A Lanthanide-Based Chemosensor for Bioavailable Fe3+ Using a Fluorescent Siderophore: An Assay Displacement Approach


Abstract
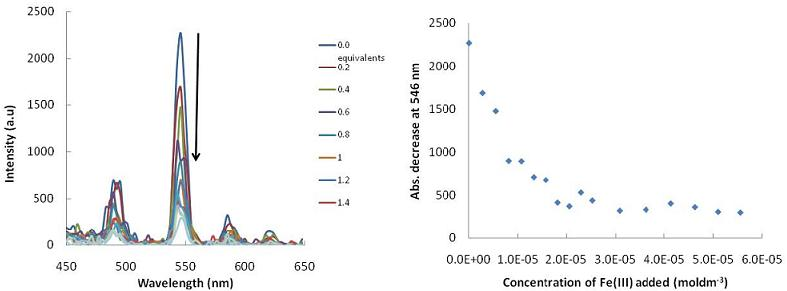
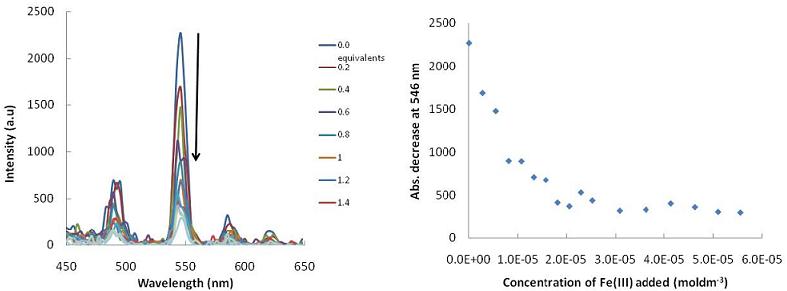
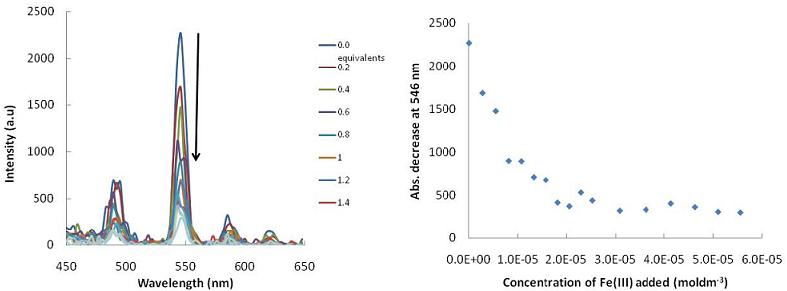
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
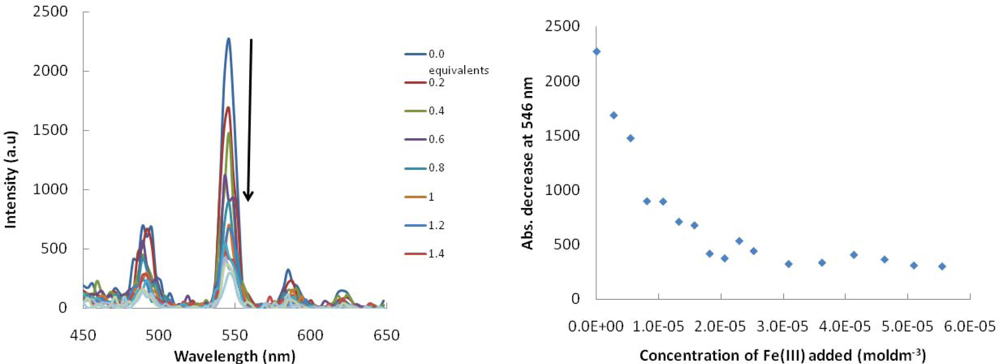
2. Results and Discussion
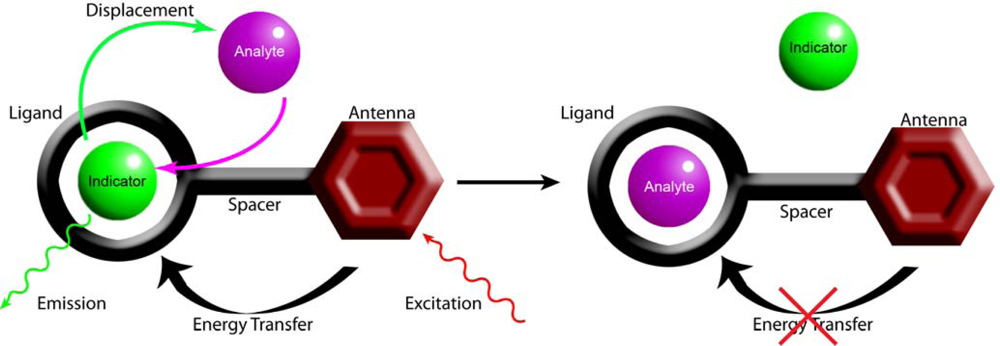
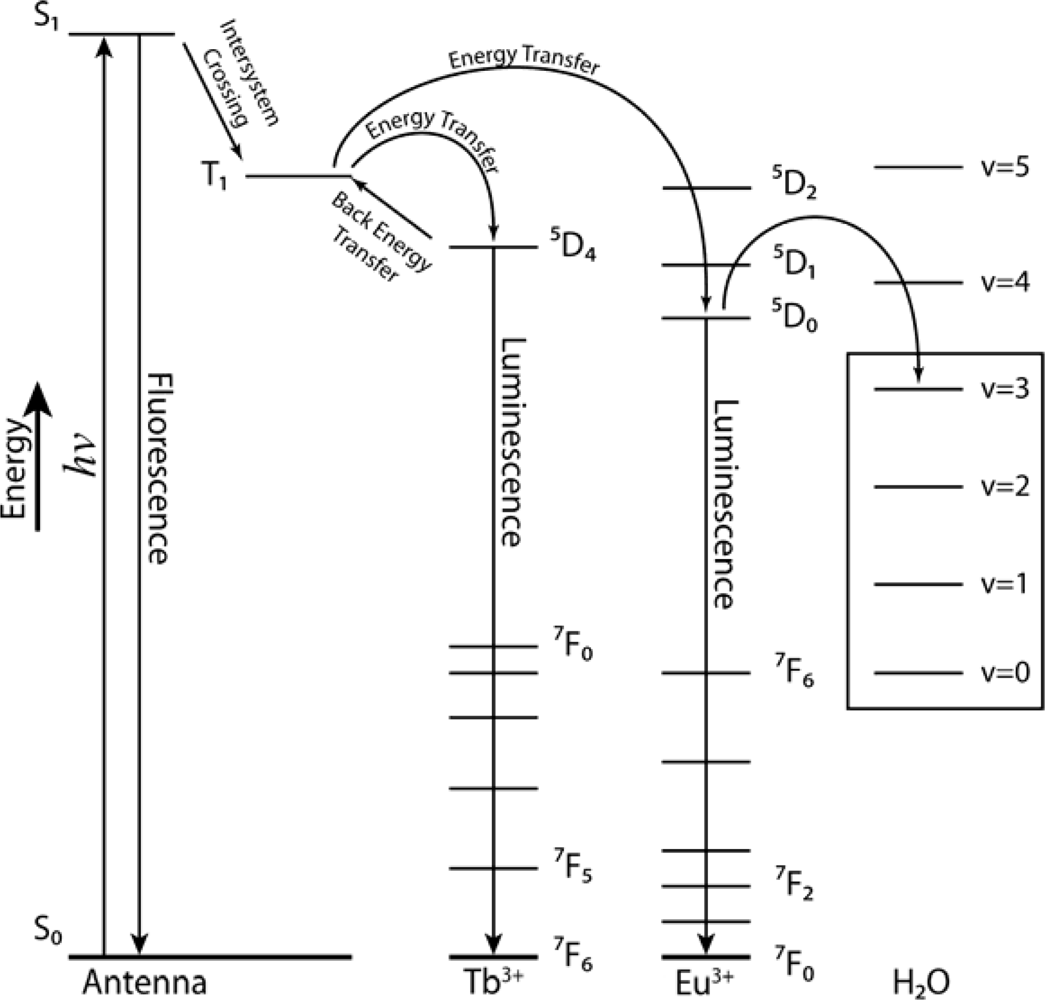
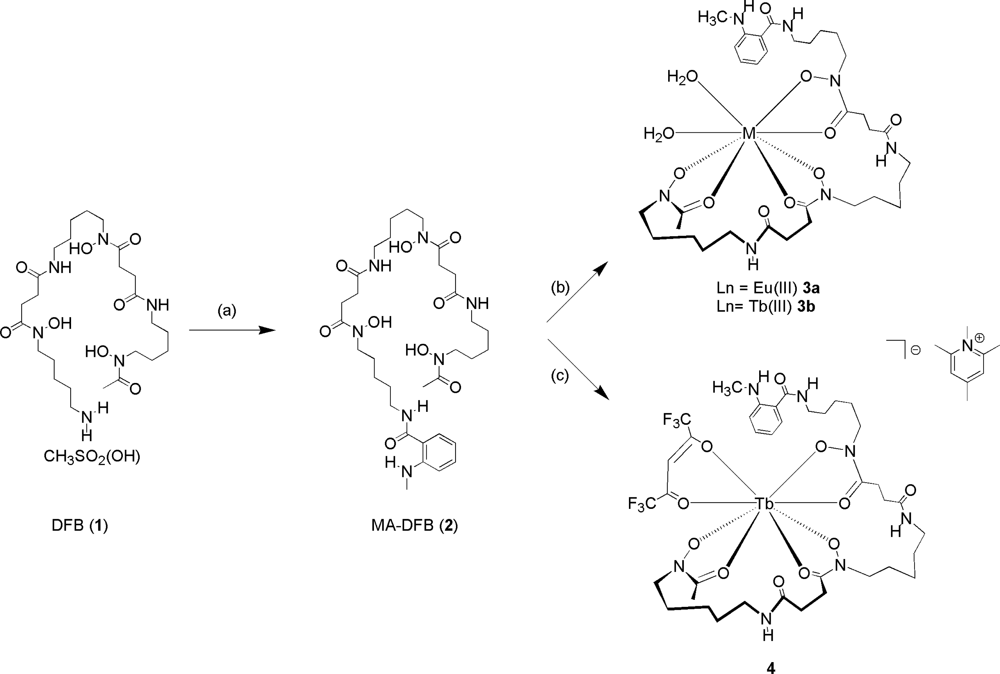
2.1. Design Criteria
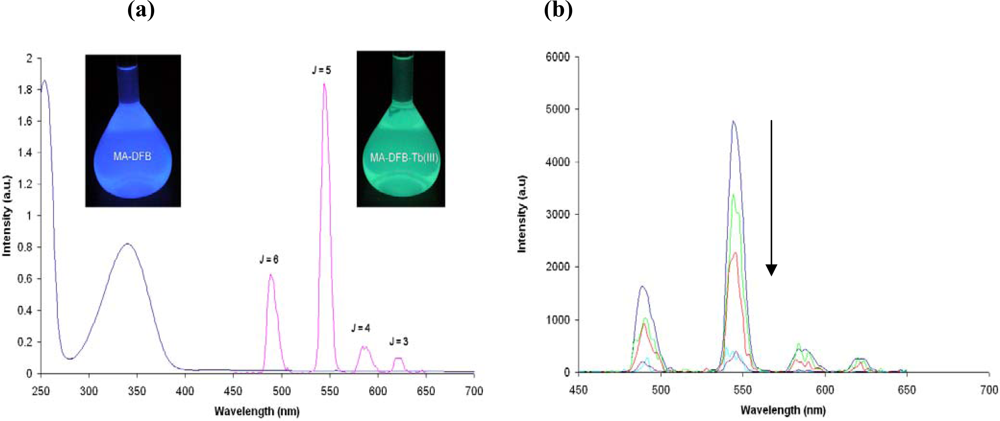
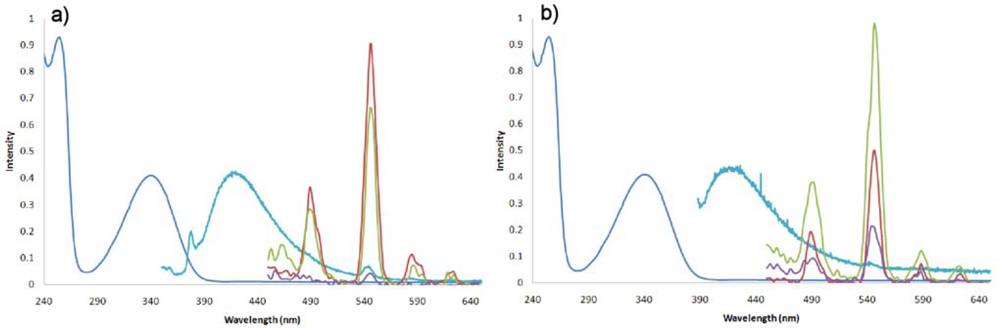
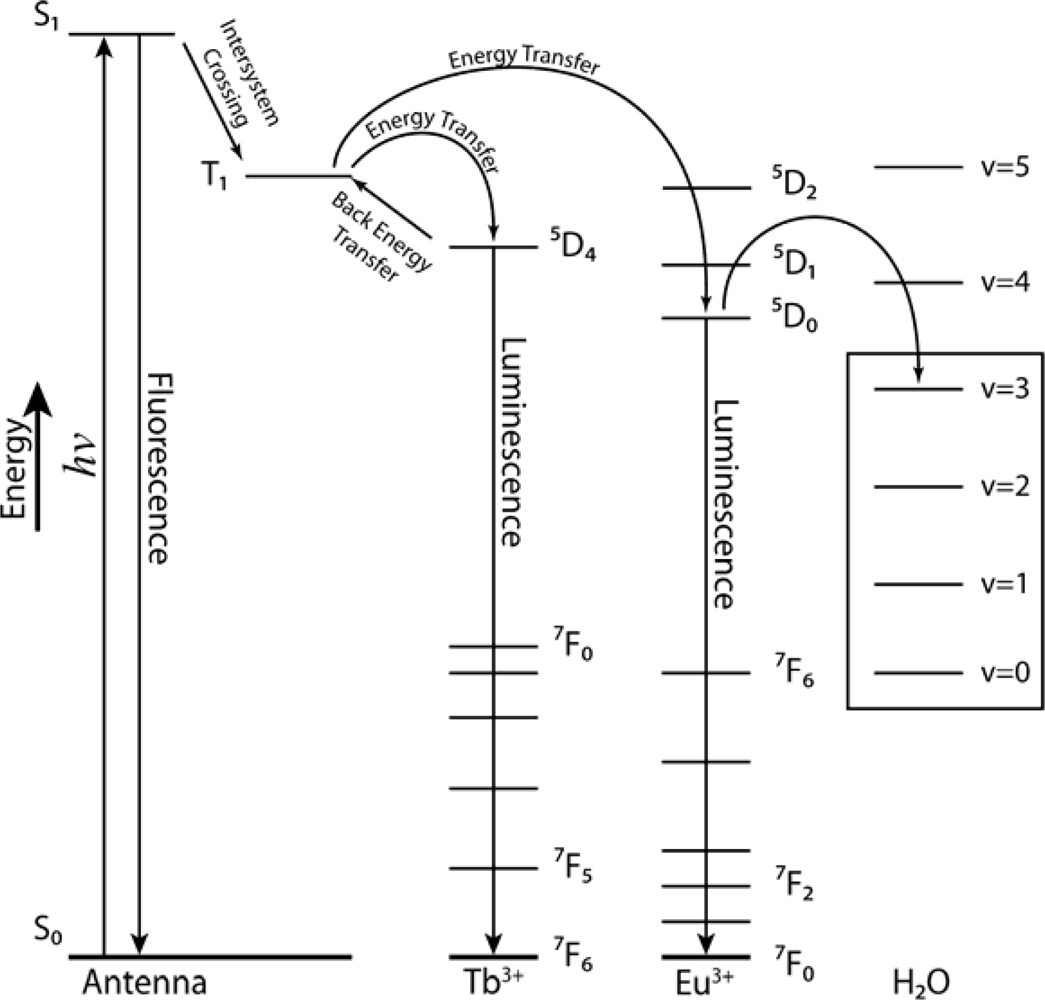
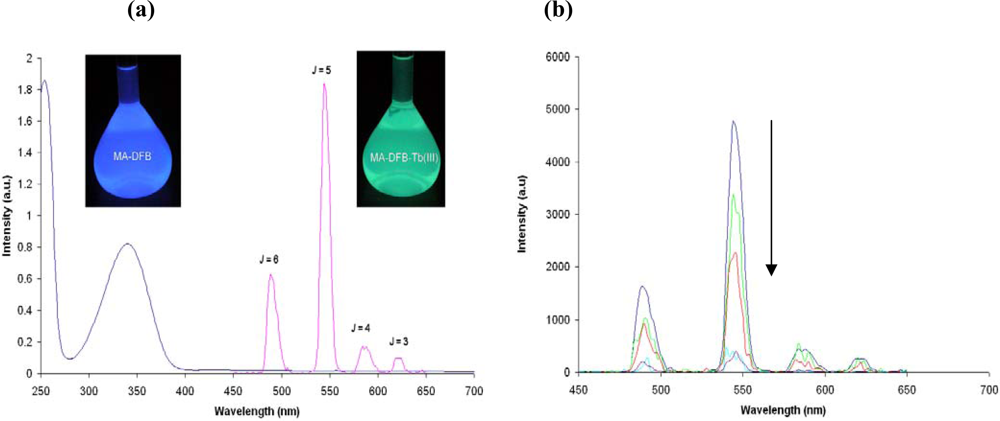
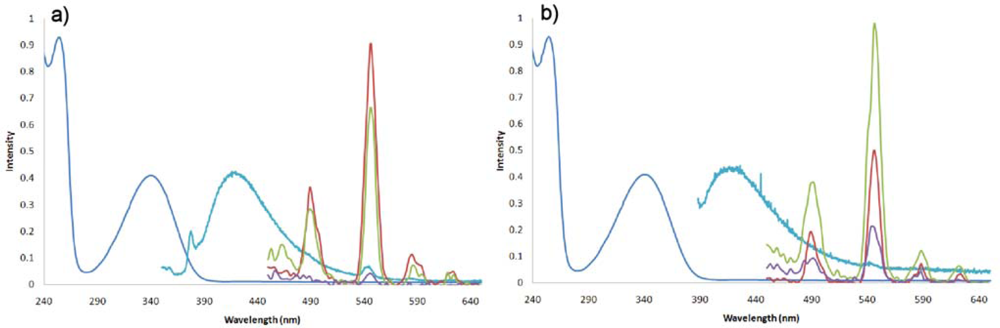
2.2. Synthesis and Photophysical Properties
3. Experimental Section
4. Conclusions
Acknowledgments
References and Notes
- Chisholm, S.W. Oceanography: Stirring times in the Southern Ocean. Nature 2000, 407, 685–687. [Google Scholar]
- Hutchins, D.A.; Franck, V.M.; Brzezinski, M.A.; Bruland, K.W. Inducing iron limitation in iron-replete coastal waters with a strong chelating ligand. Limnol. Oceanogr 1999, 44, 1009–1018. [Google Scholar]
- Wells, M.L. Manipulating iron availability in nearshore waters. Limnol. Oceanogr 1999, 44, 1002–1008. [Google Scholar]
- Wells, M.L.; Trick, C.G. Controlling iron availability to phytoplankton in iron-replete coastal waters. Mar. Chem 2004, 86, 1–13. [Google Scholar]
