Effective DNA Inhibitors of Cathepsin G by In Vitro Selection

Abstract
:1. Introduction
2. Results
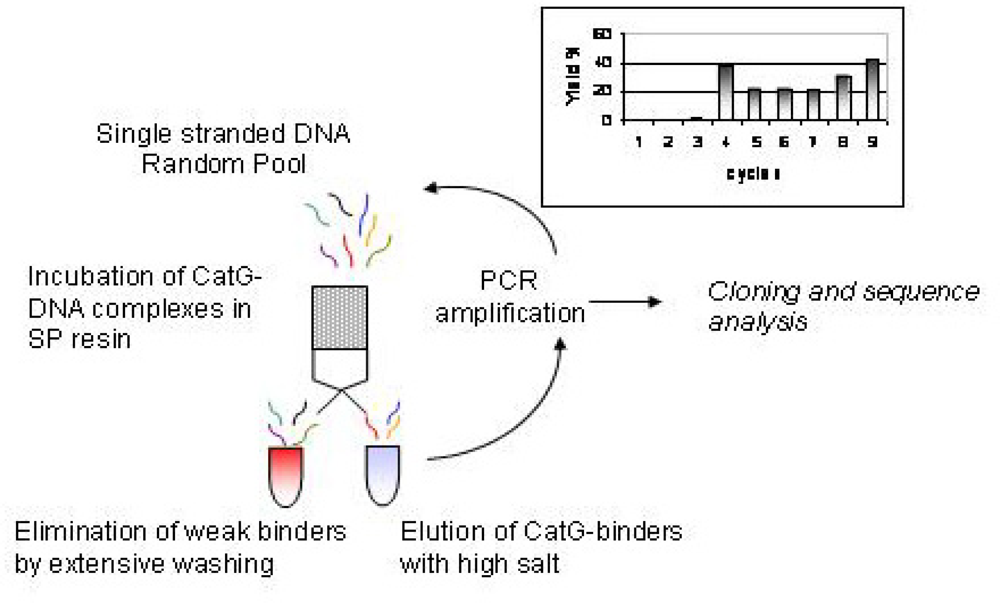
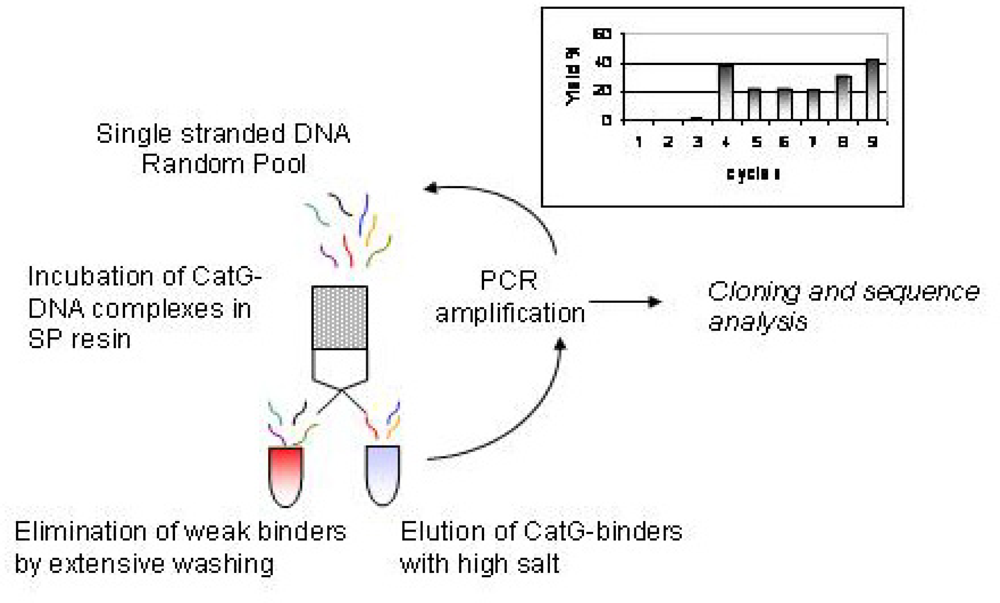
2.1 Selection of CatG binders from a random DNA pool
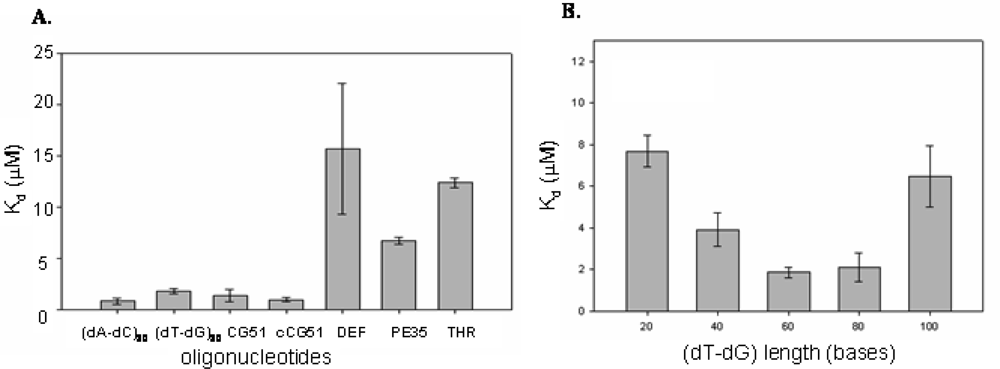
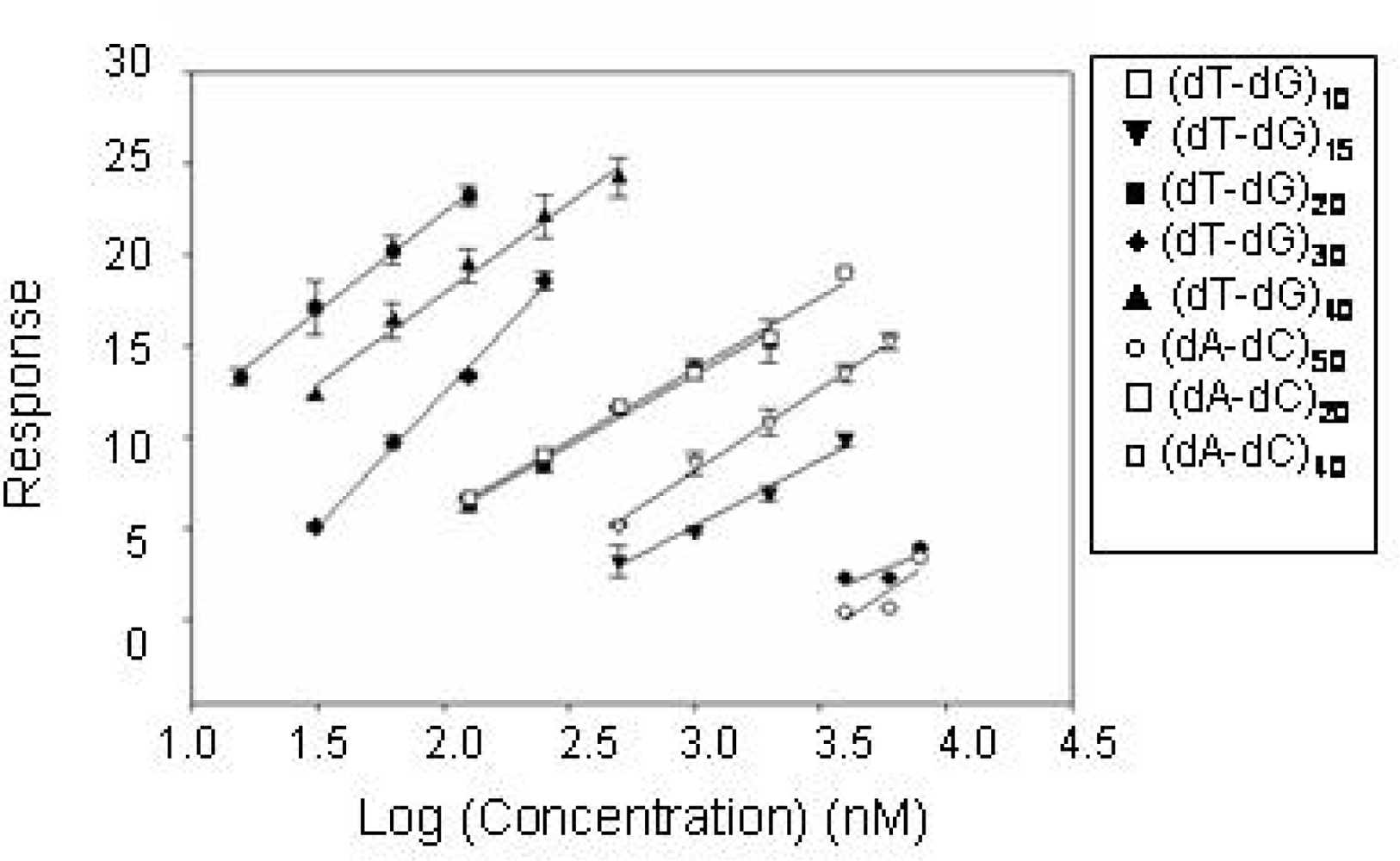
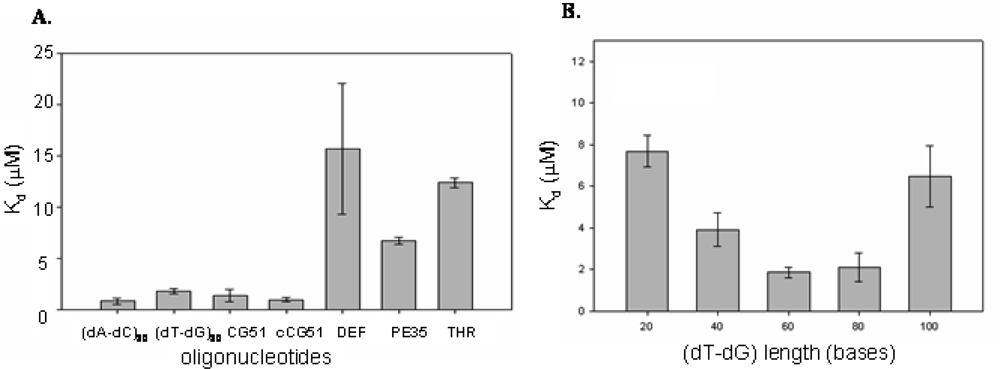
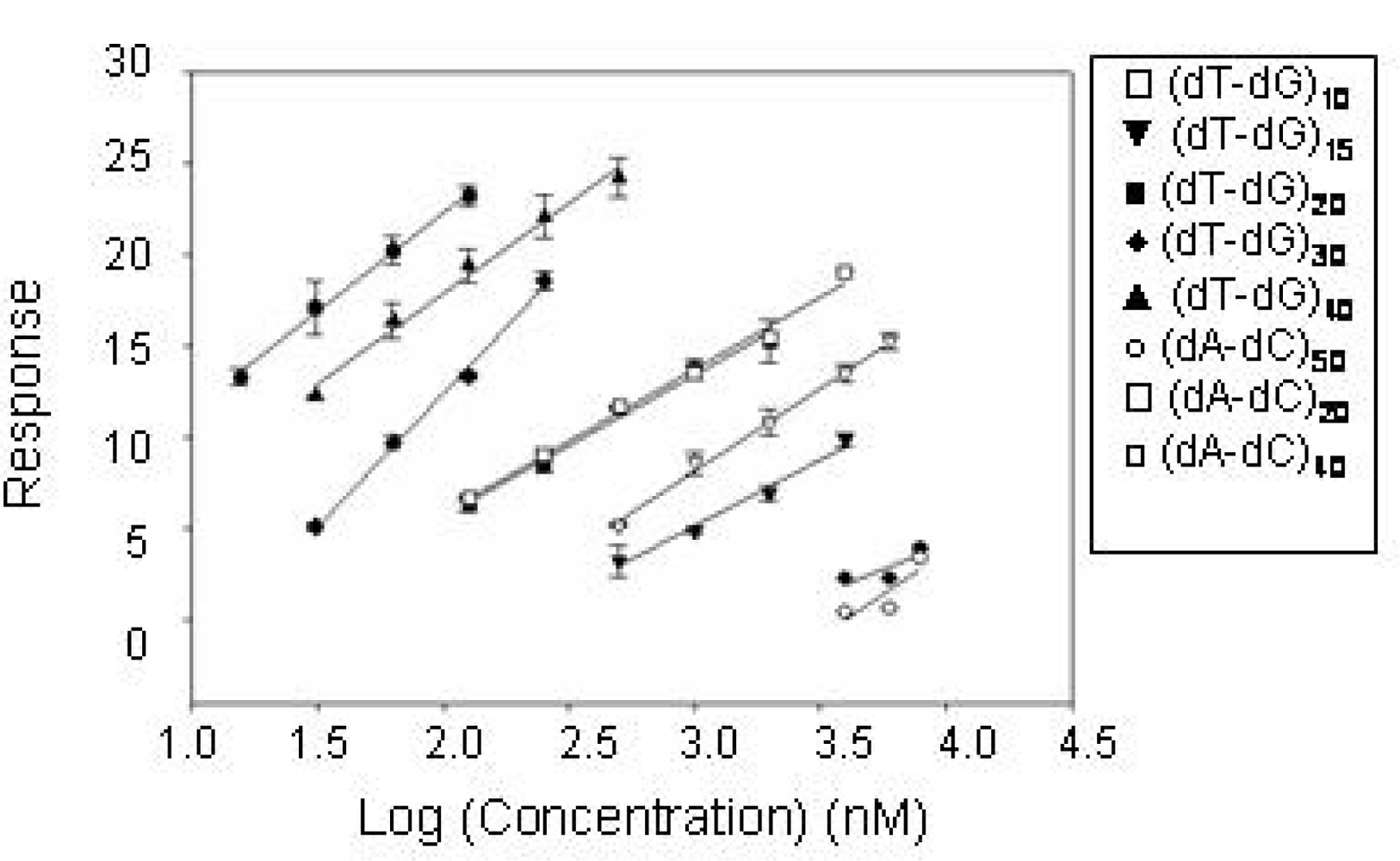
2.2 Identification of structural motifs in DNA - CatG binding
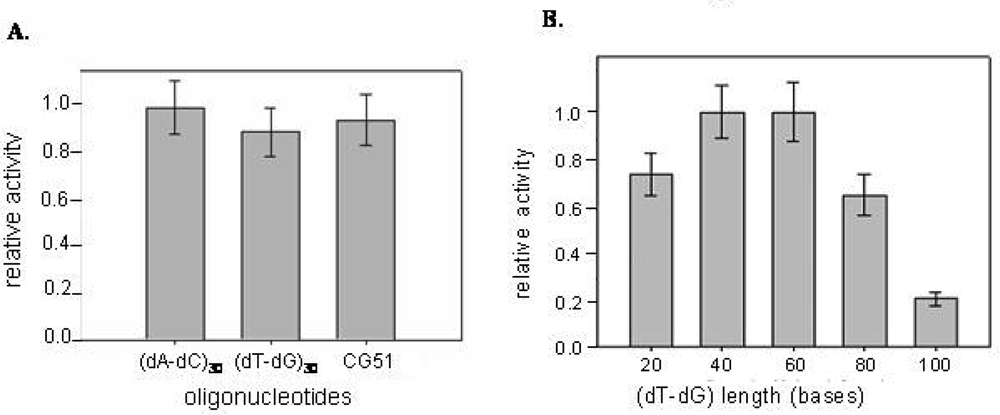
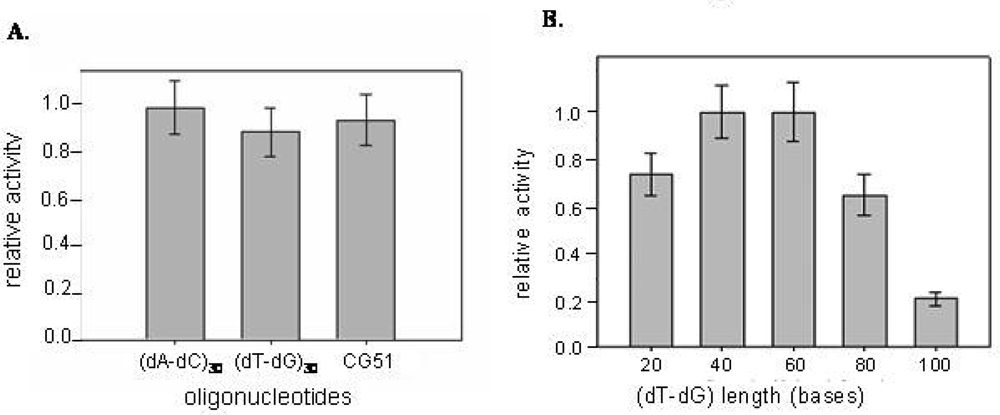
2.3 Inhibition of CatG catalytic activity
3. Discussion
4. Experimental Section
4.1 Materials
4.2 Selection of DNA-CatG binders
4.3 Cloning and sequencing
4.4 Kd determination
4.5 Surface Plasmon Resonance (SPR) experiments
4.6 Enzymatic assays
References
- Campbell, E; Silvermann, E; Campbell, M. Elastase and cathepsin G of human monocytes. Quantification of cellular content, release in response of stimuli, and heterogeneity in elastase-mediated proteolytic activity. J. Immunol. 1989, 143, 2961–2968. [Google Scholar]
- Barret, A; Rawlings, N; Woessmer, J. Handbook of proteolytic enzymes 1998.
- Plescia, J; Altieri, D. Activation of Mac-1 (CD11b/CD18)-bound factor X by released cathepsin G defines an alternative pathway of leucocyte initiation of coagulation. Biochem. J. 1996, 319, 873–879. [Google Scholar]
- Allen, D; Tracy, PB. Human coagulation factor V is activated to the functional cofactor by elastase and cathepsin G expressed at the monocyte surface. J. Biol. Chem. 1994, 1995, 1409–1415. [Google Scholar]
