3D-QSAR Study of Potent Inhibitors of Phosphodiesterase-4 Using a CoMFA Approach

College of Pharmacy, Jinan University, Guangzhou, P.R. of China
*
Author to whom correspondence should be addressed.
Int. J. Mol. Sci. 2007, 8(7), 714-722; https://doi.org/10.3390/i8070714
Submission received: 5 June 2007
/
Revised: 13 July 2007
/
Accepted: 19 July 2007
/
Published: 24 July 2007
(This article belongs to the Section Physical Chemistry, Theoretical and Computational Chemistry)
Abstract
:Phosphodiesterase-4 (PDE4) plays an important role in treatment of asthma and chronic obstructive pulmonary disease. Thirty-one analogs displaying variable inhibition of PDE4 were selected to develop models for establishing three-dimensional quantitative structure-activity relationships (3D-QSAR). Comparative molecular field analysis (CoMFA) was conducted on the group of analogs to determine the structural requirements for potency in inhibiting PDE4. The resulting model exhibited good q2 and r2 values up to 0.741 and 0.954 for CoMFA. The contributions from the steric and electrostatic fields were 0.915 and 0.085 respectively. The 3D-QSAR model should be very useful for design of novel PDE 4 inhibitors.
1. Introduction
Asthma and chronic obstructive pulmonary disease (COPD) are the two most prevalent chronic airway diseases. COPD is a treatable and preventable disease but current predictions are that it will continue to increase as an important cause of mortality and morbidity worldwide [1–2]. Phosphodiesterases (PDEs) have been classified into at least 11 families (PDE 1–11) according to their substrate sensitivity, inhibitor selectivity, Ca2+/calmodulin requirement and amino acid sequences [3–4]. Phosphodiesterase-4 (PDE4) is a key enzyme in the hydrolysis of cAMP in mast cells, basophils, eosinophils, monocytes and lymphocytes, as well as areas in the brain and airway smooth muscle [5–6].
PDE4 plays a significant role in modulating the activity of cAMP, an important second messenger that mediates the relaxation of airway smooth muscle and suppresses inflammatory cell function, thereby attenuating the inflammatory response [7]. Increasing the intracellular concentration of cAMP in the airway tissues and cells suppresses inflammatory cell function and thus should be beneficial for treatment of asthma and COPD [8]. Over the last two decades pharmaceutical companies have placed numerous PDE4 inhibitors into clinical trials for asthma or COPD. Only a small number of these drugs have the potential to be approved for market [9–10].
Comparative molecular field analysis (CoMFA) is one of the well known 3D-QSAR descriptors which has been used regularly to produce the three dimensional models to indicate the regions that affect biological activity with a change in the chemical substitution [11]. The advantages of CoMFA are the ability to predict the biological activities of the molecules and to represent the relationships between steric/electrostatic property and biological activity in the form of contour maps gives key features on not only the ligand-receptor interaction but also the topology of the receptor [12].
We present here our 3D-QSAR studies using CoMFA method on a training set of 5,6-dihydro-(9H)-pyrazolo-[4,3-c]-1,2,4-triazolo-[4,3-α]-pyridine derivatives as PDE4 inhibitors by considering the steric and electrostatic influences. The model deduced from this investigation provides underlying structural requirements and good predictive ability, which could aid new PDE4 inhibitors prior to their synthesis.
2. Computational methods
2.1 Molecular Modeling
The structures of the 5,6-dihydro-(9H)-pyrazolo-[4,3-c]-1,2,4-triazolo-[4,3-α]-pyridine derivatives and the biological activities data were obtained from the reference [8]. The negative logarithm of IC50 (pIC50) was used as the biological activity in the 3D-QSAR study (Table 1). Three-dimensional structure building and all modeling were performed using the Sybyl 7.0 program package [13] on a personal computer equipped with a Pentium IV processor. Molecular building was done with molecular sketch program. Geometry optimization was carried out using MAXIMIN molecular mechanics and Tripos force field, Gasteiger–Hückle charge supplied within Sybyl7.0, with the convergence criterion set at 0.05 kcal/(Å mol).