- Well, M.L.; Price, N.M.; Bruland, K.W. Iron chemistry in seawater and its relationship to phytoplankton: a workshop report. Mar. Chem 1995, 48, 157–182. [Google Scholar]
- Boyanapalli, R.; Bullerjahn, G.S.; Pohl, C.; Croot, P.L.; Boyd, P.W.; Michael, R.; McKay, R.M.L. Luminescent whole-cell cyanobacterial bioreporter for measuring Fe availability in diverse marine environments. Appl. Environ. Microbiol 2007, 73, 1019–1024. [Google Scholar]
- Durham, K.A.; Porta, D.; Twiss, M.R.; McKay, M.L.; Bullerjahn, G.S. Construction and initial characterization of a luminescent Synechoccus sp. PCC 7942 Fe-dependant bioreporter. FEMS Microbiol. Lett 2002, 209, 215–221. [Google Scholar]
- Hassler, C.S.; Wiss, M.R.; McKay, R.M.L.; Bullerjahn, G.S. Optimization of iron-dependant cyanobacterial (Synechoccocus, Cyanophyceae) bioreporters to measure iron availability. J. Phycol 2006, 42, 324–335. [Google Scholar]
- McKay, R.M.L.; Bullerjahn, G.S.; Porta, D.; Brown, E.T.; Sherrel, R.M.; Smutka, T.M.; Sterner, R.W.; Twiss, M.R.; Wilhelm, S.W. Consideration of the bioavailability of iron in the North American Great Lakes: Development of novel approaches toward understanding iron biogeochemistry. Aquat. Ecosyst. Health Manag 2004, 7, 475–490. [Google Scholar]