- LaRosa, C; Roher, M; Benoit, S; Rodino, L; Barnard, M; Michelso, A. Human neutrophyl cathepsin G is a potent platelet activator. J. Vasc. Surg. 1994, 19, 306–318. [Google Scholar]
- Crystal, R. Alpha 1-antitrypsin deficiency, emphysema, and liver disease. Genetic basis and strategies for therapy. J. Clin. Invest. 1990, 85, 1343–1352. [Google Scholar]
- Fujita, J; Nelson, N; Daughton, D; Dobry, C; Spurzem, J; Irino, S; Rennard, S. Evaluation of elastase and antielastase balance in patients with chronic bronchitis and pulmonary emphysema. Am. Rev. Respir. Dis. 1990, 142, 57–62. [Google Scholar]
- Boudier, C; Cadene, M; Bieth, J. Inhibition of neutrophil cathepsin G by oxidized mucus proteinase inhibitor. Effect of heparin. Biochemistry 1999, 38, 8451–8457. [Google Scholar]
- Maryanoff, B. Inhibitors of serine proteases as potential therapeutic agents: the road from thrombin to tryptase to cathepsin G. J. Med. Chem. 2004, 47, 769–787. [Google Scholar]
- Suter, S; Chevallier, I. The effect of eglin C on the function of human neutrophils in vitro. Biol. Chem. Hoppe Seyler 1988, 369, 573–578. [Google Scholar]
- Belorgey, D; Dirrig, S; Amouric, M; Figarella, C; Bieth, J. Inhibition of human pancreatic proteinases by mucus proteinase inhibitor, eglin c and aprotinin. Biochem. J. 1996, 313, 555–560. [Google Scholar]
