Recent Applications of Fluorescence Recovery after Photobleaching (FRAP) to Membrane Bio-Macromolecules

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. GUVs (Giant Unilamellar Vesicles)
2.2. The Biomimetic Sponge Phase
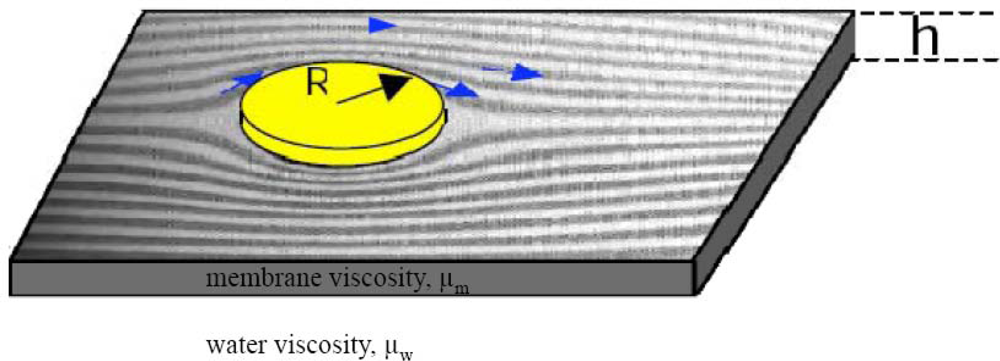
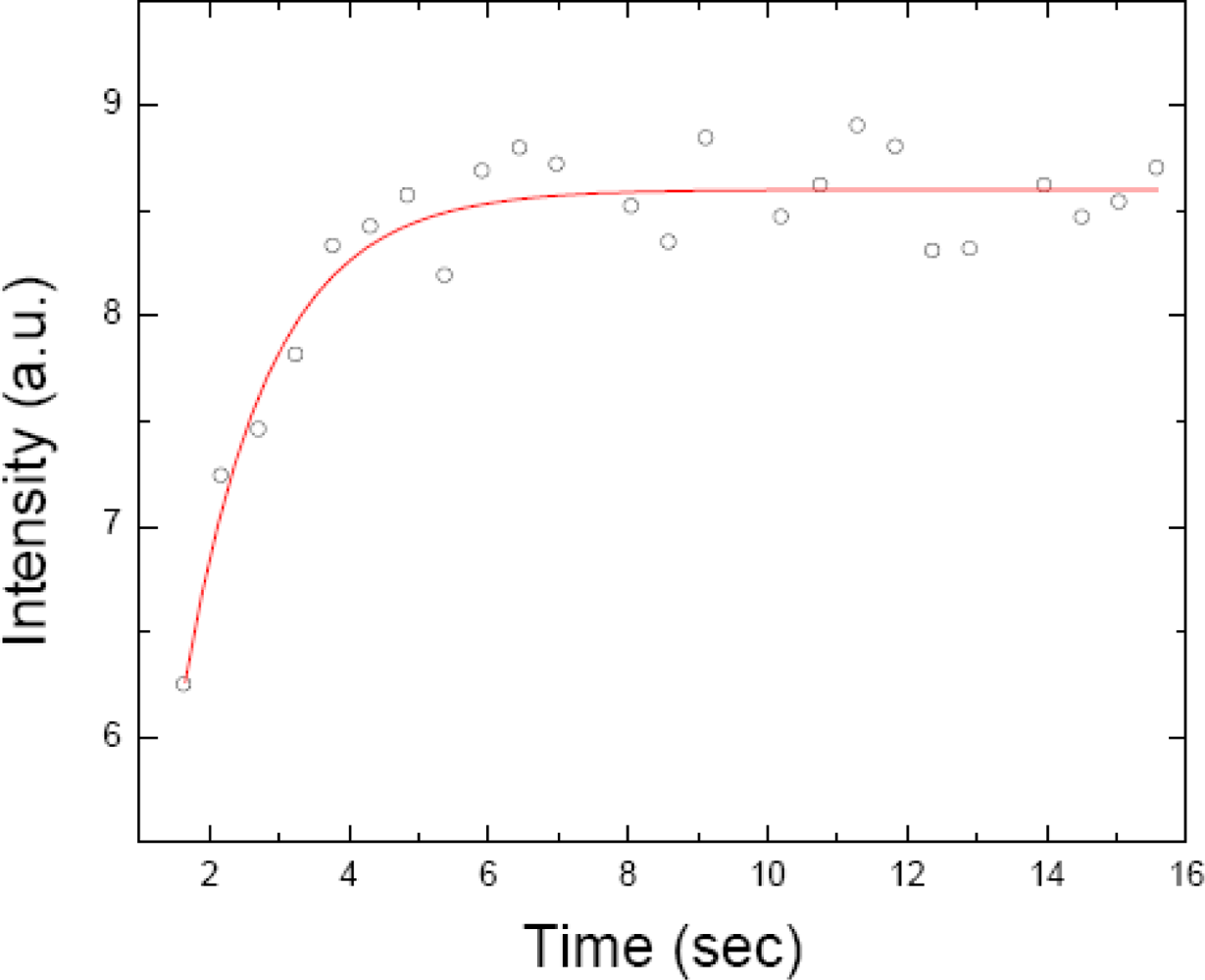
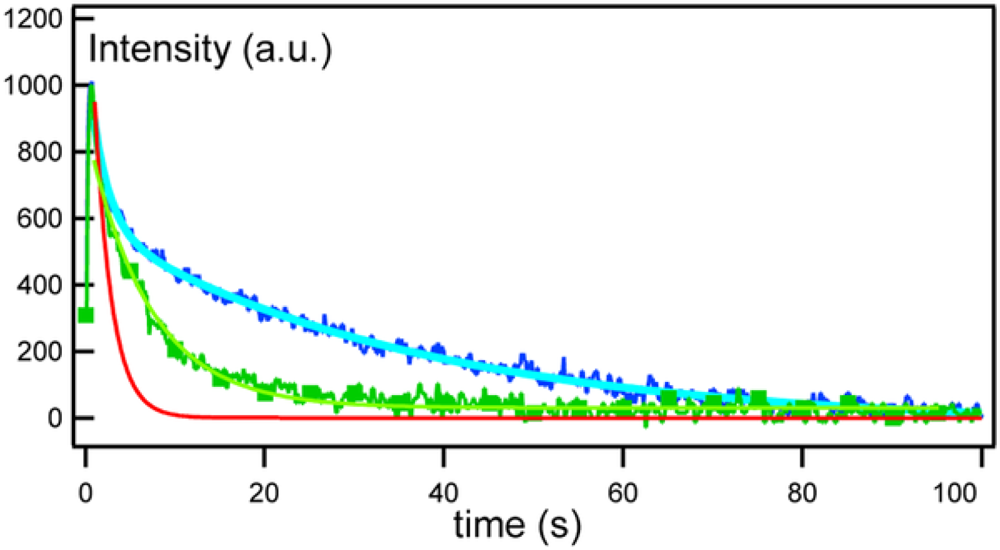
2.3. FRAP (Fluorescence Recovery after Photobleaching)
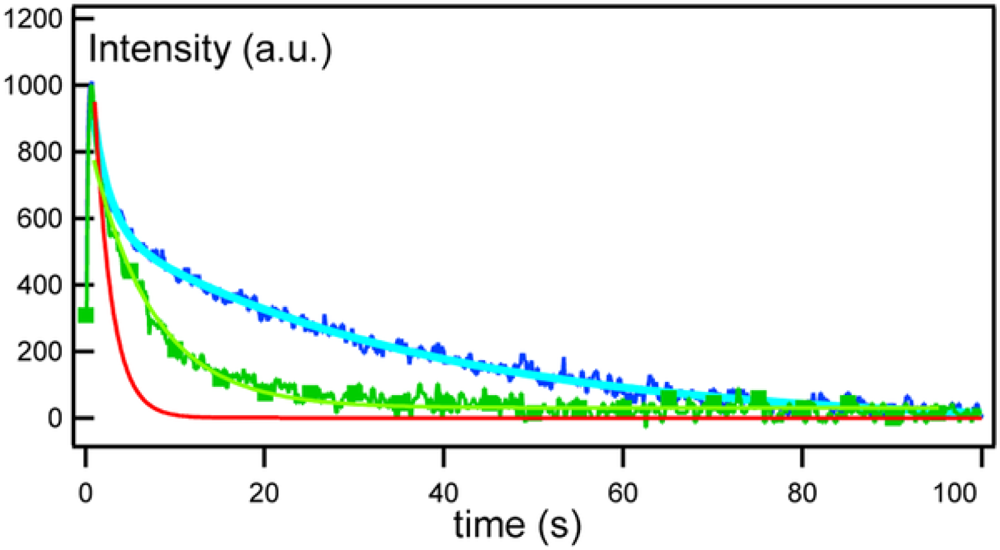
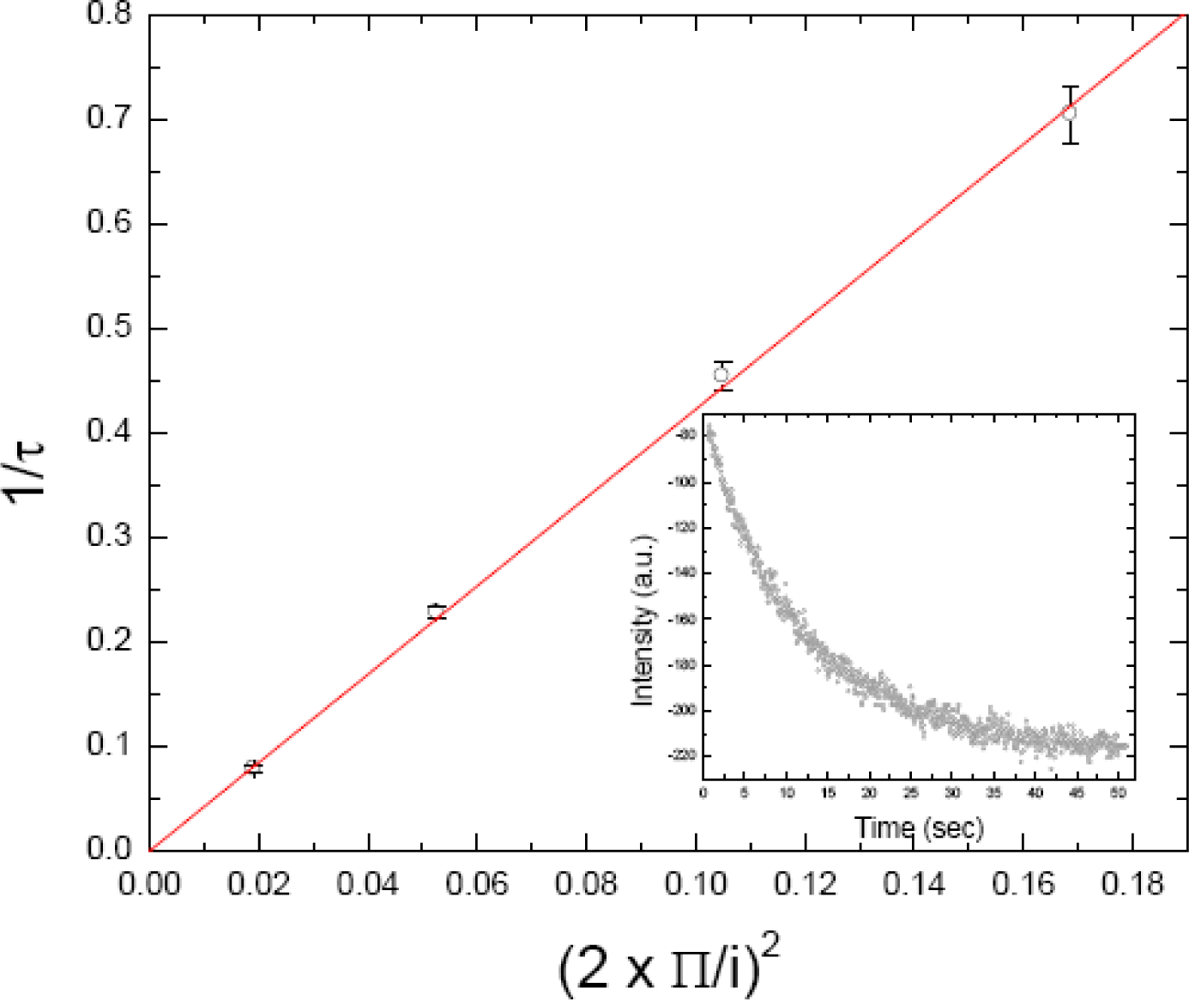
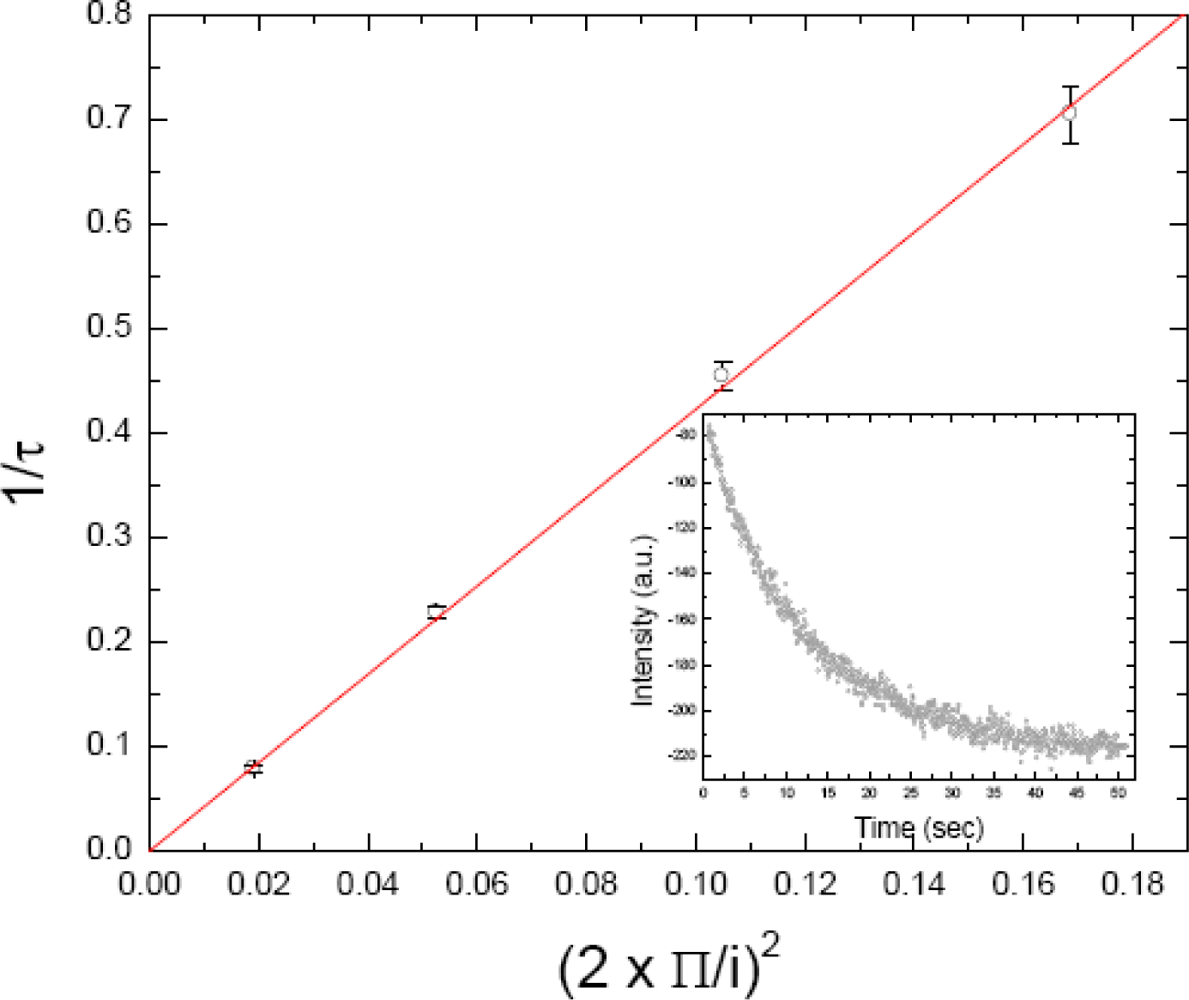
2.4. FRAPP (Fluorescence Recovery after Pattern Photobleaching)
3. Some Recent Studies
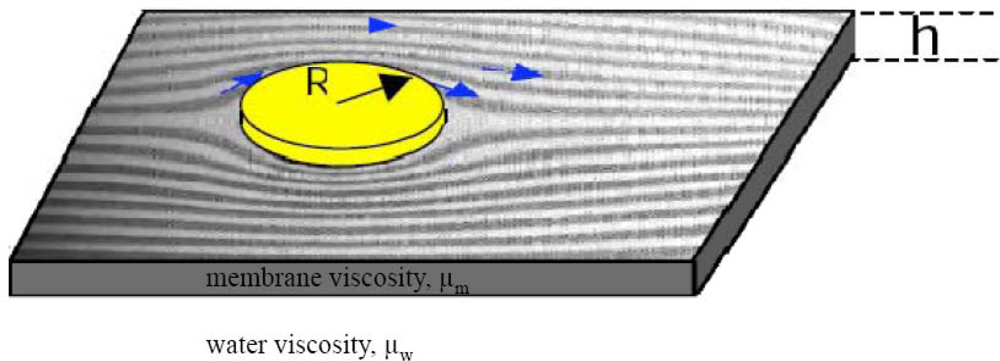
3.1. Lateral Diffusion of Membrane Proteins
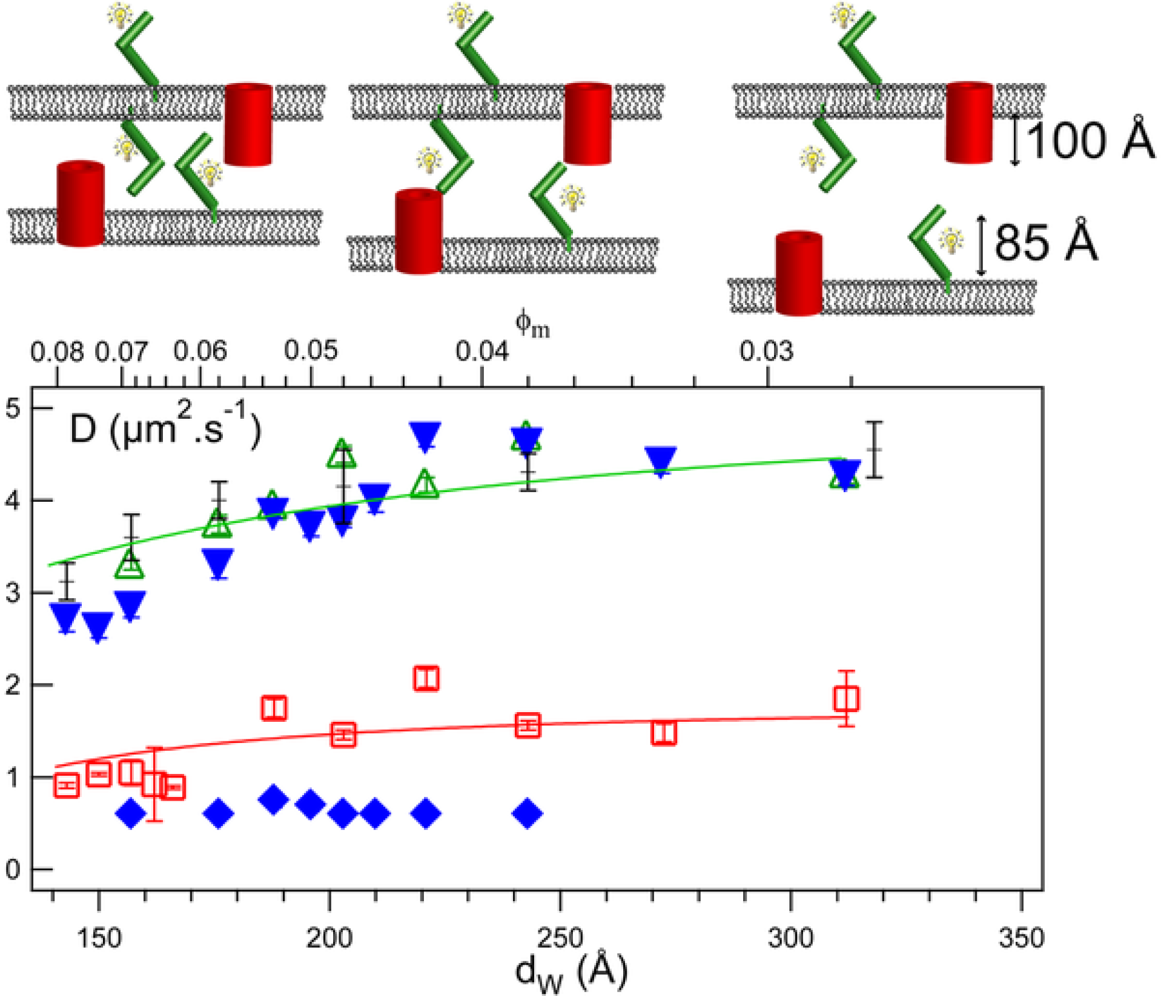
3.2. Diffusion of Lipids and Membrane Proteins under Membrane Curvature Gradients
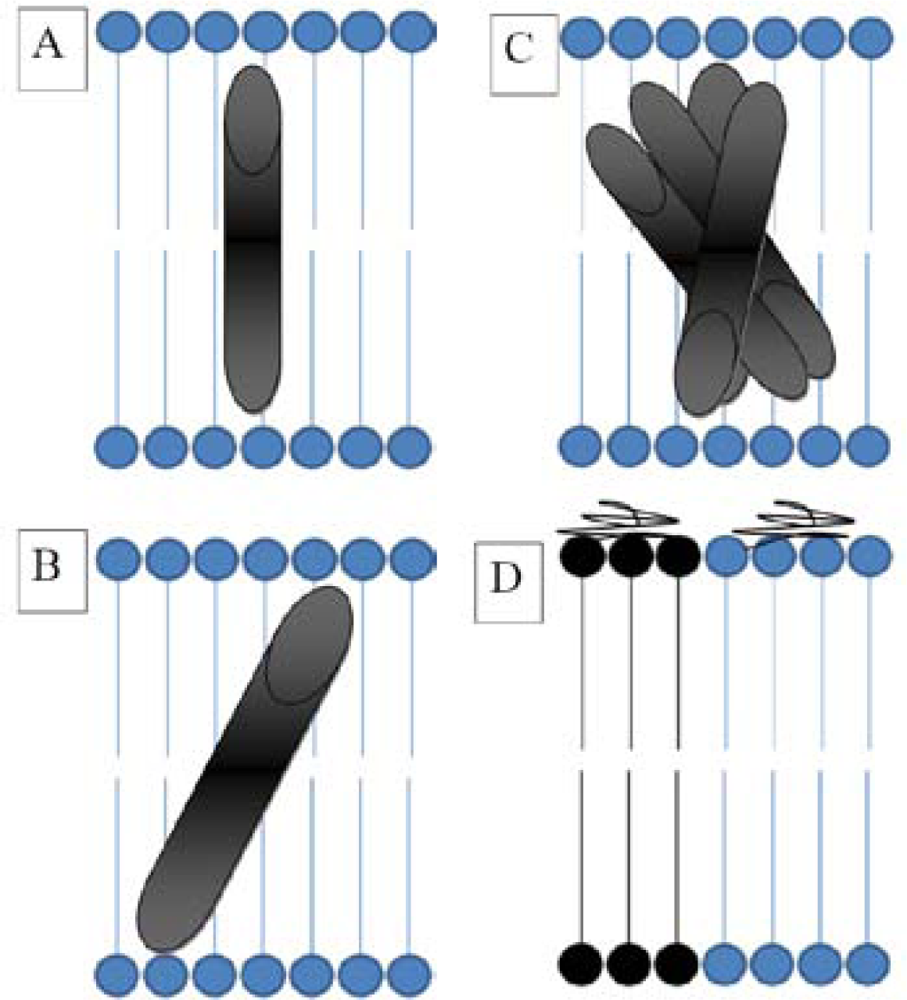
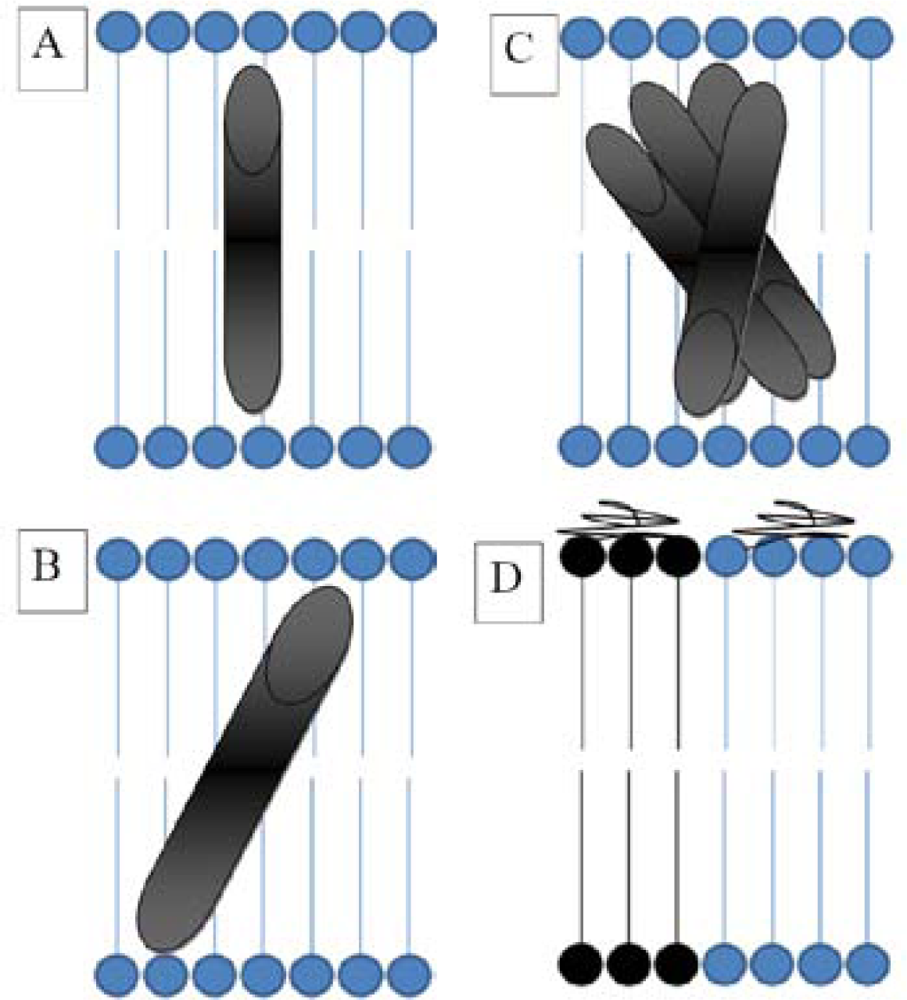
3.3. Hydrophobic Mismatch
3.4. Membrane Protein Association
3.5. Thylakoid Membrane Proteins
3.6. Green Fluorescent Protein (GFP) Diffusion in Escherichia Coli
3.7. Polyglutamine Pathogenesis in Caenorhabditis elegans and Disassembly of β-Amyloids
3.8. Lateral Diffusion of DNA and the Effect of Lipid Domains/Rafts
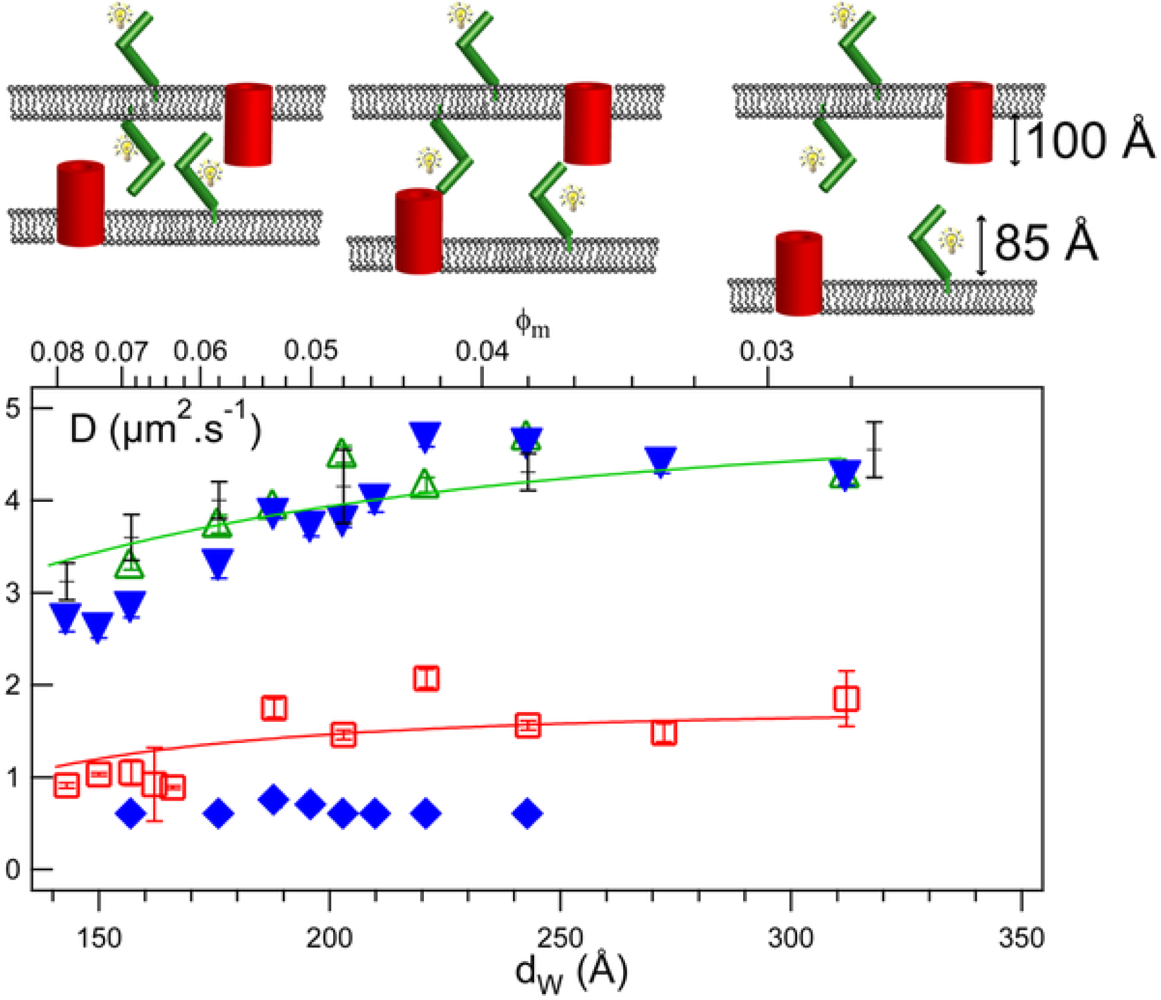
3.9. Membrane Confinement and Fluidity
4. Comparison with Some Alternative Methods
4.1. FCS (Fluorescence Correlation Spectroscopy)
4.2. SPT (Single Particle Tracking)
5. Conclusions and Future Perspectives
Abbreviations:
| FRAP | fluorescence recovery after photobleaching |
| FRAPP | fluorescence recovery after fringe-pattern photobleaching |
| FCS | fluorescence correlation spectroscopy |
| SPT | single particle tracking |
| D | diffusion coefficient |
| R | radius |
| SD | Saffman Delbrück |
| GUV | giant unilamellar vesicle |
| FFEM | freeze fracture electron microscopy |
| SAXS | small angle X-ray scattering |
| SANS | small angle neutron scattering |
| FITC | fluorescein isothyocyanate |
| MHC | major histocompatibility complex |
| DiOC16 | 3.3′-dihexadecyloxacarbocyanine iodide |
| CTB | cholera toxin subunit B |
| σ | surface tension |
| ATR-FTIR | attenuated total reflectance-Fourier transform infra red |
| Lx | a poly-leucine peptide where x is the number of leucine residues |
| dw | intermembrane distance |
| GFP | green fluorescent protein, |
| Q | glutamine |
| YFP | yellow fluorescent protein |
| Aβ | β-amyloid peptide |
| DSPC | distearoylphosphatidylcholine |