- McKay, R.M.L.; Porta, D.; Bullerjahn, G.S.; Al-Rshaidat, M.M.D.; Kilmowicz, J.A.; Sterner, R.W.; Smutka, T.M.; Brown, E.T.; Sherrel, R.M. Bioavailable iron in oligotrophic Lake Superior assessed using biological reporters. J. Plankton Res 2005, 27, 1033–1044. [Google Scholar]
- Mioni, C.E.; Handy, S.M.; Ellwood, M.J.; Twiss, M.R.; McKay, R.M.L.; Boyd, P.W.; Wilhelm, S.W. Tracking changes in bioavailable Fe within high-nutrient low-chlorophyll oceanic waters: a first estimate using a heterotrophic bacterial bioreporter. Global Biogeochem. Cycles 2005, 19, 1–10. [Google Scholar]
- Mioni, C.E.; Howard, A.M.; Debruyn, J.M.; Bright, N.G.; Twiss, M.R.; Applegate, B.M.; Wilhelm, S.W. Characterization and field trials of a bioluminescent bacterial reporter of iron availability. Mar. Chem 2003, 83, 31–46. [Google Scholar]
- Mioni, C.E.; Poorvin, L.; Wilhelm, S.W. Virus and siderophore-mediated transfer of available Fe between heterotrophic bacteria: characterization using an Fe-specific reporter. Aquat. Microb. Ecol 2005, 41, 233–245. [Google Scholar]
- Porta, D.; Bullerjahn, G.S.; Wilhelm, S.W.; Twiss, M.R.; McKay, R.M.L. Physiological characterization of Synechococcus sp. (cyanophyceae) strain PCC 7942 iron-dependant bioreporter for freshwater environments. J. Phycol 2003, 39, 64–73. [Google Scholar]
- Mioni, C.E.; Pakulski, J.D.; Poorvin, L.; Baldwin, A.; Twiss, M.R.; Jeffrey, W.H.; Wilhelm, S.W. Variability in the in situ bioavailability of Fe to bacterioplankton communities in the eastern subtropical Pacific Ocean. Aquat. Microb. Ecol 2007, 46, 239–251. [Google Scholar]
