Glucose Sensing Neurons in the Ventromedial Hypothalamus

{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Glucose Sensing Neurons
2.1. GE Neurons
2.2. GI Neurons
3. Glucose Sensing Neurons and Energy Deficit
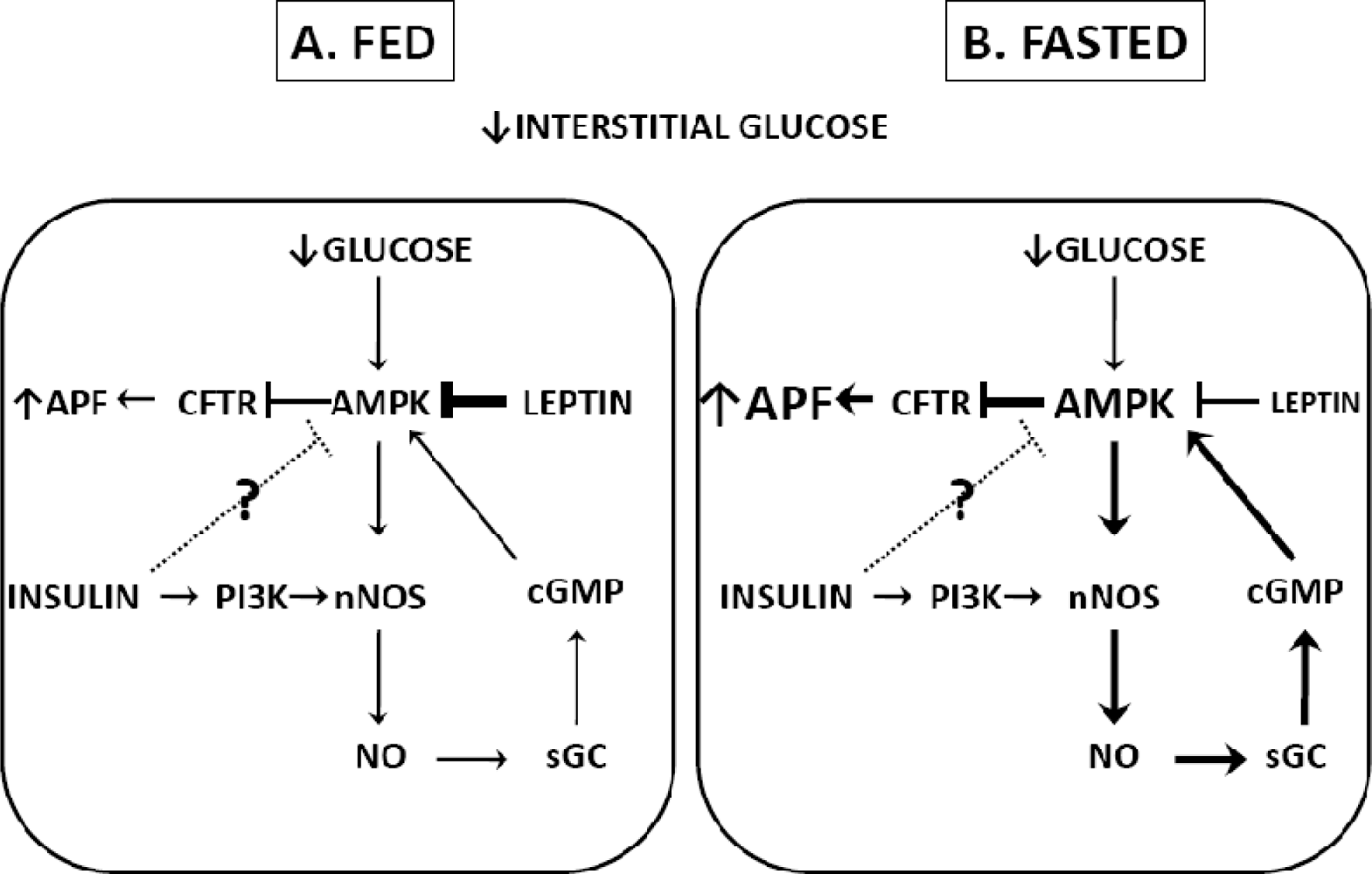
3.1. Fasting
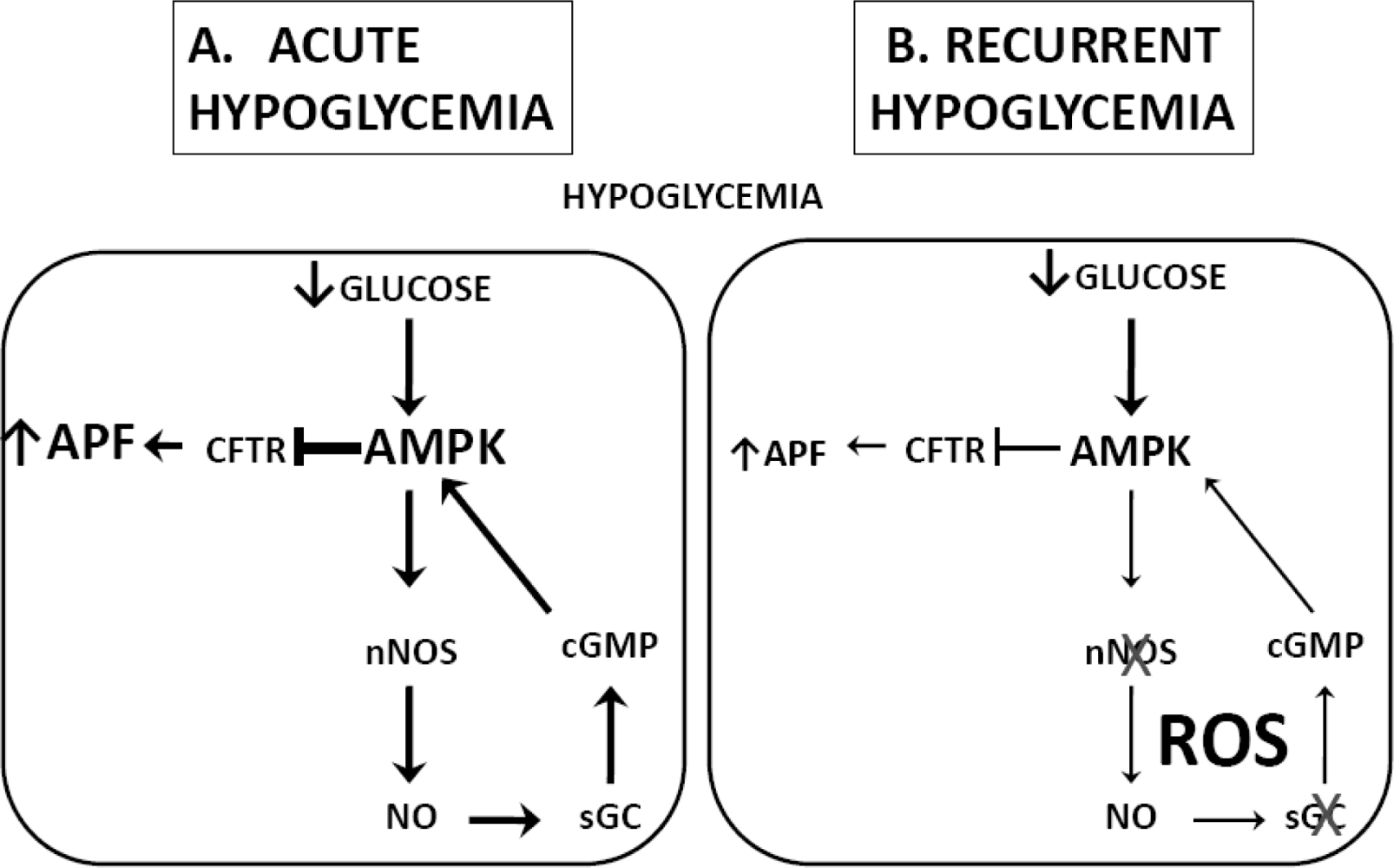
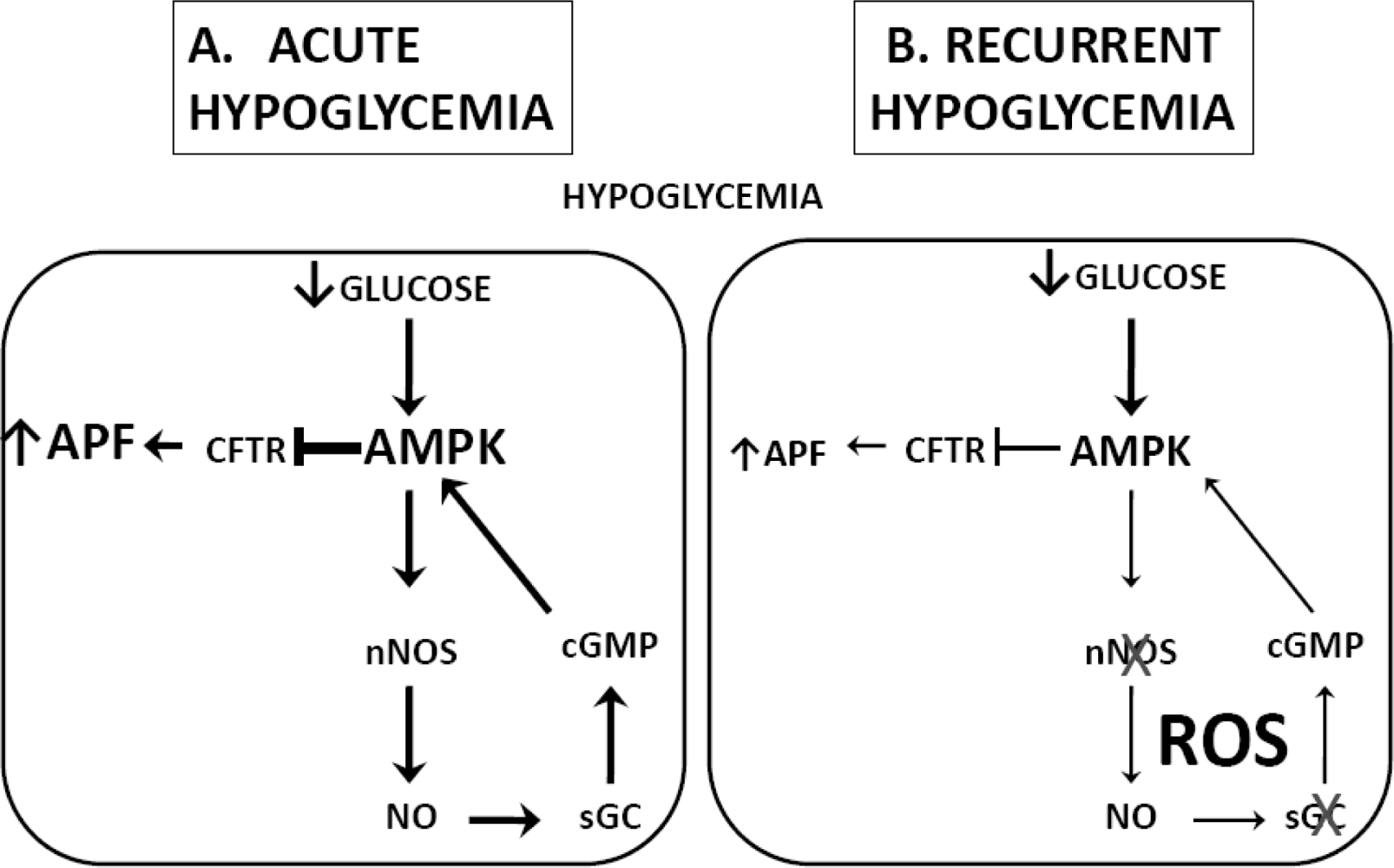
3.2. Hypoglycemia Detection and the Counterregulatory Response (CRR)
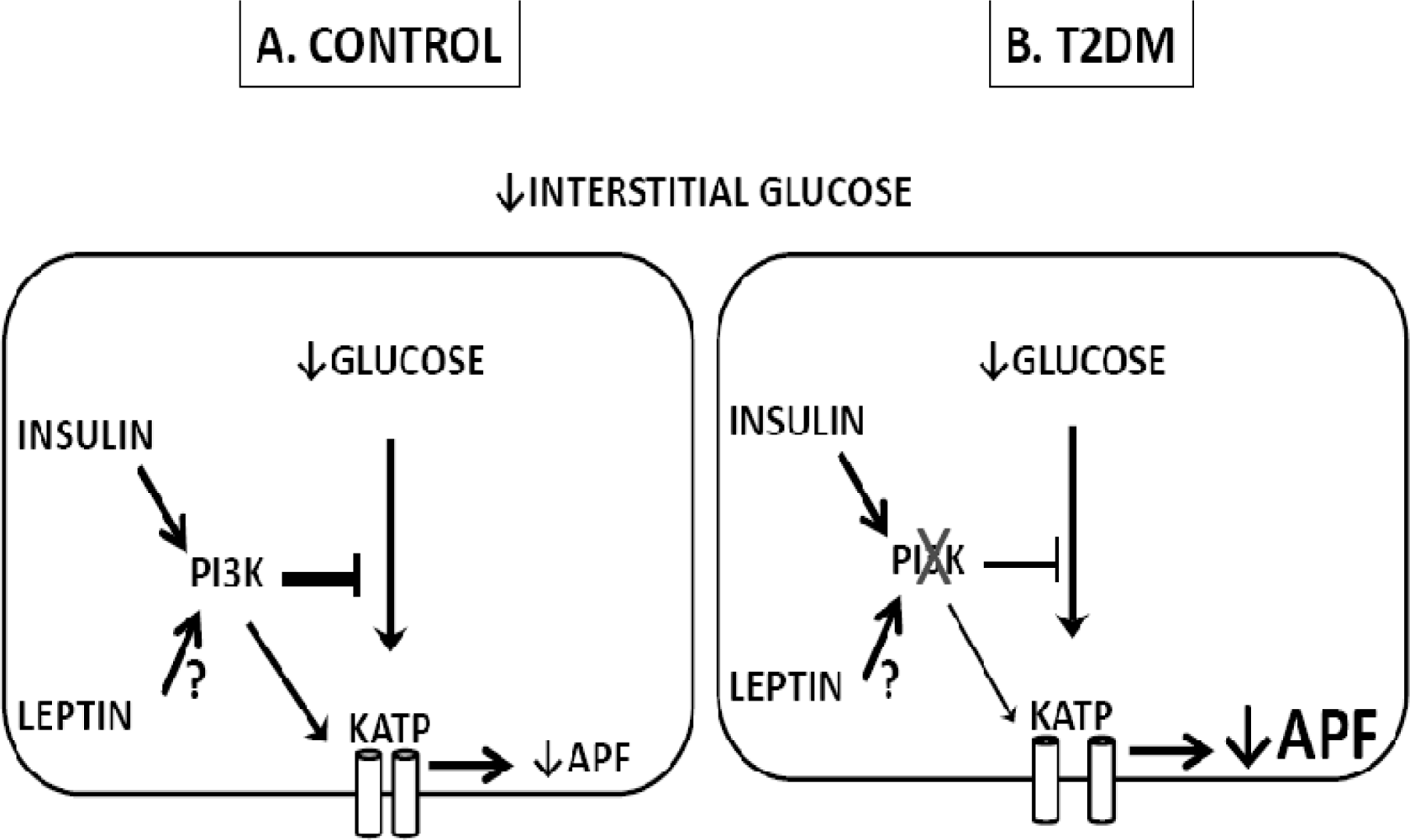
4. Glucose Sensing Neurons and T2DM
5. Summary
Acknowledgments
References
- Schwartz, MW; Woods, SC; Porte, D; Seeley, RJ; Baskin, DG. Central nervous system control of food intake. Nature 2000, 404, 661–671. [Google Scholar]
- Niswender, KD; Schwartz, MW. Insulin and leptin revisited: Adiposity signals with overlapping physiological and intracellular signaling capabilities. Front. Neuroendocrinol 2003, 24, 1–10. [Google Scholar]
- Shin, AC; Pistell, PJ; Phifer, CB; Berthoud, HR. Reversible suppression of food reward behavior by chronic mu-opioid receptor antagonism in the nucleus accumbens. Neuroscience 2010, 170, 580–588. [Google Scholar]
- King, BM. The rise, fall, and resurrection of the ventromedial hypothalamus in the regulation of feeding behavior and body weight. Physiol. Behav 2006, 87, 221–244. [Google Scholar]
- Elmquist, JK. Hypothalamic pathways underlying the endocrine, autonomic, and behavioral effects of leptin. Physiol. Behav 2001, 74, 703–708. [Google Scholar]
- Adage, T; Scheurink, AJ; de Boer, SF; de Vries, K; Konsman, JP; Kuipers, F; Adan, RA; Baskin, DG; Schwartz, MW; van Dijk, G. Hypothalamic, metabolic, and behavioral responses to pharmacological inhibition of CNS melanocortin signaling in rats. J. Neurosci 2001, 21, 3639–3645. [Google Scholar]
- Mayer, J. Regulation of energy intake and the body weight: The glucostatic theory and the lipostatic hypothesis. Ann. N. Y. Acad. Sci 1958, 63, 15–43. [Google Scholar]
- Anand, BK; China, GS; Sharma, KN; Dua, S; Singh, B. Activity of single neurons in the hypothalamus feeding centers: effect of glucose. Amer. J. Physiol 1964, 2207, 1146–1154. [Google Scholar]
- Oomura, Y; Kimura, K; Ooyama, H; Maeo, T; Iki, M; Kuniyoshi, N. Reciprocal activities of the ventromedial and lateral hypothalamic area of cats. Science 1964, 143, 484–485. [Google Scholar]
- Campfield, LA; Smith, FJ. Functional coupling between transient declines in blood glucose and feeding behavior: temporal relationships. Brain. Res. Bull 1986, 17, 427–549. [Google Scholar]
- Silver, IA; Erecinska, M. Extracellular glucose concentration in mammalian brain: continuous monitoring of changes during increased neuronal activity and upon limitation in oxygen supply in normo-, hypo-, and hyperglycemic animals. J. Neurosci 1994, 14, 5068–5076. [Google Scholar]
- Dunn-Meynell, AA; Sanders, NM; Compton, D; Becker, TC; Eiki, Ji; Zhang, BB; Levin, BE. Relationship among brain and blood glucose levels and spontaneous and glucoprivic feeding. J. Neurosci 2009, 29, 7015–7022. [Google Scholar]
- Borg, MA; Sherwin, RS; Borg, WP; Tamborlane, WV; Shulman, GI. Local ventromedial hypothalamus glucose perfusion blocks counterregulation during systemic hypoglycemia in awake rats. J. Clin. Invest 1997, 99, 361–365. [Google Scholar]
- Borg, WP; During, MJ; Sherwin, RS; Borg, MA; Brines, ML; Shulman, GI. Ventromedial hypothalamic lesions in rats suppress counterregulatory responses to hypoglycemia. J. Clin. Invest 1994, 93, 1677–1682. [Google Scholar]
- Borg, WP; Sherwin, RS; During, MJ; Borg, MA; Shulman, GI. Local ventromedial hypothalamus glucopenia triggers counterregulatory hormone release. Diabetes 1995, 44, 180–184. [Google Scholar]
