Computational Study for Protein-Protein Docking Using Global Optimization and Empirical Potentials

Abstract
:1. Introduction
2. Computational Method
2.1. Conformational Searches
2.1.1 Conformational space annealing (CSA)
2.1.2 Simulated Annealing (SA)
2.1.3 Combined CSA/SA
2.2. Energy Function
2.3. Benchmark Test Set
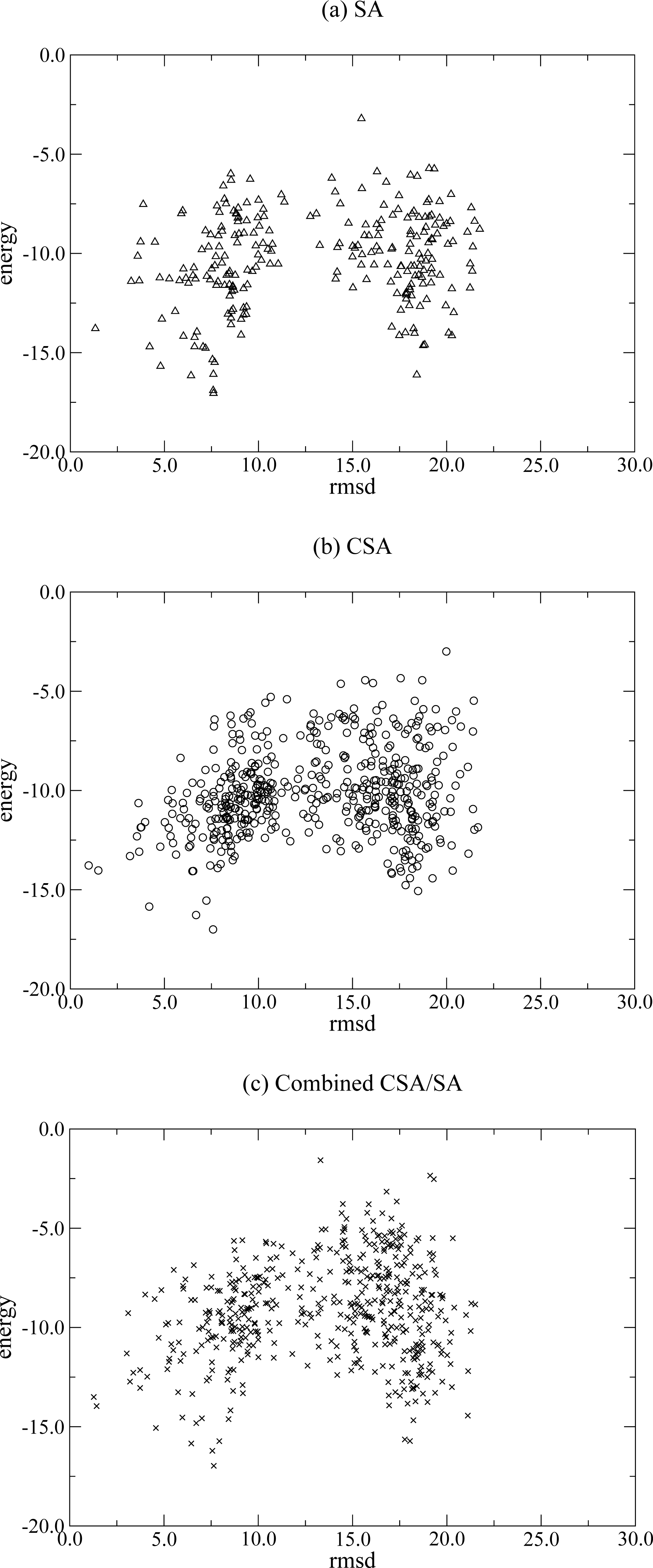
3. Results and Discussion
Acknowledgements
References and Notes
- Berman, HM; Westbrook, J; Feng, Z; Gilliland, G; Bhat, TN; Weissig, H; Shindyalov, IN; Bourne, PE. The Protein Data Bank. Nucleic Acids Research 2000, 28, 235–242, DOI 10.1093/nar/28.1.235; PubMed 10592235. [Google Scholar]
- Jones, S; Thornton, JM. Principles of protein-protein interactions. Proc Natl Acad Sci USA 1996, 93, 13–20, DOI 10.1073/pnas.93.1.13. [Google Scholar]
- Smith, GR; Sternberg, MJE. Prediction of protein-protein interactions by docking methods. Curr Opin Struct Biol 2002, 12, 28–35, DOI 10.1016/S0959-440X(02)00285-3; PubMed 11839486. [Google Scholar]
- Elcock, AH; Sept, D; McCammon, JA. Computer simulation of protein–protein interactions. J Phys Chem B 2001, 105, 1504–1518, DOI 10.1021/jp003602d. [Google Scholar]
- Bonvin, AM. Flexible protein-protein docking. Curr Opin Struct Biol 2006, 16, 194–200, DOI 10.1016/j.sbi.2006.02.002; PubMed 16488145. [Google Scholar]
- Gary, JJ. High-resolution protein-protein docking. Curr Opin Struct Biol 2006, 16, 183–193, DOI 10.1016/j.sbi.2006.03.003; PubMed 16546374. [Google Scholar]
- Sternberg, MJE; Gabb, HA; Jackson, RM. Predictive docking of protein-protein and protein-DNA complexes. Curr Opin Struct Biol 1998, 8, 250–256, DOI 10.1016/S0959-440X(98)80047-X; PubMed 9631301. [Google Scholar]
- Halperin, I; Ma, B; Wolfson, H; Nussinov, R. Principles of docking: an overview of search algorithms and a guide to scoring functions. Proteins 2002, 47, 409–443, DOI 10.1002/prot.10115; PubMed 12001221. [Google Scholar]
- Russell, RB; Alber, F; Aloyl, P; Davis, FP; Korkin, D; Pichaud, M; Topf, M; Sali, A. A structural perspective on protein–protein interactions. Curr Opin Struct Biol 2004, 14, 313–324, DOI 10.1016/j.sbi.2004.04.006; PubMed 15193311. [Google Scholar]
- Vakser, IA. Protein docking for low-resolution structures. Protein Eng 1995, 8, 371–377, DOI 10.1093/protein/8.4.371; PubMed 7567922. [Google Scholar]