- Chung Chun Lam, C.K.S.; Jickells, T.D.; Richardson, D.J.; Russell, D.A. Fluorescence-based siderophore biosensor for the determination of bioavailable iron in oceanic waters. Anal. Chem 2006, 78, 5040–5045. [Google Scholar]
- Orcutt, K.M.; L, W.M. A liposome-based nanodevice for sequestering siderophore-bound Fe. J. Membrane Sci 2007, 288, 247–254. [Google Scholar]
- Espósito, B.P.; Epsztejn, S.; Breuer, W.; Cabantchik, Z.I. A review of fluorescence methods for assessing labile iron in cells and biological fluids. Anal. Biochem 2002, 304, 1–18. [Google Scholar]
- Palanché, B.P.; Marmolle, F.; Abraham, M.A.; Shanzer, A.; Albrecht-Gray, A.M. Fluorescent siderophore-based chemosensors: iron (III) quantitative determinations. J. Biol. Inorg. Chem 1999, 4, 188–198. [Google Scholar]
- de Silva, A.P.; Gunaratne, H.Q.N.; Gunnlaugsson, T.; Huxley, A.J.M.; McCoy, C.P.; Rademacher, J.T.; Rice, T.E. Signaling recognition events with fluorescent sensors and switches. Chem. Rev 1997, 97, 1515–1566. [Google Scholar]
- Houk, R.J.T.; Wallace, K.J.; Hewage, H.S.; Anslyn, E.V. A colorimetric chemodosimeter for Pd(II): a method for detecting residual palladium in cross-coupling reactions. Tetrahedron 2008, 64, 8271–8278. [Google Scholar]