- Kow, LM; Pfaff, DW. Responses of hypothalamic paraventricular neurons in vitro to norepinephrine and other feeding-relevant agents. Physiol. Behav 1989, 46, 265–271. [Google Scholar]
- Ashford, MLJ; Boden, PR; Treherne, JM. Tolbutamide excites rat glucoreceptive ventromedial hypothalamic neurones by indirect inhibition of ATP-sensitive K+ channels. Brit. J. Pharmacol 1990, 101, 531–540. [Google Scholar]
- Song, Z; Levin, BE; McArdle, JJ; Bakhos, N; Routh, VH. Convergence of pre- and postsynaptic influences on glucosensing neurons in the ventromedial hypothalamic nucleus. Diabetes 2001, 50, 2673–2681. [Google Scholar]
- Shiraishi, T. Noradrenergic neurons modulate lateral hypothalamic chemical and electrical stimulation induced feeding. Brain. Res. Bull 1991, 27, 347–351. [Google Scholar]
- Mizuno, Y; Oomura, Y. Glucose responding neurons in the nucleus tractus solitarius of the rat, in vitro study. Brain. Res 1984, 307, 109–116. [Google Scholar]
- Nakano, Y; Oomura, Y. Feeding-related activity of glucose- and morphine-sensitive neurons in the monkey amygdala. Brain. Res 1986, 399, 167–172. [Google Scholar]
- Matveyenko, AV; Donovan, CM. Metabolic Sensors mediate hypoglycemic detection at the portal vein. Diabetes 2006, 55, 1276–1282. [Google Scholar]
- Koyama, Y; Coker, RH; Stone, EE; Lacy, DB; Jabbour, K; Williams, PE; Wasserman, DH. Evidence that carotid bodies play an important role in glucoregulation in vivo. Diabetes 2000, 49, 1434–1442. [Google Scholar]
- Raybould, HE. Sensing of glucose in the gastrointestinal tract. Auton. Neurosci—Basic. Clin 2007, 133, 86–90. [Google Scholar]
- Ashford, MLJ; Boden, PR; Treherne, JM. Glucose-induced excitation of hypothalamic neurones is mediated by ATP-sensitive K+ channels. Pfugers. Arch—Eur. J. Physiol 1990, 415, 479–483. [Google Scholar]
- Fioramonti, X; Lorsignol, A; Taupignon, A; Penicaud, L. A new ATP-Sensitive K+ channel-independent mechanism is involved in glucose-excited neurons of mouse arcuate nucleus. Diabetes 2004, 53, 2767–2775. [Google Scholar]
- Kang, L; Dunn-Meynell, AA; Routh, VH; Gaspers, LD; Nagata, Y; Nishimura, T; Eiki, J; Zhang, BB; Levin, BE. Glucokinase is a critical regulator of ventromedial hypothalamic neuronal glucosensing. Diabetes 2006, 55, 412–420. [Google Scholar]
- Kang, L; Routh, VH; Kuzhikandathil, EV; Gaspers, L; Levin, BE. Physiological and molecular properties of rat hypothalamic ventromedial nucleus glucosensing neurons. Diabetes 2004, 53, 559. [Google Scholar]
- González, JA; Jensen, LT; Fugger, L; Burdakov, D. Metabolism-Independent sugar sensing in central orexin neurons. Diabetes 2008, 57, 2569–2576. [Google Scholar]
- Wang, R; Liu, X; Hentges, ST; Dunn-Meynell, AA; Levin, BE; Wang, W; Routh, VH. The regulation of glucose-excited neurons in the hypothalamic arcuate nucleus by glucose and feeding-relevant peptides. Diabetes 2004, 53, 1959–1965. [Google Scholar]
- Song, Z; Routh, VH. Differential effects of glucose and lactate on glucosensing neurons in the ventromedial hypothalamic nucleus. Diabetes 2005, 54, 15–22. [Google Scholar]
- Song, Z; Routh, VH. Recurrent hypoglycemia reduces the glucose sensitivity of glucose-inhibited neurons in the ventromedial hypothalamus nucleus (VMN). Amer. J. Physiol.—Regul. Integr. C 2006, 291, 1283–1287. [Google Scholar]
- Murphy, BA; Fioramonti, X; Jochnowitz, N; Fakira, K; Gagen, K; Contie, S; Lorsignol, A; Penicaud, L; Martin, WJ; Routh, VH. Fasting enhances the response of arcuate neuropeptide Y (NPY)-glucose-inhibited (GI) neurons to decreased extracellular glucose. Amer. J. Physiol.—Cell. Physiol 2009, 296, 746–756. [Google Scholar]