- Beatty, K; Bieth, J; Travis, J. Kinetic of association of serine proteinases with native and oxidized alpha-(1)-proteinase inhibitor and alpha-(1)-antichymotrypsin. J. Biol. Chem. 1980, 255, 3931–3934. [Google Scholar]
- Hof, P; Mayr, I; Huber, R; Korzus, E; Potempa, J; Travis, J; Powers, J; Bode, W. The 1.8 A crystal structure of human cathepsin G in complex with Suc-Val-Pro-PheP-(OPh)2: a Janus-faced proteinase with two opposite specificities. EMBO J. 1996, 15, 5481–5491. [Google Scholar]
- Ermolieff, J; Boudier, C; Laine, A; Meyer, B; Bieth, J. Heparin protects cathepsin G against inhibition by protein proteinase inhibitors. J. Biol. Chem. 1994, 269, 29502–29508. [Google Scholar]
- Evangelista, V; Piccardoni, P; Maugeri, N; De Gaetano, G; Cerletti, C. Inhibition by heparin of platelet activation induced by neutrophil-derived cathepsin G. Eur. J. Pharmacol. 1992, 216, 401–5. [Google Scholar]
- Ledoux, D; Merciris, D; Barritault, D; Caruelle, J. Heparin-like dextran derivatives as well as glycosaminoglycans inhibit the enzymatic activity of human cathepsin G. FEBS Lett. 2003, 537, 23–9. [Google Scholar]
- Peplow, PV. Glycosaminoglycan: a candidate to stimulate the repair of chronic wounds. Thromb. Haemost. 2005, 94, 4–16. [Google Scholar]
- Belorgey, D; Bieth, JG. DNA binds neutrophil elastase and mucus proteinase inhibitor and impairs their functional activity. FEBS Lett. 1995, 361, 265–268. [Google Scholar]
- Belorgey, D; Bieth, JG. Effect of polynucleotides on the inhibition of neutrophil elastase by mucus proteinase inhibitor and alpha 1-proteinase inhibitor. Biochemistry 1998, 37, 16416–22. [Google Scholar]