- Wallace, K.J.; Nguyen, B.T.; Anslyn, E.V. Indicator Displacement Assay (IDA). In Encyclopedia of Supramolecular Chemistry, 1st ed; Atwood, J.L., Steed, J.W., Eds.; Taylor & Francis: Baca Raton, FL, USA, 2006. [Google Scholar]
- Wiskur, S.L.; Ait-Haddou, H.; Lavigne, J.J.; Anslyn, E.V. Teaching old indicators new tricks. Acc. Chem. Res 2001, 34, 963–972. [Google Scholar]
- Graeppi, N.; Powell, G.H.; Laurenczy, G.; Zékány, L.; Merbach, A. Coordination equilibria and water exchange kinetics of lanthanide(III) propylenediaminetetraacetates and other magnetic resonance imaging related complexes. Inorg Chimi. Acta 1995, 235, 311–326. [Google Scholar]
- Goshe, A.J.; Steele, I.M.; Ceccarelli, C.; Rheingold, A.L.; Bosnich, B. Supramolecular recognition: On the kinetic lability of thermodynamically stable host-guest association complexes. Proc. Natl. Acad. Sci 2002, 99, 4823–4829. [Google Scholar]
- Bunzli, J.C.G.; Piguet, C. Lanthanide-Containing molecular and supramolecular polymetallic functional assemblies. Chem. Rev 2002, 102, 1897–1928. [Google Scholar]
- Tsukube, H.; Shinoda, S. Lanthanide complexes in molecular recognition and chirality sensing of biological substrates. Chem. Rev 2002, 102, 2389–2403. [Google Scholar]
- Selvin, P.R. Principles and biophysical applications of lanthanide-based probes. Annu. Rev. Biophys. Biomol. Struct 2002, 31, 275–302. [Google Scholar]