- Silver, IA; Erecinska, M. Glucose-induced intracellular ion changes in sugar-sensitive hypothalamic neurons. J. Neurophysiol 1998, 79, 1733–1745. [Google Scholar]
- De Vries, MG; Arseneau, LM; Lawson, ME; Beverly, JL. Extracellular glucose in rat ventromedial hypothalamus during acute and recurrent hypoglycemia. Diabetes 2003, 52, 2767–2773. [Google Scholar]
- Fan, X; Ding, Y; Brown, S; Zhou, L; Shaw, M; Vella, MC; Cheng, H; McNay, EC; Sherwin, RS; McCrimmon, RJ. Hypothalamic AMP-activated protein kinase activation with AICAR amplifies counterregulatory responses to hypoglycemia in a rodent model of type 1 diabetes. Amer. J. Physiol.—Regul. Integr. C 2009, 296, 1702–1708. [Google Scholar]
- McNay, EC; Fries, TM; Gold, PE. Decreases in rat extracellular hippocampal glucose concentration associated with cognitive demand during a spatial task. Proc. Nat. Acad. Sci 2000, 97, 2881–2885. [Google Scholar]
- McNay, EC; Gold, PE. Extracellular glucose concentrations in the rat hippocampus measured by zero-net-flux: effects of microdialysis flow rate, strain, and age. J. Neurochem 1999, 72, 785–790. [Google Scholar]
- McNay, EC; McCarty, RC; Gold, PE. Fluctuations in brain glucose concentration during behavioral testing: dissociations between brain areas and between brain and blood. Neurobiol. Learn. Memory 2001, 75, 325–337. [Google Scholar]
- VanGilder, R; Kelly, K; Chua, M; Ptachcinski, R; Huber, J. Administration of sesamol improved blood-brain barrier function in streptozotocin-induced diabetic rats. Exp. Brain. Res 2009, 197, 23–34. [Google Scholar]
- Colombani, AL; Carneiro, L; Benani, A; Galinier, A; Jaillard, T; Duparc, T; Offer, GR; Lorsignol, A; Magnan, C; Casteilla, L; Pénicaud, L; Leloup, C. Enhanced hypothalamic glucose sensing in obesity: alteration of redox wignaling. Diabetes 2009, 58, 2189–2197. [Google Scholar]
- Spanswick, D; Smith, MA; Mirshamsi, S; Routh, VH; Ashford, MLJ. Insulin activates ATP-sensitive K+ channels in hypothalamic neurons of lean, but not obese rats. Nat. Neurosci 2000, 3, 757–758. [Google Scholar]
- Ashford, MLJ; Sturgess, NC; Trout, NJ; Gardner, NJ; Hales, CN. Adenosine-5′-triphosphate-sensitive ion channels in neonatal rat cultured central neurons. Pfugers. Arch.—Eur. J. Physiol 1988, 412, 297–304. [Google Scholar]
- Miki, T; Liss, B; Minami, K; Shiuchi, T; Saraya, A; Kashima, Y; Horiuchi, M; Ashcroft, FM; Minokoshi, Y; Roeper, J; Seino, S. ATP-sensitive potassium channels in hypothalamic neurons play an essential role in the maintenance of glucose homeostasis by controlling glucagon release and food intake. Nat. Neurosci 2001, 5, 507–512. [Google Scholar]
- Lee, K; Dixon, AK; Richardson, PJ; Pinnock, RD. Glucose-receptive neurones in the rat ventromedial hypothalamus express KATP channels composed of Kir6.1 and SUR1 subunits. J. Physiol 1999, 515, 439–452. [Google Scholar]
- Dunn-Meynell, AA; Routh, VH; Kang, L; Gaspers, L; Levin, BE. Glucokinase is the likely mediator of glucosensing in both glucose-excited and glucose-inhibited central neurons. Diabetes 2002, 51, 2056–2065. [Google Scholar]
- Claret, M; Smith, MA; Batterham, RL; Selman, C; Choudury, AI; Fryer, LG; Clements, M; Al-Qassab, H; Heffron, H; Xu, AW; Speakman, JR; Barsh, GS; Viollet, B; Vaulont, S; Ashford, ML; Carling, D; Withers, DJ. AMPK is essential for energy homeostasis regulation and glucose sensing by POMC and AgRP neurons. J. Clin. Invest 2007, 117, 2325–2336. [Google Scholar]