- Duranton, J; Boudier, C; Belorgey, D; Mellet, P; Bieth, J. DNA strongly impairs the inhibition of cathepsin G by alpha(1)-antichymotrypsin and alpha(1)-proteinase inhibitor. J. Biol. Chem. 2000, 275, 3787–3792. [Google Scholar]
- Duranton, J; Belorgey, D; Carrere, J; Donato, L; Moritz, T; Bieth, J. Effect of DNase on the activity of neutrophil elastase, cathepsin G and proteinase 3 in the presence of DNA. FEBS Lett. 2000, 473, 154–156. [Google Scholar]
- Violi, F; Ferro, D; Alessandri, C; Quintarelli, C; Saliola, M; Balsano, F. Inhibition of tissue plasminogen activator inhibitor by defibrotide in atherosclerotic patients. Semin. Thromb. Hemost. 1989, 15, 226–9. [Google Scholar]
- Violi, F; Ferro, D; Saliola, M; Quintarelli, C; Basili, S; Balsano, F. Effect of oral defibrotide on tissue-plasminogen activator and tissue-plasminogen activator inhibitor balance. Eur. J. Clin. Pharmacol. 1992, 42, 379–83. [Google Scholar]
- Richardson, PG; Murakami, C; Jin, Z; Warren, D; Momtaz, P; Hoppensteadt, D; Elias, AD; Antin, JH; Soiffer, R; Spitzer, T; et al. Multi-institutional use of defibrotide in 88 patients after stem cell transplantation with severe veno-occlusive disease and multisystem organ failure: response without significant toxicity in a high-risk population and factors predictive of outcome. Blood 2002, 100, 4337–43. [Google Scholar]