- Gabb, HA; Jackson, RM; Sternberg, MJE. Modeling protein docking using shape complementarity, electrostatics and biochemical information. J Mol Biol 1997, 272, 106–120, DOI 10.1006/jmbi.1997.1203; PubMed 9299341. [Google Scholar]
- Chen, R; Weng, Z. Docking unbound proteins using shape complementarity desolvation, and electrostatics. Proteins 2002, 47, 281–294, DOI 10.1002/prot.10092; PubMed 11948782. [Google Scholar]
- Mandell, JG; Roberts, VA; Pique, ME; Kotlovyi, V; Mitchell, JC; Nelson, E; Tsigelny, I; Eyck, TLF. Protein docking using continuum electrostatics and geometric fit. Protein Eng 2001, 14, 105–113, DOI 10.1093/protein/14.2.105; PubMed 11297668. [Google Scholar]
- Harrison, RW; Kourinov, IV; Andrews, LC. The Fourier-Greens function and the rapid evaluation of molecular potentials. Protein Eng 1994, 7, 359–369, DOI 10.1093/protein/7.3.359; PubMed 8177885. [Google Scholar]
- Metropolis, N; Rosenbluth, AW; Rosenbluth, MN; Teller, AH; Teller, E. Equation of state calculations by fast computing machines. J Chem Phys 1953, 21, 1087–1092, DOI 10.1063/1.1699114. [Google Scholar]
- Kirkpatrick, S; Gelatt, CD; Vecchi, MP. Optimization by simulated annealing. Science 1983, 220, 671–680, PubMed 17813860. [Google Scholar]
- Hwang, J; Liao, W. Side-chain prediction by neural networks and simulated annealing optimization. Protein Eng 1995, 8, 363–370, DOI 10.1093/protein/8.4.363; PubMed 7567921. [Google Scholar]
- Cerny, V. Thermodynamical approach to the traveling salesman problem: an efficient simulation algorithm. J Optim Theory Appl 1985, 45, 41–51, DOI 10.1007/BF00940812. [Google Scholar]
- de Vicente, J; Lanchares, J; Hermida, R. Placement by thermodynamic simulated annealing. Phys Lett A 2003, 317, 415–423, DOI 10.1016/j.physleta.2003.08.070. [Google Scholar]
- Lee, J; Scheraga, HA; Rackovsky, S. New optimization method for conformational energy calculations on polypeptide: conformational space annealing. J Comput Chem 1997, 18, 1222–1232, DOI 10.1002/(SICI)1096-987X(19970715)18:9<1222::AID-JCC10>3.0.CO;2–7. [Google Scholar]
- Lee, K; Czaplewski, C; Kim, S-Y; Lee, J. An efficient molecular docking using conformational space annealing. J Comput Chem 2005, 26, 78–87, PubMed 15538770. [Google Scholar]
- Lee, K; Sim, J; Lee, J. Study of protein-protein interaction using conformational space annealing. Proteins 2005, 60, 257–262, DOI 10.1002/prot.20567; PubMed 15981254. [Google Scholar]