- Cotero, VE; Routh, VH. Insulin blunts the response of glucose-excited (GE) neurons in the ventrolateral-ventromedial hypothalamic nucleus (VL-VMN) to decreased glucose. Amer. J. Physiol.—Endocrinol. Metab 2009, 296, 1101–1109. [Google Scholar]
- Plum, L; Ma, X; Hampel, B; Balthasar, N; Coppari, R; Munzberg, H; Shanabrough, M; Burdakov, D; Rother, E; Janoschek, R; Alber, J; Belgardt, BF; Koch, L; Seibler, J; Schwenk, F; Fekete, C; Suzuki, A; Mak, TW; Krone, W; Horvath, TL; Ashcroft, FM; Bruning, JC. Enhanced PIP3 signaling in POMC neurons causes KATP channel activation and leads to diet-sensitive obesity. J. Clin. Invest 2006, 116, 1886–1901. [Google Scholar]
- Ibrahim, N; Bosch, MA; Smart, JL; Qiu, J; Rubinstein, M; Ronnekleiv, OK; Low, MJ; Kelly, MJ. Hypothalamic proopiomelanocortin neurons are glucose responsive and express K(ATP) channels. Endocrinology 2003, 144, 1331–1340. [Google Scholar]
- Fioramonti, X; Contie, S; Song, Z; Routh, VH; Lorsignol, A; Penicaud, L. Characterization of glucosensing neuron subpopulations in the arcuate nucleus: integration in NPY and POMC networks? Diabetes 2007, 56, 1219–1227. [Google Scholar]
- Hetherington, AW; Ranson, SW. The relation of various hypothalamic lesions to adiposity in the rat. J. Comp. Neurol 1942, 76, 475–499. [Google Scholar]
- Kishi, E; Takahashi, A; Ishimaru, H; Ikarashi, Y; Maruyama, Y. Development of obesity and neurochemical backing in aurothioglucose-treated mice. Auton. Neurosci.—Basic. Clin 2001, 92, 21–27. [Google Scholar]
- Spanswick, D; Smith, MA; Groppi, VE; Logan, SD; Ashford, MLJ. Leptin inhibits hypothalamic neurons by activation of ATP-sensitive potassium channels. Nature 1997, 390, 521–525. [Google Scholar]
- Irani, BG; Le Foll, C; Dunn-Meynell, A; Levin, BE. Effects of leptin on rat ventromedial hypothalamic neurons. Endocrinology 2008, 149, 5146–5154. [Google Scholar]
- Elmquist, JK; Ahima, RS; Elias, CF; Flier, JS; Saper, CB. Leptin activates distinct projections from the dorsomedial and ventromedial hypothalamic nuclei. Proc. Natl. Acad. Sci 1998, 95, 741–746. [Google Scholar]
- Mirshamsi, S; Laidlaw, H; Ning, K; Anderson, E; Burgess, L; Gray, A; Sutherland, C; Ashford, M. Leptin and insulin stimulation of signalling pathways in arcuate nucleus neurones: PI3K dependent actin reorganization and KATP channel activation. BMC. Neurosci 2004, 5, 54. [Google Scholar]
- Murphy, BA; Fakira, KA; Song, Z; Beuve, A; Routh, VH. AMP-activated Protein Kinase (AMPK) and Nitric Oxide (NO) regulate the glucose sensitivity of ventromedial hypothalamic (VMH) glucose-inhibited (GI) neurons. Amer. J. Physiol.—Cell. Physiol 2009, 297, C750–C758. [Google Scholar]
- Anderson, KA; Ribar, TJ; Lin, F; Noeldner, PK; Green, MF; Muehlbauer, MJ; Witters, LA; Kemp, BE; Means, AR. Hypothalamic CaMKK2 contributes to the regulation of energy balance. Cell. Metab 2008, 7, 377–388. [Google Scholar]
- Xue, B; Kahn, BB. AMPK integrates nutrient and hormonal signals to regulate food intake and energy balance through effects in the hypothalamus and peripheral tissues. J. Physiol 2006, 574, 73–83. [Google Scholar]
- Hegyi, K; Fnlöp, K; Kovács, K; Tóth, S; Falus, A. Leptin-induced signal transduction pathways. Cell. Biol. Int 2004, 28, 159–169. [Google Scholar]
- Kohno, D; Sone, H; Minokoshi, Y; Yada, T. Ghrelin raises [Ca2+] i via AMPK in hypothalamic arcuate nucleus NPY neurons. Biochem. Biophys. Res. Commun 2008, 366, 388–392. [Google Scholar]