- Pescador, R; Porta, R; Ferro, L. An integrated view of the activities of defibrotide. Semin. Thromb. Hemost. 1996, 22 Suppl 1, 71–5. [Google Scholar]
- Violi, F; Marubini, E; Coccheri, S; Nenci, GG. Improvement of walking distance by defibrotide in patients with intermittent claudication--results of a randomized, placebo-controlled study (the DICLIS study). Defibrotide Intermittent CLaudication Italian Study. Thromb. Haemost. 2000, 83, 672–7. [Google Scholar]
- Evangelista, V; Piccardoni, P; de Gaetano, G; Cerletti, C. Defibrotide inhibits platelet activation by cathepsin G released from stimulated polymorphonuclear leukocytes. Thromb. Haemost. 1992, 67, 660–4. [Google Scholar]
- Tuerk, C; Gold, L. Systematic evolution of ligands by exponential enrichment: RNA ligands to bacteriophage T4 DNA polymerase. Science 1990, 249, 505–510. [Google Scholar]
- Gatto, B; Cavalli, M. From proteins to nucleic acid-based drugs: the role of biotech in anti-VEGF therapy. Anticancer Agents Med. Chem. 2006, 6, 287–301. [Google Scholar]
- Hud, N; Smith, F; Anet, F; Feigon, J. The selectivity for K+ versus Na+ in DNA quadruplex is dominated by relative free energies of hydration: a thermodinamic analysis by 1H NMR. Biochemistry 1996, 35, 15383–15390. [Google Scholar]
- Pearson, WR. Rapid and sensitive sequence comparison with FASTP and FASTA. Meth. Enzymol. 1990, 183, 63–98. [Google Scholar]
- Thompson, JD; Gibson, TJ; Plewniak, F; Jeanmougin, F; Higgins, DG. The CLUSTAL_X windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res. 1997, 25, 4876–82. [Google Scholar]