| DOATP | 1,2-dioleoyl-3-trimethylammonium propane |
| NBD-PE | N-(7-nitrobenz-2-oxa-1,3-diazol-4-yl)-1,2-dihexadecanoyl-sn-glycero-3-phospho-ethanolamine |
| BSA | bovine serum albumin |
| C12E5 | pentaethylene glycol monododecyl ether AOT/ aerosol OT - sodium 1,4-bis(2-ethyl-hexoxy)-1,4-dioxobutane-2-sulfonate |
| ICS | image correlation spectroscopy |
| CFTR | cystic fibrosis conductance regulator |
Acknowledgments
References
- Singer, S.J.; Nicolson, G.L. The fluid mosaic model of the structure of cell membranes. Science 1972, 175, 720–731. [Google Scholar]
- Gorter, E.; Grendel, F. On bimolecular layers of lipoids on the chromocytes of the blood. J. Exp. Med 1925, 41, 439–443. [Google Scholar]
- Danielli, J.F.; Davson, H. A contribution to the theory of permeability of thin films. J. Cell. Comp. Physiol 1935, 5, 495–508. [Google Scholar]
- Mouritsen, O.G.; Bloom, M. Mattress model of lipid-protein interactions in membranes. Biophys. J 1984, 46, 141–153. [Google Scholar]
- Johannsson, A.; Keightley, C.A.; Smith, G.A.; Richards, C.D.; Hesketh, T.R.; Metcalfe, J.C. The effect of bilayer thickness and n-alkanes on the activity of the (Ca2++ Mg2+)-dependent ATPase of sarcoplasmic reticulum. J. Biol. Chem 1981, 256, 1643–1650. [Google Scholar]
- Caffrey, M.; Feigenson, G.W. Fluorescence quenching in model membranes. 3. Relationship between calcium adenosinetriphosphatase enzyme activity and the affinity of the protein for phosphatidylcholines with different acyl chain characteristics. Biochemistry 1981, 20, 1949–1961. [Google Scholar]
- Dumas, F.; Tocanne, J.F.; Leblanc, G.; Lebrun, M.C. Consequences of hydrophobic mismatch between lipids and melibiose permease on melibiose transport. Biochemistry 2000, 39, 4846–4854. [Google Scholar]
- Cornelius, F. Modulation of Na, K-ATPase and Na-ATPase activity by phospholipids and cholesterol. I. Steady-state kinetics. Biochemistry 2001, 40, 8842–8851. [Google Scholar]
- Munro, S. An investigation of the role of transmembrane domains in Golgi protein retention. EMBO J 1995, 14, 4695–4704. [Google Scholar]
- Andersen, O.S.; Koeppe, R.E., 2nd. Bilayer thickness and membrane protein function: an energetic perspective. Annu. Rev. Biophys. Biomol. Struct 2007, 36, 107–130. [Google Scholar]
- Heimburg, T. Thermal Biophysics of Membrane; WILEY-VCH: Weinheim, Germany, 2007. [Google Scholar]
- Luckey, M. Membrane Structural Biology: With Biochemical and Biophysical Foundations; Cambridge University Press: Cambridge, UK, 2008. [Google Scholar]