- Kola, B; Hubina, E; Tucci, SA; Kirkham, TC; Garcia, EA; Mitchell, SE; Williams, LM; Hawley, SA; Hardie, DG; Grossman, AB; Korbonits, M. Cannabinoids and ghrelin have both central and peripheral metabolic and cardiac effects via AMP-activated protein kinase. J. Biol. Chem 2005, 280, 25196–25201. [Google Scholar]
- Minokoshi, Y; Alquier, T; Furukawa, N; Kim, YB; Lee, A; Xue, B; Mu, J; Foufelle, F; Ferre, P; Birnbaum, MJ; Stuck, BJ; Kahn, BB. AMP-kinase regulates food intake by responding to hormonal and nutrient signals in the hypothalamus. Nature 2004, 428, 569–574. [Google Scholar]
- Kahn, BB; Alquier, T; Carling, D; Hardie, DG. AMP-activated protein kinase: Ancient energy gauge provides clues to modern understanding of metabolism. Cell. Metab 2005, 1, 15–25. [Google Scholar]
- Andersson, U; Filipsson, K; Abbott, CR; Woods, A; Smith, K; Bloom, SR; Carling, D; Small, CJ. AMP-activated protein kinase plays a role in the control of food intake. J. Biol. Chem 2004, 279, 12005–12008. [Google Scholar]
- Canabal, DD; Song, Z; Potian, JG; Beuve, A; McArdle, JJ; Routh, VH. Glucose, insulin and leptin signaling pathways modulate nitric oxide (NO) synthesis in glucose-inhibited (GI) neurons in the ventromedial hypothalamus (VMH). Amer. J. Physiol. Regul. Integr. C 2007, 292, 1418–1428. [Google Scholar]
- Lira, VA; Soltow, QA; Long, JHD; Betters, JL; Sellman, JE; Criswell, DS. Nitric oxide increases GLUT4 expression and regulates AMPK signaling in skeletal muscle. Amer. J. Physiol. Endocrinol. Metab 2007, 293, 1062–1068. [Google Scholar]
- Zhang, J; Xie, Z; Dong, Y; Wang, S; Liu, C; Zou, MH. Identification of nitric oxide as an endogenous activator of the AMP-activated protein kinase in vascular endothelial cells. J. Biol. Chem 2008, 283, 27452–27461. [Google Scholar]
- Alquier, T; Kawashima, J; Tsuji, Y; Kahn, BB. Role of hypothalamic AMP kinase in the impaired counterregulatory response induced by repetitive neuroglucopenia. Endocrinology 2006, 148, 1367–1375. [Google Scholar]
- Kahn, BB; Myers, MG. MTOR tells the brain that the body is hungry. Nature. Med 2006, 12, 615–617. [Google Scholar]
- Patti, ME; Kahn, BB. Nutrient sensor links obesity with diabetes risk. Nature Med 2004, 10, 1049–1050. [Google Scholar]
- Jaffrey, SR; Snyder, SH. Nitric oxide: A neural messenger. Annu. Rev. Cell. Dev. Biol 1995, 11, 417–440. [Google Scholar]
- Boehning, D; Snyder, SH. Novel neural modulators. Annu. Rev. Neurosci 2003, 26, 105–131. [Google Scholar]
- Farr, SA; Banks, WA; Kumar, VB; Morley, JE. Orexin-A-induced feeding is dependent on nitric oxide. Peptides 2005, 26, 759–765. [Google Scholar]
- Gaskin, FS; Farr, SA; Banks, WA; Kumar, VB; Morley, JE. Ghrelin-induced feeding is dependent on nitric oxide. Peptides 2003, 24, 913–918. [Google Scholar]
- Morley, JE; Farr, SA. Cachexia and neuropeptide Y. Nutrition 2008, 24, 815–819. [Google Scholar]
- Morley, JE; Kumar, VB; Mattammal, M; Villareal, DT. Measurement of nitric oxide synthase and its mRNA in genetically obese (ob/ob) mice. Life Sci 1995, 57, 1327–1331. [Google Scholar]
- Hallows, KR; Raghuram, V; Kemp, BE; Witters, LA; Foskett, JK. Inhibition of cystic fibrosis transmembrane conductance regulator by novel interaction with the metabolic sensor AMP-activated protein kinase. J. Clin. Invest 2000, 105, 1711–1721. [Google Scholar]
- Conte-Camerino, D; Mambrini, M; DeLuca, A; Tricarico, D; Bryant, SH; Tortorella, V; Bettoni, G. Enantiomers of clofibric acid analogs have opposite actions on rat skeletal muscle chloride channels. Pflugers. Arch.—Eur. J. Physiol 1988, 413, 105–107. [Google Scholar]
- Gadsby, DC; Nairn, AC. Control of CFTR channel gating by phosphorylation and nucleotide hydrolysis. Physiolo. Rev 1999, 79, 77–107. [Google Scholar]