- Jeanmougin, F; Thompson, JD; Gouy, M; Higgins, DG; Gibson, TJ. Multiple sequence alignment with Clustal X. Trends Biochem. Sci. 1998, 23, 403–5. [Google Scholar]
- Zuker, M. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res. 2003, 31, 3406–3415. [Google Scholar]
- Widmer, U; Yang, Z; van Deventer, S; Manogue, KR; Sherry, B; Cerami, A. Genomic structure of murine macrophage inflammatory protein-1 alpha and conservation of potential regulatory sequences with a human homolog, LD78. J. Immunol. 1991, 146, 4031–40. [Google Scholar]
- Hamada, H; Petrino, MG; Kakunaga, T. A novel repeated element with Z-DNA-forming potential is widely found in evolutionarily diverse eukaryotic genomes. Proc. Natl. Acad. Sci. USA 1982, 79, 6465–9. [Google Scholar]
- Hamada, H; Petrino, MG; Kakunaga, T; Seidman, M; Stollar, BD. Characterization of genomic poly(dT-dG).poly(dC-dA) sequences: structure, organization, and conformation. Mol. Cell. Biol. 1984, 4, 2610–21. [Google Scholar]
- Vianini, E; Palumbo, M; Gatto, B. In vitro selection of DNA aptamers that bind L-Tyrosinamide. Bioorg. Med. Chem. 2001, 9, 2543–2548. [Google Scholar]
- Michaud, M; Jourdan, E; Ravelet, C; Villet, A; Ravel, A; Grosset, C; Peyrin, E. Immobilized DNA aptamers as target-specific chiral stationary phases for resolution of nucleoside and amino acid derivative enantiomers. Anal. Chem. 2004, 76, 1015–1020. [Google Scholar]