- De Silva, C.R.; Maeyer, J.R.; Wang, R.; Nichol, G.S.; Zheng, Z. Adducts of europium b-diketonates with nitrogen p,p′-disubstituted bipyridine and phenanthroline ligands: Synthesis structural characterization, and luminescence studies. Inorg. Chimi. Acta 2007, 360, 3543–3552. [Google Scholar]
- Lytton, S.D.; Cabantchik, Z.I.; Libman, J.; Shanzer, A. Reversed siderophores as antimalarial agents. II. Selective scavenging of Fe (III) from parasitized erythrocytes by a fluorescent derivitive of Desferal. Mol. Pharmacol 1991, 40, 584–590. [Google Scholar]
- Moore, E.G.; Xu, J.; Jocher, C.J.; Werner, E.J.; Raymond, K.N. “Cymothoe sangaris”: An extremely stable and highly luminescent 1,2-hydroxypyridinonate chelate of Eu(III). J. Am. Chem. Soc 2006, 128, 10648–10649. [Google Scholar]
- Bassett, A.P.; Magennis, S.W.; Glover, P.B.; Lewis, D.J.; Spencer, N.; Parsons, S.; Williams, R.M.; De Cola, L.; Pikramenou, Z. Hghly Luminescent, Triple- and quadruple-stranded dinuclear Eu, Nd, and Sm(III) lanthanide complexes based on bis-diketonates? J. Am. Chem. Soc 2004, 126, 9413–9424. [Google Scholar]
- Batista, H.J.; de Andrade, A.V.M.; Longo, R.L.; Simas, M.; de Sá, G.D.; Ito, N.K.; Thompson, L.C. Synthesis, X-ray structure, spectroscopic characterization, and theoretical prediction of the Structure and Electronic Spectrum of Eu (btfa)3·bipy and an assesessment of the effect of fluorine as β-diketone substituent on the ligand-metal energy transfer proces. Inorg. Chem 1998, 37, 3542–3547. [Google Scholar]