- Hadjiliadis, D; Madill, J; Chaparro, C; Tsang, A; Waddell, TK; Singer, LG; Hutcheon, MA; Keshavjee, S; Elizabeth Tullis, D. Incidence and prevalence of diabetes mellitus in patients with cystic fibrosis undergoing lung transplantation before and after lung transplantation. Clin. Transplant 2005, 19, 773–778. [Google Scholar]
- Lanng, S. Glucose intolerance in cystic fibrosis patients. Paed. Resp. Rev. Online 2001, 2, 253–259. [Google Scholar]
- Reali, MF; Festini, F; Neri, AS; Taccetti, G; Repetto, T; Chiarelli, F; Toni, S. Use of continuous subcutaneous insulin infusion in cystic fibrosis patients with cystic fibrosis-related diabetes awaiting transplantation. J. Cystic. Fibrosis 2006, 5, 67–68. [Google Scholar]
- Siahanidou, T; Mandyla, H; Doudounakis, S; Anagnostakis, D. Hyperglycaemia and insulinopenia in a neonate with cystic fibrosis. Acta. Paediatr 2005, 94, 1837–1840. [Google Scholar]
- Bai, FL; Yamano, M; Shiotani, Y; Emson, PC; Smith, AD; Powell, JF; Tohyama, M. An arcuato-paraventricular and -dorsomedial hypothalamic neuropeptide Y-containing system which lacks noradrenaline in the rat. Brain. Res 1985, 331, 172–175. [Google Scholar]
- Muroya, S; Yada, T; Shioda, S; Takigawa, M. Glucose-sensitive neurons in the rat arcuate nucleus contain neuropeptide Y. Neurosci. Lett 1999, 264, 113–116. [Google Scholar]
- Amiel, SA; Tamborlane, WV; Simonson, DC; Sherwin, RS. Defective glucose counterregulation after strict glycemic control of insulin-dependent diabetes mellitus. N. Engl. J. Med 1987, 316, 1376–1383. [Google Scholar]
- Cryer, PE. Glucose counterregulation in man. Diabetes 1981, 30, 261–264. [Google Scholar]
- The Diabetes Control and Complications Trial Research Group. The effect of intensive treatment of diabetes on the development and progression of long-term complications in insulin-dependent diabetes mellitus. The diabetes control and complications trial research group. N. Engl. J. Med 1993, 329, 977–986. [Google Scholar]
- Henderson, JN; Allen, KV; Deary, IJ; Frier, BM. Hypoglycaemia in insulin-treated Type 2 diabetes: frequency, symptoms and impaired awareness. Diabetic. Med 2003, 20, 1016–1021. [Google Scholar]
- Donnelly, LA; Morris, AD; Frier, BM; Ellis, JD; Donnan, PT; Durrant, R; Band, MM; Reekie, G; Leese, GP. Frequency and predictors of hypoglycaemia in Type 1 and insulin-treated Type 2 diabetes: A population-based study. Diabetic. Med 2005, 22, 749–755. [Google Scholar]
- Saberi, M; Bohland, M; Donovan, CM. The locus for hypoglycemic detection shifts with the rate of fall in glycemia: The role of portal-superior mesenteric vein glucose sensing. Diabetes 2008, 57, 1380–1386. [Google Scholar]
- Ritter, S; Bugarith, K; Dinh, TT. Immunotoxic destruction of distinct catecholamine subgroups produces selective impairment of glucoregulatory responses and neuronal activation. J. Comp. Neurol 2001, 432, 197–216. [Google Scholar]
- McCrimmon, RJ; Song, Z; Cheng, H; McNay, EC; Weikart-Yeckel, C; Fan, X; Routh, VH; Sherwin, RS. Corticotrophin-releasing factor receptors within the ventromedial hypothalamus regulate hypoglycemia- induced hormonal counterregulation. J. Clin. Invest 2006, 116, 1723–1730. [Google Scholar]
- Watts, AG; Donovan, CM. Sweet talk in the brain: Glucosensing, neural networks, and hypoglycemic counterregulation. Front. Neuroendocrinol 2010, 31, 32–43. [Google Scholar]
- Kang, L; Sanders, NM; Dunn-Meynell, AA; Gaspers, LD; Routh, VH; Thomas, AP; Levin, BE. Prior hypoglycemia enhances glucose responsiveness in some ventromedial hypothalamic glucosensing neurons. Amer. J. Physiol. Regul. Integr. C 2008, 294, R784–R792. [Google Scholar]
