tseng, H.R. Nanostructure embedded microchips for detection, isolation, and characterization of circulating tumor cells. Accounts Chem. Res. 2014, 47, 2941–2950. [Google Scholar] [CrossRef] [PubMed]
- Qiu, X.; De Jesus, J.; Pennell, M.; Troiani, M.; Haun, J.B. Microfluidic device for mechanical dissociation of cancer cell aggregates into single cells. Lab Chip 2015, 15, 339–350. [Google Scholar] [CrossRef] [PubMed]
- Ting, D.T.; Wittner, B.S.; Ligorio, M.; Jordan, N.V.; Shah, A.M.; Miyamoto, D.T.; Aceto, N.; Bersani, F.; Brannigan, B.W.; Xega, K.; et al. Single-cell RNA sequencing identifies extracellular matrix gene expression by pancreatic circulating tumor cells. Cell Rep. 2014, 8, 1905–1918. [Google Scholar] [CrossRef] [PubMed]
- Miyamoto, D.T.; Zheng, Y.; Wittner, B.S.; Lee, R.J.; Zhu, H.; Broderick, K.T.; Desai, R.; Fox, D.B.; Brannigan, B.W.; Trautwein, J.; et al. RNA-Seq of single prostate CTCs implicates noncanonical Wnt signaling in antiandrogen resistance. Science 2015, 349, 1351–1356. [Google Scholar] [CrossRef] [PubMed]
- Rival, A.; Jary, D.; Delattre, C.; Fouillet, Y.; Castellan, G.; Bellemin-Comte, A.; Gidrol, X. An EWOD-based microfluidic chip for single-cell isolation, mRNA purification and subsequent multiplex qPCR. Lab Chip 2014, 19, 3739–3749. [Google Scholar] [CrossRef] [PubMed]
- Stumpf, F.; Schoendube, J.; Gross, A.; Rath, C.; Niekrawietz, S.; Koltay, P.; Roth, G. Single-cell PCR of genomic DNA enabled by automated single-cell printing for cell isolation. Biosens. Bioelectron. 2015, 69, 301–306. [Google Scholar] [CrossRef] [PubMed]
- Sardesai, N.P.; Kadimisetty, K.; Faria, R.; Rusling, J.F. A microfluidic electrochemiluminescent device for detecting cancer biomarker proteins. Anal. Bioanal. Chem. 2013, 405, 3831–3838. [Google Scholar] [CrossRef] [PubMed]
- Blazek, M.; Santisteban, T.S.; Zengerle, R.; Meier, M. Analysis of fast protein phosphorylation kinetics in single cells on a microfluidic chip. Lab Chip 2015, 15, 726–734. [Google Scholar] [CrossRef] [PubMed]
- Thege, F.I.; Lannin, T.B.; Saha, T.N.; Tsai, S.; Kochman, M.L.; Hollingsworth, M.A.; Rhim, A.D.; Kirby, B.J. Microfluidic immunocapture of circulating pancreatic cells using parallel EpCAM and MUC1 capture: characterization, optimization and downstream analysis. Lab Chip 2014, 10, 1775–1784. [Google Scholar] [CrossRef] [PubMed]
- Pratt, E.D.; Huang, C.; Hawkins, B.G.; Gleghorn, J.P.; Kirby, B.J. Rare cell capture in microfluidic devices. Chem. Eng. Sci. 2011, 66, 1508–1522. [Google Scholar] [CrossRef] [PubMed]
- Gossett, D.R.; Weaver, W.M.; Mach, A.J.; Hur, S.C.; Tse, H.T.K.; Lee, W.; Amini, H.; Di Carlo, D. Label-free cell separation and sorting in microfluidic systems. Anal. Bioanal. Chem. 2010, 397, 3249–3267. [Google Scholar] [CrossRef] [PubMed]
- Sarioglu, A.F.; Aceto, N.; Kojic, N.; Donaldson, M.C.; Zeinali, M.; Hamza, B.; Engstrom, A.; Zhu, H.; Sundaresan, T.K.; et al. A microfluidic device for label-free, physical capture of circulating tumor cell clusters. Nat. Methods 2015, 12, 685–691. [Google Scholar] [CrossRef] [PubMed]
- Volpatti, L.R.; Yetisen, A.K. Commercialization of microfluidic devices. Trends Biotechnol. 2014, 32, 347–350. [Google Scholar] [CrossRef] [PubMed]
- Yin, H.; Marshall, D. Microfluidics for single cell analysis. Curr. Opin. Biotech. 2012, 23, 110–119. [Google Scholar] [CrossRef] [PubMed]
- Citri, A.; Pang, Z.P.; Südhof, T.C.; Wernig, M.; Malenka, R.C. Comprehensive qPCR profiling of gene expression in single neuronal cells. Nat. Prot. 2012, 7, 118–127. [Google Scholar] [CrossRef] [PubMed]
- White, A.K.; VanInsberghe, M.; Petriv, I.; Hamidi, M.; Sikorski, D.; Marra, M.A.; Piret, J.; Aparicio, S.; Hansen, C.L. High-throughput microfluidic single-cell RT-qPCR. Proc. Natl. Acad. Sci. USA 2011, 108, 13999–14004. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.; Olsen, T.; Zhu, J.; Tao, J.; Ponnaiya, B.; Amundson, S.A.; Brenner, D.J.; Lin, Q. A bead-based microfluidic approach to integrated single-cell gene expression analysis by quantitative RT-PCR. RSC Adv. 2015, 5, 4886–4893. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.; Olsen, T.; Zhu, J.; Tao, J.; Ponnaiya, B.; Amundson, S.A.; Brenner, D.J.; Lin, Q. A microfluidic approach to parallelized transcriptional profiling of single cells. Microfluid. Nanofluid. 2015, 19, 1429–1440. [Google Scholar] [CrossRef] [PubMed]