2.2 CoMFA analysis
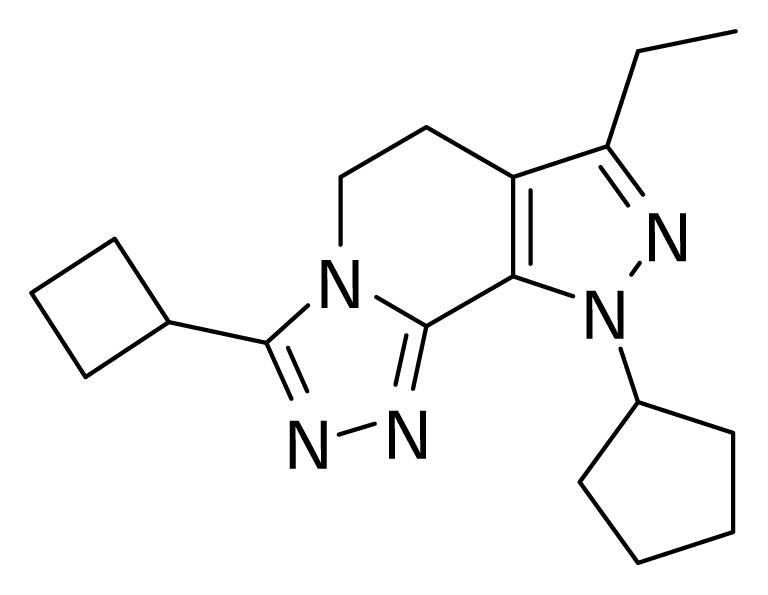
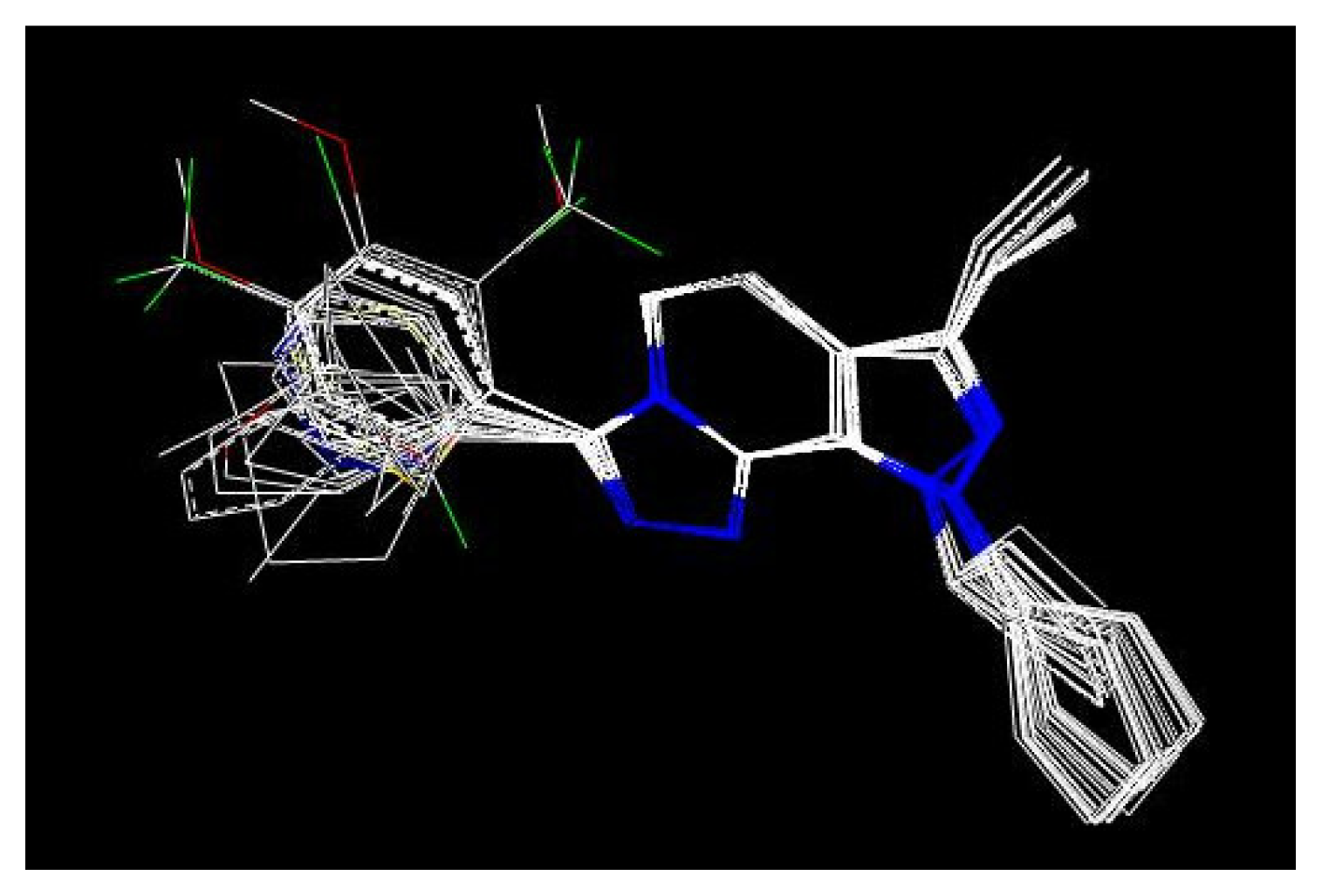
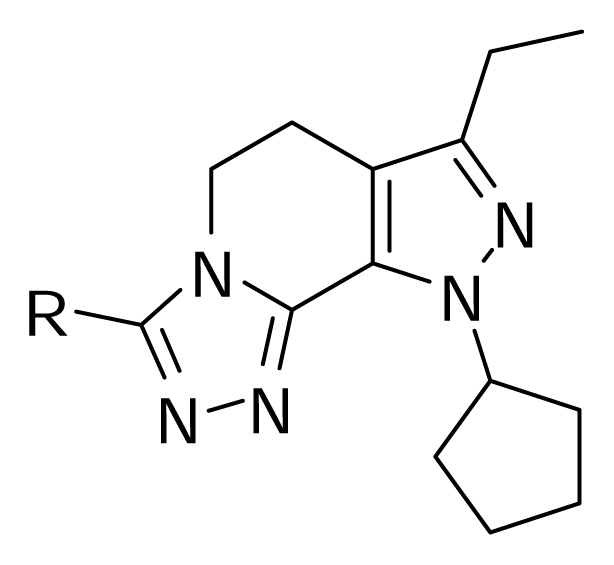
QSAR models were random derived from a training set of 27 molecules. An external test set consisting of four molecules was used to validate the CoMFA models. The most active molecule 24 was used as a template molecule for alignment. A common substructure-based alignment was adopted in the present study, which attempted to align molecules to the template molecule on a common backbone. Molecule 24 is shown in Figure 1. The alignment of the training set molecules was derived by Sybyl 7.0 (Figure 2).
CoMFA of these molecules was carried out on the steric and electrostatic fields using the default values. The steric and electrostatic CoMFA potential fields were calculated at each lattice intersection of a regularly spaced grid of 2.0 Å. The grid box dimensions were determined automatically in such a way that the region boundaries were extended beyond 4 Å in each direction from the coordinates of each molecule. The steric and electrostatic fields were calculated separately for each molecule using sp3 carbon atom probe with a charge of 1 (default probe atom in SYBYL) and energy cut-off values of 30 kcal/mol for both steric and electrostatic fields.
2.3. Partial least squares (PLS) analysis
The relationship between the structural parameters (CoMFA interaction energies) and the biological activities has been quantified by the PLS algorithm. PLS regression technique is especially useful in quite common case where the number of descriptors (independent variables) is comparable to or greater than the number of compounds (data points) and/or there exist other factors leading to correlations between variables [14]. The cross-validation analysis was carried out using Leave-One-Out (LOO) method