- Mouritsen, O.G. Life-As a Matter of Fat; Springer-Verlag: Berlin, Germany, 2005. [Google Scholar]
- Shechter, E. Biochimie et Biophysique des Membranes, 2nd ed; DUNOD: Paris, France, 2004. [Google Scholar]
- Cherry, R.J. Membrane Protein Dynamics: Rotational Dynamics. In The Structure of Biological Membranes, 2nd ed; Yeagle, P., Ed.; CRC Press: Boca Raton, FL, USA, 2004; pp. 389–409. [Google Scholar]
- Peters, R.; Cherry, R.J. Lateral and rotational diffusion of bacteriorhodopsin in lipid bilayers: experimental test of the Saffman-Delbruck equations. Proc. Natl. Acad. Sci. USA 1982, 79, 4317–4321. [Google Scholar]
- Fooksman, D.R.; Edidin, M.; Barisas, B.G. Measuring rotational diffusion of MHC class I on live cells by polarized FPR. Biophys. Chem 2007, 130, 10–16. [Google Scholar]
- Frye, L.D.; Edidin, M. The rapid intermixing of cell surface antigens after formation of mouse-human heterokaryons. J. Cell Sci 1970, 7, 319–335. [Google Scholar]
- Saffman, P.G.; Delbruck, M. Brownian motion in biological membranes. Proc. Natl. Acad. Sci. USA 1975, 72, 3111–3113. [Google Scholar]
- Chen, Y.; Lagerholm, B.C.; Yang, B.; Jacobson, K. Methods to measure the lateral diffusion of membrane lipids and proteins. Methods 2006, 39, 147–153. [Google Scholar]
- Marguet, D.; Lenne, P.F.; Rigneault, H.; He, H.T. Dynamics in the plasma membrane: how to combine fluidity and order. EMBO J 2006, 25, 3446–3457. [Google Scholar]
- Destainville, N.; Dumas, F.; Salome, L. What do diffusion measurements tell us about membrane compartmentalisation? Emergence of the role of interprotein interactions. J. Chem. Biol 2008, 1, 37–48. [Google Scholar]
- Bates, I.R.; Wiseman, P.W.; Hanrahan, J.W. Investigating membrane protein dynamics in living cells. Biochem. Cell Biol 2006, 84, 825–831. [Google Scholar]
- Owen, D.M.; Williamson, D.; Rentero, C.; Gaus, K. Quantitative microscopy: protein dynamics and membrane organisation. Traffic 2009, 10, 962–971. [Google Scholar]
- Gambin, Y.; Lopez-Esparza, R.; Reffay, M.; Sierecki, E.; Gov, N.S.; Genest, M.; Hodges, R.S.; Urbach, W. Lateral mobility of proteins in liquid membranes revisited. Proc. Natl. Acad. Sci. USA 2006, 103, 2098–2102. [Google Scholar]
- Ramadurai, S.; Holt, A.; Krasnikov, V.; van den Bogaart, G.; Killian, J.A.; Poolman, B. Lateral diffusion of membrane proteins. J. Am. Chem. Soc 2009, 131, 12650–12656. [Google Scholar]