- Sun, H. A multi-layer microchip for high-throughput single-cell gene expression profiling. Anal. Biochem. 2016, 508, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Ellinger-Ziegelbauer, H.; Fostel, J.M.; Aruga, C.; Bauer, D.; Boitier, E.; Deng, S.; Dickinson, D.; Le Fevre, A.C.; Fornace, A.J.; Grenet, O.; et al. Characterization and interlaboratory comparison of a gene expression signature for differentiating genotoxic mechanisms. Toxicol. Sci. 2009. [Google Scholar] [CrossRef] [PubMed]
- Lee, W.J.; Kim, S.C.; Lee, S.J.; Lee, J.; Park, J.H.; Yu, K.S.; Lim, J.; Kwon, S.W. Investigating the different mechanisms of genotoxic and non-genotoxic carcinogens by a gene set analysis. PLoS ONE 2014, 9, e86700. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.-S.J.; Haga, J.H.; Chien, S. Molecular basis of the effects of shear stress on vascular endothelial cells. J. Biomech. 2015, 38, 1949–1971. [Google Scholar] [CrossRef] [PubMed]
- Rutkowski, J.M.; Swartz, M.A. A driving force for change: Interstitial flow as a morphoregulator. Trends Cell Biol. 2007, 17, 44–50. [Google Scholar] [CrossRef] [PubMed]
- Unger, M.A.; Chou, H.P.; Thorsen, T.; Scherer, A.; Quake, S.R. Monolithic microfabricated valves and pumps by multilayer soft lithography. Science 2000, 288, 113–116. [Google Scholar] [CrossRef] [PubMed]
- Nilsson, J.; Evander, M.; Hammarström, B.; Laurell, T. Review of cell and particle trapping in microfluidic systems. Anal. Chim. Acta. 2009, 649, 141–157. [Google Scholar] [CrossRef] [PubMed]
- Karimi, A.; Yazdi, S.; Ardekani, A.M. Hydrodynamic mechanisms of cell and particle trapping in microfluidics. Biomicrofluidics 2013, 7, 021501. [Google Scholar] [CrossRef] [PubMed]
- Bustin, S.A.; Benes, V.; Garson, J.A.; Hellemans, J.; Huggett, J.; Kubista, M.; Mueller, R.; Nolan, T.; Pfaffl, M.W.; Shipley, G.L.; Vandesompele, J. The MIQE guidelines: Minimum information for publication of quantitative real-time PCR experiments. Clin. Chem. 2009, 55, 611–622. [Google Scholar] [CrossRef] [PubMed]
- Marinov, G.K.; Williams, B.A.; McCue, K.; Schroth, G.P.; Gertz, J.; Myers, R.M.; Wold, B.J. From single-cell to cell-pool transcriptomes: Stochasticity in gene expression and RNA splicing. Genome Res. 2014, 24, 496–510. [Google Scholar] [CrossRef] [PubMed]
- Lundin, C.; North, M.; Erixon, K.; Walters, K.; Jenssen, D.; Goldman, A.S.; Helleday, T. Methyl methanesulfonate (MMS) produces heat-labile DNA damage but no detectable in vivo DNA double-strand breaks. Nucleic Acids Res. 2005, 33, 3799–3811. [Google Scholar] [CrossRef] [PubMed]
- Ocker, M.; Schneider-Stock, R. Histone deacetylase inhibitors: Signalling towards p21cip1/waf1. Int. J. Biochem. Cell Biol. 2007, 39, 1367–1374. [Google Scholar] [CrossRef] [PubMed]
- Hernández-Vargas, H.; Palacios, J.; Moreno-Bueno, G. Molecular profiling of docetaxel cytotoxicity in breast cancer cells: Uncoupling of aberrant mitosis and apoptosis. Oncogene 2007, 26, 2902–2913. [Google Scholar] [CrossRef] [PubMed]
- List of Cancer Drugs. Available online: http://www.cancer.gov/about-cancer/treatment/drugs/docetaxel (accessed on 5 October 2006).
- Kruman, I.I.; Wersto, R.P.; Cardozo-Pelaez, F.; Smilenov, L.; Chan, S.L.; Chrest, F.J.; Emokpae, R.; Gorospe, M.; Mattson, M.P. Cell cycle activation linked to neuronal cell death initiated by DNA damage. Neuron 2004, 41, 549–561. [Google Scholar] [CrossRef]
- Hata, T.; Furukawa, T.; Sunamura, M.; Egawa, S.; Motoi, F.; Ohmura, N.; Marumoto, T.; Saya, H.; Horii, A. RNA interference targeting aurora kinase a suppresses tumor growth and enhances the taxane chemosensitivity in human pancreatic cancer cells. Cancer Res. 2005, 65, 2899–2905. [Google Scholar] [CrossRef] [PubMed]



© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dong, H.; Sun, H. A Microchip for Integrated Single-Cell Gene Expression Profiling and Genotoxicity Detection. Sensors 2016, 16, 1489. https://doi.org/10.3390/s16091489
Dong H, Sun H. A Microchip for Integrated Single-Cell Gene Expression Profiling and Genotoxicity Detection. Sensors. 2016; 16(9):1489. https://doi.org/10.3390/s16091489
Chicago/Turabian StyleDong, Hui, and Hao Sun. 2016. "A Microchip for Integrated Single-Cell Gene Expression Profiling and Genotoxicity Detection" Sensors 16, no. 9: 1489. https://doi.org/10.3390/s16091489
APA StyleDong, H., & Sun, H. (2016). A Microchip for Integrated Single-Cell Gene Expression Profiling and Genotoxicity Detection. Sensors, 16(9), 1489. https://doi.org/10.3390/s16091489