- Fioramonti, X; Song, Z; Vazirani, RP; Beuve, A; Routh, VH. Hypothalamic NO in hypoglycemia detection and counter-regulation: A two edged sword. Antioxid. Redox. Signal 2010, 0, 1. [Google Scholar]
- Borg, MA; Tamborlane, WV; Shulman, GI; Sherwin, RS. Local lactate perfusion of the ventromedial hypothalamus suppresses hypoglycemic counterregulation. Diabetes 2003, 52, 663–666. [Google Scholar]
- Fioramonti, X; Marsollier, N; Song, Z; Fakira, KA; Patel, RM; Brown, S; Duparc, T; Pica-Mendez, A; Sanders, NM; Knauf, C; Valet, P; McCrimmon, RJ; Beuve, A; Magnan, C; Routh, VH. Ventromedial Hypothalamic nitric oxide production is necessary for hypoglycemia detection and counterregulation. Diabetes 2010, 59, 519–528. [Google Scholar]
- Canabal, DD; Potian, JG; Duran, RG; McArdle, JJ; Routh, VH. Hyperglycemia impairs glucose and insulin regulation of nitric oxide production in glucose-inhibited neurons in the ventromedial hypothalamus. Amer. J. Physiol. Regul. Integr. C 2007, 293, 592–600. [Google Scholar]
- Cidad, P; Almeida, A; Bolanos, JP. Inhibition of mitochondrial respiration by nitric oxide rapidly stimulates cytoprotective GLUT-3mediated glucose uptake through 5′-AMP-activated protein kinase. Biochem. J 2004, 384, 629–636. [Google Scholar]
- Tsacopoulos, M; Magistretti, PJ. Metabolic coupling between glia and neurons. Eur. J. Neurosci 1996, 16, 877–885. [Google Scholar]
- Bolanos, JP; Almeida, A. Modulation of astroglial energy metabolism by nitric oxide. Antioxid. Redox. Signal 2006, 8, 955–965. [Google Scholar]
- Page, KA; Arora, J; Qiu, M; Relwani, R; Constable, RT; Sherwin, RS. Small decrements in systemic glucose provoke increases in hypothalamic blood flow prior to the release of counterregulatory Hormones. Diabetes 2009, 58, 448–452. [Google Scholar]
- Kennan, RP; Takahashi, K; Pan, C; Shamoon, H; Pan, JW. Human cerebral blood flow and metabolism in acute insulin-induced hypoglycemia. J. Cerebr. Blood. Flow. Metabol 2005, 25, 527–534. [Google Scholar]
- Singh, P; Jain, A; Kaur, G. Impact of hypoglycemia and diabetes on CNS: Correlation of mitochondrial oxidative stress with DNA damage. Mol. Cell. Biochem 2004, 260, 153–159. [Google Scholar]
- Jaffrey, SR; Erdjument-Bromage, H; Ferris, CS; Tempst, P; Snyder, SH. Protein S-nitrosylation: a physiological signal for neuronal nitric oxide. Nat. Cell. Biol 2001, 3, 193–197. [Google Scholar]
- Yu, HM; Xu, J; Li, C; Zhou, C; Zhang, F; Han, D; Zhang, GY. Coupling between neuronal nitric oxide synthase and glutamate receptor 6-mediated c-Jun N-terminal kinase signaling pathway via S-nitrosylation contributes to ischemia neuronal death. Neuroscience 2008, 155, 1120–1132. [Google Scholar]
- Sayed, N; Baskaran, P; Ma, X; van den Akker, F; Beuve, A. Desensitization of soluble guanylyl cyclase, the NO receptor, by S-nitrosylation. Proc. Nat. Amer. Sci 2007, 104, 12312–12317. [Google Scholar]
- Fioramonti, X; Fakira, K; Beluch, N; Leloup, C; Saba, N; Penicaud, L; Beuve, A; Routh, VH. Role of hypothalamic S-nitrosylation in the development of hypoglycemia-associated autonomic failure. Amer Diabetes Assoc 69th Sci Session 2009. Abstract # 129-OR. [Google Scholar]
- Beck, B. Neuropeptides and obesity. Nutrition 2000, 16, 916–923. [Google Scholar]
- Sindelar, DK; Marie, L; Miura, GI; Palmiter, RD; McMinn, JE; Morton, GJ; Schwartz, MW. Neuropeptide Y Is required for hyperphagic feeding in response to neuroglucopenia. Endocrinology 2004, 145, 3363–3368. [Google Scholar]