- Kriegsmann, J.; Gregor, I.; von der Hocht, I.; Klare, J.; Engelhard, M.; Enderlein, J.; Fitter, J. Translational diffusion and interaction of a photoreceptor and its cognate transducer observed in giant unilamellar vesicles by using dual-focus FCS. Chembiochem 2009, 10, 1823–1829. [Google Scholar]
- Korlach, J.; Schwille, P.; Webb, W.W.; Feigenson, G.W. Characterization of lipid bilayer phases by confocal microscopy and fluorescence correlation spectroscopy. Proc. Natl. Acad. Sci. USA 1999, 96, 8461–8466. [Google Scholar]
- Doeven, M.K.; Folgering, J.H.; Krasnikov, V.; Geertsma, E.R.; van den Bogaart, G.; Poolman, B. Distribution, lateral mobility and function of membrane proteins incorporated into giant unilamellar vesicles. Biophys. J 2005, 88, 1134–1142. [Google Scholar]
- Rodriguez, N.; Pincet, F.; Cribier, S. Giant vesicles formed by gentle hydration and electroformation: a comparison by fluorescence microscopy. Colloids Surf. B: Biointerfaces 2005, 42, 125–130. [Google Scholar]
- Reffay, M.; Gambin, Y.; Benabdelhak, H.; Phan, G.; Taulier, N.; Ducruix, A.; Hodges, R.S.; Urbach, W. Tracking membrane protein association in model membranes. PLoS One 2009, 4, e5035. [Google Scholar]
- Soumpasis, D.M. Theoretical analysis of fluorescence photobleaching recovery experiments. Biophys. J 1983, 41, 95–97. [Google Scholar]
- Braeckmans, K.; Peeters, L.; Sanders, N.N.; De Smedt, S.C.; Demeester, J. Three-dimensional fluorescence recovery after photobleaching with the confocal scanning laser microscope. Biophys. J 2003, 85, 2240–2252. [Google Scholar]
- Saxton, M.J.; Jacobson, K. Single-particle tracking: applications to membrane dynamics. Annu. Rev. Biophys. Biomol. Struct 1997, 26, 373–399. [Google Scholar]
- Malchus, N.; Weiss, M. elucidating anomalous protein diffusion in living cells with fluorescence correlation spectroscopy-facts and pitfalls. J. Fluoresc 2009, 20, 19–26. [Google Scholar]
- Davoust, J.; Devaux, P.F.; Leger, L. Fringe pattern photobleaching, a new method for the measurement of transport coefficients of biological macromolecules. EMBO J 1982, 1, 1233–1238. [Google Scholar]
- Munnelly, H.M.; Roess, D.A.; Wade, W.F.; Barisas, B.G. Interferometric fringe fluorescence photobleaching recovery interrogates entire cell surfaces. Biophys. J 1998, 75, 1131–1138. [Google Scholar]
- Jacobson, K.; Hou, Y.; Wojcieszyn, J. Evidence for lack of damage during photobleaching measurements of the lateral mobility of cell surface components. Exp. Cell. Res 1978, 116, 179–189. [Google Scholar]