- dos Santos, C.M.G.; Harte, A.J.; Quinn, S.J.; Gunnlaugsson, T. “Recent developments in the field of supramolecular lanthanide luminescent sensors and self-assemblies”. Coord. Chem. Rev 2008, 252, 2512–2527. [Google Scholar]
- Tsukube, H.; Shinoda, S.; Uenishi, J.; Kanatani, T.; Itoch, H.; Shiode, M.; Iwachido, T.; Yonemitsu, O. “Molecular Recognition with lanthanide(III) tris(β-diketonate) complexes: extraction, transport, and chiral recognition of unprotected amino acids”. Inorg. Chem 1998, 37, 1585–1591. [Google Scholar]
- Thibon, A.; Pierre, V.C. A highly selective luminescent sensor for the time-gated detection of potassium. J. Am. Chem. Soc 2009, 131, 434–435. [Google Scholar]
- Terenghi, M.; Elviri, L.; Careri, M.; Mangia, A.; Lobinski, R. Multiplexed determination of protein biomarkers using metal-tagged antibodies and size exclusion chromatography-inductively coupled plasma mass spectrometry. Anal. Chem 2009, 81, 9440–9448. [Google Scholar]
- Binnemans, K. Luminescence of metallomesogens in the liquid crystal state. J. Mater. Chem 2009, 19, 448–453. [Google Scholar]
- Guo, X.; Guo, H.; Fu, L.; Carlos, L.D.; Ferreira, R.A.S.; Sun, L.; Deng, R.; Zhang, H. Novel near-infrared luminescent hybrid materials covalently linking with lanthanide [Nd(III), Er(III), Yb(III), and Sm(III)] complexes via a primary β-Diketone ligand: synthesis and photophysical studies. J. Phys. Chem. C 2009, 113, 12538–12545. [Google Scholar]