- Janin, J; Hendrick, K; Moult, J; Eyck, LT; Sternberg, MJE; Vajda, S; Vakser, IA; Wodak, SJ. CAPRI: A Critical Assessment of PRedicted Interactions. Proteins 2003, 52, 2–9, DOI 10.1002/prot.10381; PubMed 12784359. [Google Scholar]
- Camacho, CJ; Zhang, C. FastContact: rapid estimate of contact and binding free energies. Bioinformatics 2005, 21, 2534–2536, DOI 10.1093/bioinformatics/bti322; PubMed 15713734. [Google Scholar]
- Champ, PC; Camacho, CJ. FastContact: a free energy scoring tool for protein–protein complex structures. Nucleic Acids Research 2007, 35, W556–W560, PubMed 17537824. [Google Scholar]
- Dunbrack, RL; Cohen, FE. Bayesian statistical analysis of protein side-chain rotamer preferences. Protein Sci 1997, 6, 1661–1681, PubMed 9260279. [Google Scholar]
- Mintseris, J; Wiehe, K; Pierce, B; Anderson, R; Chen, R; Janin, J; Weng, Z. Protein-protein docking benchmark 20: an update. Proteins 2005, 60, 214–216. [Google Scholar]
- Goldberg, DE. Genetic Algorithms in Search, Optimization and Machine Learning; Kluwer Academic Publishers: Boston, MA, 1989. [Google Scholar]
- Nayeem, A; Vila, J; Scheraga, HA. A comparative study of the simulated annealing and Monte Carlo-with-minimization approaches to the minimum-energy structures of polypeptides: [met]-enkephalin. J Comput Chem 1991, 12, 594–605, DOI 10.1002/jcc.540120509. [Google Scholar]
- Lee, J; Liwo, A; Ripoll, DR; Pillardy, J; Scheraga, HA. Calculation of protein conformation by global optimization of a potential energy function. Proteins Suppl 1999, 3, 204–208, DOI 10.1002/(SICI)1097-0134(1999)37:3+<204::AID-PROT26>3.0.CO;2-F. [Google Scholar]
- Lee, J; Kim, S-Y; Joo, K; Kim, I; Lee, J. Prediction of protein tertiary structure using profesy, a novel method based on fragment assembly and conformational space annealing. Proteins 2004, 56, 704–714, PubMed 15281124. [Google Scholar]
- Brooks, BR; Bruccoleri, RE; Olafson, BD; States, DJ; Karplus, M. CHARMM: a program for macromolecular energy, minimization, and molecular dynamics calculations. J Comput Chem 1983, 4, 187–217, DOI 10.1002/jcc.540040211. [Google Scholar]
- Miyazawa, S; Jernigan, RL. Estimation of effective interresidue contact energies from protein crystal structures: quasi-chemical approximation. Macromolecules 1985, 18, 534–552, DOI 10.1021/ma00145a039. [Google Scholar]