- Axelrod, D. Cell surface heating during fluorescence photobleaching recovery experiments. Biophys. J 1977, 18, 129–131. [Google Scholar]
- Axelrod, D.; Koppel, D.E.; Schlessinger, J.; Elson, E.; Webb, W.W. Mobility measurement by analysis of fluorescence photobleaching recovery kinetics. Biophys. J 1976, 16, 1055–1069. [Google Scholar]
- Elson, E.L.; Schlessinger, J.; Koppel, D.E.; Axelrod, D.; Webb, W.W. Measurement of lateral transport on cell surfaces. Prog. Clin. Biol. Res 1976, 9, 137–147. [Google Scholar]
- Schlessinger, J.; Koppel, D.E.; Axelrod, D.; Jacobson, K.; Webb, W.W.; Elson, E.L. Lateral transport on cell membranes: mobility of concanavalin A receptors on myoblasts. Proc. Natl. Acad. Sci. USA 1976, 73, 2409–2413. [Google Scholar]
- Axelrod, D.; Ravdin, P.; Koppel, D.E.; Schlessinger, J.; Webb, W.W.; Elson, E.L.; Podleski, T.R. Lateral motion of fluorescently labeled acetylcholine receptors in membranes of developing muscle fibers. Proc. Natl. Acad. Sci. USA 1976, 73, 4594–4598. [Google Scholar]
- Koppel, D.E.; Axelrod, D.; Schlessinger, J.; Elson, E.L.; Webb, W.W. Dynamics of fluorescence marker concentration as a probe of mobility. Biophys. J 1976, 16, 1315–1329. [Google Scholar]
- Houtsmuller, A.B. Fluorescence recovery after photobleaching: application to nuclear proteins. Adv. Biochem. Eng. Biotechnol 2005, 95, 177–199. [Google Scholar]
- White, J.; Stelzer, E. Photobleaching GFP reveals protein dynamics inside live cells. Trends Cell Biol 1999, 9, 61–65. [Google Scholar]
- Zimmer, M. Green fluorescent protein (GFP): applications, structure, and related photophysical behavior. Chem. Rev 2002, 102, 759–781. [Google Scholar]
- Swaminathan, R.; Hoang, C.P.; Verkman, A.S. Photobleaching recovery and anisotropy decay of green fluorescent protein GFP-S65T in solution and cells: cytoplasmic viscosity probed by green fluorescent protein translational and rotational diffusion. Biophys. J 1997, 72, 1900–7907. [Google Scholar]
- Patterson, G.H.; Knobel, S.M.; Sharif, W.D.; Kain, S.R.; Piston, D.W. Use of the green fluorescent protein and its mutants in quantitative fluorescence microscopy. Biophys. J 1997, 73, 2782–2790. [Google Scholar]
- Morrot, G.; Cribier, S.; Devaux, P.F.; Geldwerth, D.; Davoust, J.; Bureau, J.F.; Fellmann, P.; Herve, P.; Frilley, B. Asymmetric lateral mobility of phospholipids in the human erythrocyte membrane. Proc. Natl. Acad. Sci. USA 1986, 83, 6863–6867. [Google Scholar]