- Levin, BE; Dunn-Meynell, AA. Reduced central leptin sensitivity in rats with diet-induced obesity. Amer. J. Physiol 2002, 283, R941–R948. [Google Scholar]
- Orosco, M; Rouch, C; Nicolaidis, S. Resistance of the obese zucker rat to insulin-induced feeding and to satiety induced by coinfusion of insulin and glucose. Appetite 1994, 23, 209–218. [Google Scholar]
- Cotero, VE; Zhang, BB; Routh, VH. The response of glucose-excited neurones in the ventromedial hypothalamus to decreased glucose is enhanced in a murine model of Type 2 diabetes mellitus. J. Neuroendocrinol 2009, 22, 65–74. [Google Scholar]
- Irani, BG; Dunn-Meynell, AA; Levin, BE. Altered hypothalamic leptin, insulin, and melanocortin binding associated with moderate-fat diet and predisposition to obesity. Endocrinology 2007, 148, 310–316. [Google Scholar]
- Irani, BG; Le Foll, C; Dunn-Meynell, AA; Levin, BE. Ventromedial nucleus neurons are less sensitive to leptin excitation in rats bred to develop diet-induced obesity. Amer. J. Physiol. Regul. Integr. C 2009, 296, 521–527. [Google Scholar]
- Hill, JW; Xu, Y; Preitner, F; Fukuda, M; Cho, YR; Luo, J; Balthasar, N; Coppari, R; Cantley, LC; Kahn, BB; Zhao, JJ; Elmquist, JK. Phosphatidyl inositol 3-kinase signaling in hypothalamic proopiomelanocortin neurons contributes to the regulation of glucose homeostasis. Endocrinology 2009, 150, 4874–4882. [Google Scholar]
- Levin, BE; Govek, EK; Dunn-Meynell, AA. Reduced glucose-induced neuronal activation in the hypothalamus of diet-induced obese rats. Brain. Res 1998, 808, 317–319. [Google Scholar]
- Diggs-Andrews, KA; Zhang, X; Song, Z; Daphna-Iken, D; Routh, VH; Fisher, SJ. Brain insulin action regulates hypothalamic glucose sensing and the counterregulatory response to hypoglycemia. Diabetes 2010, 59, 2271–2280. [Google Scholar]
- Guillod-Maximin, E; Lorsignol, A; Alquier, T; Penicaud, L. Acute intracarotid glucose injection towards the brain induces specific c-fos activation in hypothalamic nuclei: involvement of astrocytes in cerebral glucose-sensing in rats. J. Neuroendocrinol 2004, 16, 464–471. [Google Scholar]
- Bady, I; Marty, N; Dallaporta, M; Emery, M; Gyger, J; Tarussio, D; Foretz, M; Thorens, B. Evidence from glut2-null mice that glucose is a critical physiological regulator of feeding. Diabetes 2006, 55, 988–995. [Google Scholar]
- Stolarczyk, E; Guissard, C; Michau, A; Even, PC; Grosfeld, A; Serradas, P; Lorsignol, A; Penicaud, L; Brot-Laroche, E; Leturque, A; Le Gall, M. Detection of extracellular glucose by GLUT2 contributes to hypothalamic control of food intake. Amer. J. Physiol.—Endocrinol. Met 2010, 298, 1078–1087. [Google Scholar]
- Marty, N; Dallaporta, M; Foretz, M; Emery, M; Tarussio, D; Bady, I; Binnert, C; Beermann, F; Thorens, B. Regulation of glucagon secretion by glucose transporter type 2 (glut2) and astrocyte-dependent glucose sensors. J. Clin. Invest 2005, 115, 3545–3553. [Google Scholar]
© 2010 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Routh, V.H. Glucose Sensing Neurons in the Ventromedial Hypothalamus. Sensors 2010, 10, 9002-9025. https://doi.org/10.3390/s101009002
Routh VH. Glucose Sensing Neurons in the Ventromedial Hypothalamus. Sensors. 2010; 10(10):9002-9025. https://doi.org/10.3390/s101009002
Chicago/Turabian StyleRouth, Vanessa H. 2010. "Glucose Sensing Neurons in the Ventromedial Hypothalamus" Sensors 10, no. 10: 9002-9025. https://doi.org/10.3390/s101009002
APA StyleRouth, V. H. (2010). Glucose Sensing Neurons in the Ventromedial Hypothalamus. Sensors, 10(10), 9002-9025. https://doi.org/10.3390/s101009002