- Bock, L; Griffin, L; Latham, J; Vermaas, E; Toole, J. Selection of single-stranded DNA molecules that bind and inhibit human thrombin. Nature 1992, 355, 564–566. [Google Scholar]
- Zon, G; Gallo, K; Samson, C; Shao, K; Summers, M; Byrd, R. Analytical studies of “mixed sequences” oligodeoxyribonucleotides synthesised by competitive coupling of either methyl- or β- cyanoethyl-N,N diisopropylamino phosphoramidite reagents, including 2′-deoxysine. Nucleic Acids Res. 1985, 13, 8181–8196. [Google Scholar]
- Nakajima, K; Powers, J; Ashe, B; Zimmerman, M. Mapping the extended substrate binding site of cathepsin G and human leukocyte elastase. Studies with peptide substrates related to the alpha 1-protease inhibitor reactive site. J. Biol. Chem. 1979, 254, 4027–4023. [Google Scholar]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Clone | DNA sequence |
|---|---|
| CG1 | GGGTGGCCCCCTAGTCGCGCACTGGAAGCGGTAGTGTCGTGAGATTCGTATCTGGGGTAT |
| CG3 | CAACGAGTCAGGGCGTGATTGGTGAAGATGTGTGGTTTGGCCAGAAAGGGCGATGGTGGA |
| CG11 | AGAGCTGAGACGGACATGCTGCCCATGGAGACTGTTCGAGAGGGTGAGCGGGAGTGGG |
| CG16 | ACCCCTAGGTCAGCACGTAGTGTAGGGCGATGTGTTCATGGCGGGAATGTGAGTTGTGGG |
| CG20 | GGGCGGCTCGCGTTGTGGAACATTCGTGGTGCCAATGCGTACCAGGGATTGCCTCCTGT |
| CG25 | GGGCGATTGGCGAATGCAAGGGTAAGGTTGGGCGATTGATGTGCACGTAGCGCAGAGCAT |
| CG28 | XXGGAACGTGGTAGGTGTGTCTGCTGTGTGTGGCTCGGGCAGGTTGTCAGGGTGTTT |
| CG32 | GGGCATAGGGCGTCGTAGCCTGAAGGTGTGATTCGTGCGTTAGATGGGGGGCAGTCTGC |
| CG43 | CAACGTGTGATATGTGGGTATACGCTTGGGTGTTACGCTGAGCACAGAGGGTATTCGTGT |
| CG45 | GGCGGGCGGTATGGGCTGCAGGATATGCAGGGGCGCAGAGGACAGTCTGGCCATGTACTA |