- el Hage Chahine, J.M.; Cribier, S.; Devaux, P.F. Phospholipid transmembrane domains and lateral diffusion in fibroblasts. Proc. Natl. Acad. Sci. USA 1993, 90, 447–451. [Google Scholar]
- Vaz, W.L.; Criado, M.; Madeira, V.M.; Schoellmann, G.; Jovin, T.M. Size dependence of the translational diffusion of large integral membrane proteins in liquid-crystalline phase lipid bilayers. A study using fluorescence recovery after photobleaching. Biochemistry 1982, 21, 5608–5612. [Google Scholar]
- Naji, A.; Levine, A.J.; Pincus, P.A. Corrections to the Saffman-Delbruck mobility for membrane bound proteins. Biophys. J 2007, 93, L49–51. [Google Scholar]
- Guigas, G.; Weiss, M. Size-dependent diffusion of membrane inclusions. Biophys. J 2006, 91, 2393–2398. [Google Scholar]
- Guigas, G.; Weiss, M. Influence of hydrophobic mismatching on membrane protein diffusion. Biophys. J 2008, 95, L25–27. [Google Scholar]
- Bates, I.R.; Hebert, B.; Luo, Y.; Liao, J.; Bachir, A.I.; Kolin, D.L.; Wiseman, P.W.; Hanrahan, J.W. Membrane lateral diffusion and capture of CFTR within transient confinement zones. Biophys. J 2006, 91, 1046–1058. [Google Scholar]
- Tian, A.; Baumgart, T. Sorting of lipids and proteins in membrane curvature gradients. Biophys. J 2009, 96, 2676–2688. [Google Scholar]
- Daniels, D.R.; Turner, M.S. Diffusion on membrane tubes: a highly discriminatory test of the Saffman-Delbruck theory. Langmuir 2007, 23, 6667–6670. [Google Scholar]
- Vaz, W.L.; Goodsaid-Zalduondo, F.; Jacobson, K. Lateral diffusion of lipids and proteins in bilayer membranes. FEBS Lett 1984, 174, 199–207. [Google Scholar]
- Weiss, T.M.; van der Wel, P.C.; Killian, J.A.; Koeppe, R.E., 2nd; Huang, H.W. Hydrophobic mismatch between helices and lipid bilayers. Biophys. J 2003, 84, 379–385. [Google Scholar]
- Killian, J.A.; Nyholm, T.K. Peptides in lipid bilayers: the power of simple models. Curr. Opin. Struct. Biol 2006, 16, 473–479. [Google Scholar]
- Gambin, Y.; Reffay, M.; Sierecki, E.; Homble, F.; Hodges, R.S.; Gov, N.S.; Taulier, N.; Urbach, W. Variation of the Lateral Mobility of Transmembrane Peptides with Hydrophobic Mismatch. J. Phys. Chem. B 2010, 114, 3559–3566. [Google Scholar]
- Kirchhoff, H.; Haferkamp, S.; Allen, J.F.; Epstein, D.B.; Mullineaux, C.W. Protein diffusion and macromolecular crowding in thylakoid membranes. Plant Physiol 2008, 146, 1571–1578. [Google Scholar]