- Binnemans, K. Lanthanide-Based Luminescent Hybrid Materials. Chem. Rev 2009, 109, 4283–4374. [Google Scholar]
- Loyevsky, M.; Lytton, S.D.; Mester, B.; Libman, J.; Shanzer, A.; Cabantchik, Z.I. The Antimalarial action of desferal involves a direct access route to erythrocytic (Plasmodium Falciparum) parasites. J. Clin. Invest 1993, 91, 218–224. [Google Scholar]



© 2010 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Orcutt, K.M.; Jones, W.S.; McDonald, A.; Schrock, D.; Wallace, K.J. A Lanthanide-Based Chemosensor for Bioavailable Fe3+ Using a Fluorescent Siderophore: An Assay Displacement Approach. Sensors 2010, 10, 1326-1337. https://doi.org/10.3390/s100201326
Orcutt KM, Jones WS, McDonald A, Schrock D, Wallace KJ. A Lanthanide-Based Chemosensor for Bioavailable Fe3+ Using a Fluorescent Siderophore: An Assay Displacement Approach. Sensors. 2010; 10(2):1326-1337. https://doi.org/10.3390/s100201326
Chicago/Turabian StyleOrcutt, Karen M., W. Scott Jones, Andrea McDonald, David Schrock, and Karl J. Wallace. 2010. "A Lanthanide-Based Chemosensor for Bioavailable Fe3+ Using a Fluorescent Siderophore: An Assay Displacement Approach" Sensors 10, no. 2: 1326-1337. https://doi.org/10.3390/s100201326