| CG49 | GGCCTGGGTGATGTACTATGTATGCGTCGTGGTGGCTGGTAAAGGGGGTCTGCTATGGGT |
| CG51 | CAACGTGTGATATGTGGGTATACGCTTGGGTGTTACGCTGAGCACAGAGGGTATTCGTGT |
| CG2 | CCACGGACGCTGTGAGCGGCCAACGGATGGGAATCACGATCTGGCCCGAACCACATACCG |
| CG31 | TCACACTAGGGCACTTGCTAAGTAGCTATGTAACTCGATCATACTTATTAGGCTTG |
| CG23 | AATCGATGGACACTTCAACGCAACTTGACATGGCGGTACGTGGACTCTTGTGGCGACAGTT |
| CG34 | AACCCGTGTGATAAGGATATGGTGACTTCGTGGCACAGCGTCGACGGACTGCCCATTCCA |
| CG40 | GGCAGGGACGTTCCCAGGAATGCGGCACAGGCAGACAGCTCCCGACGAGTACCAGGGTG |
| CG48 | AGXGGGCAGCAGCACACCACACATGTACGTGGGGGATTGCATTGTGTACTTAGACGGTAT |
| CG39 | CGGTGGAGAGGTCGCAATGACACGGTTGACGATAGGCCCCTTGCTAACATCGGTTGGTG |
Share and Cite
Gatto, B.; Vianini, E.; Lucatello, L.; Sissi, C.; Moltrasio, D.; Pescador, R.; Porta, R.; Palumbo, M. Effective DNA Inhibitors of Cathepsin G by In Vitro Selection. Int. J. Mol. Sci. 2008, 9, 1008-1023. https://doi.org/10.3390/ijms9061008
Gatto B, Vianini E, Lucatello L, Sissi C, Moltrasio D, Pescador R, Porta R, Palumbo M. Effective DNA Inhibitors of Cathepsin G by In Vitro Selection. International Journal of Molecular Sciences. 2008; 9(6):1008-1023. https://doi.org/10.3390/ijms9061008
Chicago/Turabian StyleGatto, Barbara, Elena Vianini, Lorena Lucatello, Claudia Sissi, Danilo Moltrasio, Rodolfo Pescador, Roberto Porta, and Manlio Palumbo. 2008. "Effective DNA Inhibitors of Cathepsin G by In Vitro Selection" International Journal of Molecular Sciences 9, no. 6: 1008-1023. https://doi.org/10.3390/ijms9061008