- Kirchhoff, H.; Mukherjee, U.; Galla, H.J. Molecular architecture of the thylakoid membrane: lipid diffusion space for plastoquinone. Biochemistry 2002, 41, 4872–4882. [Google Scholar]
- Vladimirou, E.; Li, M.; Aldridge, C.P.; Frigerio, L.; Kirkilionis, M.; Robinson, C. Diffusion of a membrane protein, Tat subunit Hcf106, is highly restricted within the chloroplast thylakoid network. FEBS Lett 2009, 583, 3690–3696. [Google Scholar]
- Mullineaux, C.W.; Nenninger, A.; Ray, N.; Robinson, C. Diffusion of green fluorescent protein in three cell environments in Escherichia coli. J. Bacteriol 2006, 188, 3442–3448. [Google Scholar]
- Brignull, H.R.; Morley, J.F.; Garcia, S.M.; Morimoto, R.I. Modeling polyglutamine pathogenesis in C. elegans. Methods Enzymol 2006, 412, 256–282. [Google Scholar]
- Satyal, S.H.; Schmidt, E.; Kitagawa, K.; Sondheimer, N.; Lindquist, S.; Kramer, J.M.; Morimoto, R.I. Polyglutamine aggregates alter protein folding homeostasis in Caenorhabditis elegans. Proc. Natl. Acad. Sci. USA 2000, 97, 5750–5755. [Google Scholar]
- Brignull, H.R.; Moore, F.E.; Tang, S.J.; Morimoto, R.I. Polyglutamine proteins at the pathogenic threshold display neuron-specific aggregation in a pan-neuronal Caenorhabditis elegans model. J. Neurosci 2006, 26, 7597–7606. [Google Scholar]
- Edwin, N.J.; Hammer, R.P.; McCarley, R.L.; Russo, P.S. Reversibility of beta-Amyloid Self-Assembly: Effects of pH and Added Salts Assessed by Fluorescence Photobleaching Recovery. Biomacromolecules 2010, 11, 341–347. [Google Scholar]
- Athmakuri, K.; Padala, C.; Litt, J.; Cole, R.; Kumar, S.; Kane, R.S. Controlling DNA adsorption and diffusion on lipid bilayers by the formation of lipid domains. Langmuir 2010, 26, 397–401. [Google Scholar]
- Moreau, P.; van Effenterre, D.; Navailles, L.; Nallet, F.; Roux, D. Confined diffusion of hydrophilic probes inserted in lyotropic lamellar phases. Eur. Phys. J. E Soft Matter 2008, 26, 225–234. [Google Scholar]
- Ries, J.; Schwille, P. New concepts for fluorescence correlation spectroscopy on membranes. Phys. Chem. Chem. Phys 2008, 10, 3487–3497. [Google Scholar]
- Garcia-Saez, A.J.; Schwille, P. Fluorescence correlation spectroscopy for the study of membrane dynamics and protein/lipid interactions. Methods 2008, 46, 116–122. [Google Scholar]
- Enderlein, J.; Gregor, I.; Patra, D.; Dertinger, T.; Kaupp, U.B. Performance of fluorescence correlation spectroscopy for measuring diffusion and concentration. Chemphyschem 2005, 6, 2324–2336. [Google Scholar]