- Miyazawa, S; Jernigan, RL. Residue-residue potentials with a favorable contact pair term and an unfavorable high packing density term, for simulation and threading. J Mol Biol 1996, 256, 623–644, DOI 10.1006/jmbi.1996.0114; PubMed 8604144. [Google Scholar]
- Mendez, R; Leplae, R; de Maria, L; Wodak, S. Assessment of blind predictions of protein-protein interactions: current status of docking methods. Proteins 2003, 52, 51–67, PubMed 12784368. [Google Scholar]
- Ma, B; Kumar, S; Tsai, CJ; Nussinov, R. Folding funnels and binding mechanisms. Protein Eng 1999, 12, 713–720, PubMed 10506280. [Google Scholar]
- Tovchigrechko, A; Vakser, IA. How common is the funnel-like energy landscape in protein-protein interactions. Protein Science 2001, 10, 1572–1583, PubMed 11468354. [Google Scholar]

{kind=link}
| complex pdbA | SA
| CSA
| combined CSA/SA
| |||
|---|---|---|---|---|---|---|
| smallest RMSDB | no. of acceptableC | smallest RMSD | no. of acceptable | smallest RMSD | no. of acceptable | |
| 1A0O | 4.10 | 0 | 4.16 | 0 | 3.08 | 1 |
| 1ACB | 1.33D | 6 | 0.97 | 9 | 1.28 | 8 |
| 1AVZ | 5.73 | 0 | 4.79 | 0 | 4.90 | 0 |
| 1BRC | 5.29 | 0 | 4.04 | 0 | 5.03 | 0 |
| 1BRS | 10.47 | 0 | 4.96 | 0 | 7.67 | 0 |
| 1CGI | 3.46 | 3 | 2.73 | 5 | 2.94 | 1 |
| 1CHO | 4.02 | 0 | 1.11 | 3 | 1.45 | 2 |
| 1CSE | 3.29 | 2 | 1.27 | 2 | 1.62 | 3 |
| 1MEL | 9.38 | 0 | 3.66 | 1 | 7.31 | 0 |
| 1PPE | 4.07 | 0 | 3.11 | 6 | 2.40 | 8 |
| 1STF | 4.98 | 0 | 4.95 | 0 | 4.96 | 0 |
| 1TAB | 5.86 | 0 | 4.98 | 0 | 5.97 | 0 |
| 1TGS | 1.64 | 1 | 5.87 | 0 | 5.24 | 0 |
| 1UDI | 2.14 | 4 | 4.05 | 0 | 2.25 | 4 |
| 2KAI | 5.66 | 0 | 5.28 | 0 | 5.55 | 0 |
| 2PTC | 5.29 | 0 | 4.98 | 0 | 5.61 | 0 |
| 2TEC | 2.63 | 4 | 2.60 | 1 | 2.37 | 3 |
| 4HTC | 6.14 | 0 | 7.54 | 0 | 3.57 | 1 |
Share and Cite
Lee, K. Computational Study for Protein-Protein Docking Using Global Optimization and Empirical Potentials. Int. J. Mol. Sci. 2008, 9, 65-77. https://doi.org/10.3390/ijms9010065
Lee K. Computational Study for Protein-Protein Docking Using Global Optimization and Empirical Potentials. International Journal of Molecular Sciences. 2008; 9(1):65-77. https://doi.org/10.3390/ijms9010065
Chicago/Turabian StyleLee, Kyoungrim. 2008. "Computational Study for Protein-Protein Docking Using Global Optimization and Empirical Potentials" International Journal of Molecular Sciences 9, no. 1: 65-77. https://doi.org/10.3390/ijms9010065