- Hess, S.T.; Webb, W.W. Focal volume optics and experimental artifacts in confocal fluorescence correlation spectroscopy. Biophys. J 2002, 83, 2300–2317. [Google Scholar]
- Magde, D.; Elson, E.L.; Webb, W.W. Thermodynamic fluctuations in a reacting system: measurement by fluorescence correlation spectroscopy. Phys. Rev. Lett 1972, 29, 705–708. [Google Scholar]
- Elson, E.L.; Magde, D. Fluorescence correlation spectroscopy. I. Conceptual basis and theory. Biopolymers 1974, 13, 1–27. [Google Scholar]
- Magde, D.; Elson, E.L.; Webb, W.W. Fluorescence correlation spectroscopy. II. An experimental realization. Biopolymers 1974, 13, 29–61. [Google Scholar]
- Schwille, P. Fluorescence correlation spectroscopy and its potential for intracellular applications. Cell Biochem. Biophys 2001, 34, 383–408. [Google Scholar]
- Kohl, T.; Schwille, P. Fluorescence correlation spectroscopy with autofluorescent proteins. Adv. Biochem. Eng. Biotechnol 2005, 95, 107–142. [Google Scholar]
- Martin, D.S.; Forstner, M.B.; Kas, J.A. Apparent subdiffusion inherent to single particle tracking. Biophys. J 2002, 83, 2109–2117. [Google Scholar]
- Barak, L.S.; Webb, W.W. Diffusion of low density lipoprotein-receptor complex on human fibroblasts. J. Cell. Biol 1982, 95, 846–852. [Google Scholar]
- Kusumi, A.; Nakada, C.; Ritchie, K.; Murase, K.; Suzuki, K.; Murakoshi, H.; Kasai, R.S.; Kondo, J.; Fujiwara, T. Paradigm shift of the plasma membrane concept from the two-dimensional continuum fluid to the partitioned fluid: high-speed single-molecule tracking of membrane molecules. Annu. Rev. Biophys. Biomol. Struct 2005, 34, 351–378. [Google Scholar]


© 2010 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Rayan, G.; Guet, J.-E.; Taulier, N.; Pincet, F.; Urbach, W. Recent Applications of Fluorescence Recovery after Photobleaching (FRAP) to Membrane Bio-Macromolecules. Sensors 2010, 10, 5927-5948. https://doi.org/10.3390/s100605927
Rayan G, Guet J-E, Taulier N, Pincet F, Urbach W. Recent Applications of Fluorescence Recovery after Photobleaching (FRAP) to Membrane Bio-Macromolecules. Sensors. 2010; 10(6):5927-5948. https://doi.org/10.3390/s100605927
Chicago/Turabian StyleRayan, Gamal, Jean-Erik Guet, Nicolas Taulier, Frederic Pincet, and Wladimir Urbach. 2010. "Recent Applications of Fluorescence Recovery after Photobleaching (FRAP) to Membrane Bio-Macromolecules" Sensors 10, no. 6: 5927-5948. https://doi.org/10.3390/s100605927