where one compound is removed from the dataset and its activity is predicted using the model derived from the rest of the dataset. The cross-validated q2 and the optimum number of components were obtained. To speed up the analysis and reduce noise, a minimum column filtering value (σ) of 2.00 kcal/mol was used for the cross-validation. Finally, non-cross-validated analysis was performed to calculate non-cross-validated r2 using the optimal number of previously identified components and was employed to analyze the result of the CoMFA.
3. Results and Discussion
Thirty-one molecules were randomly partitioned into a training set of 27 and a test set of four compounds with bias given to both chemical and biological diversity in both the training set and the test set molecules. Despite the ambiguity of drug-receptor interactions in general, a statistically significant model was obtained from the CoMFA study. A “cross-validated q2” may then be defined, completely analogously to the definition of the conventional q2, as
where press is the standard errors of the cross-validated predictions and SD is the sum of squared deviations of each biological property value from their mean and press, or predictive sum of squares, is the sum, over all compounds, of the squared differences between the actual and “predicted” biological property values[15].
Often for QSARs developed with CoMFA a shift in the q2 values is observed as the grid spacing is altered. To examine this possibility with these data, the different grid boxes with 1.0, 1.5, 2.0, 2.5, and 3.0 Å grid spacing, respectively, were used for the CoMFA calculations. The influence of the different grid spacing to CoMFA model is obvious. Only from the q2 after leave-one-out cross-validation, the model with the grid spacing of 2.0 Å was selected as the best model (see Table 2).
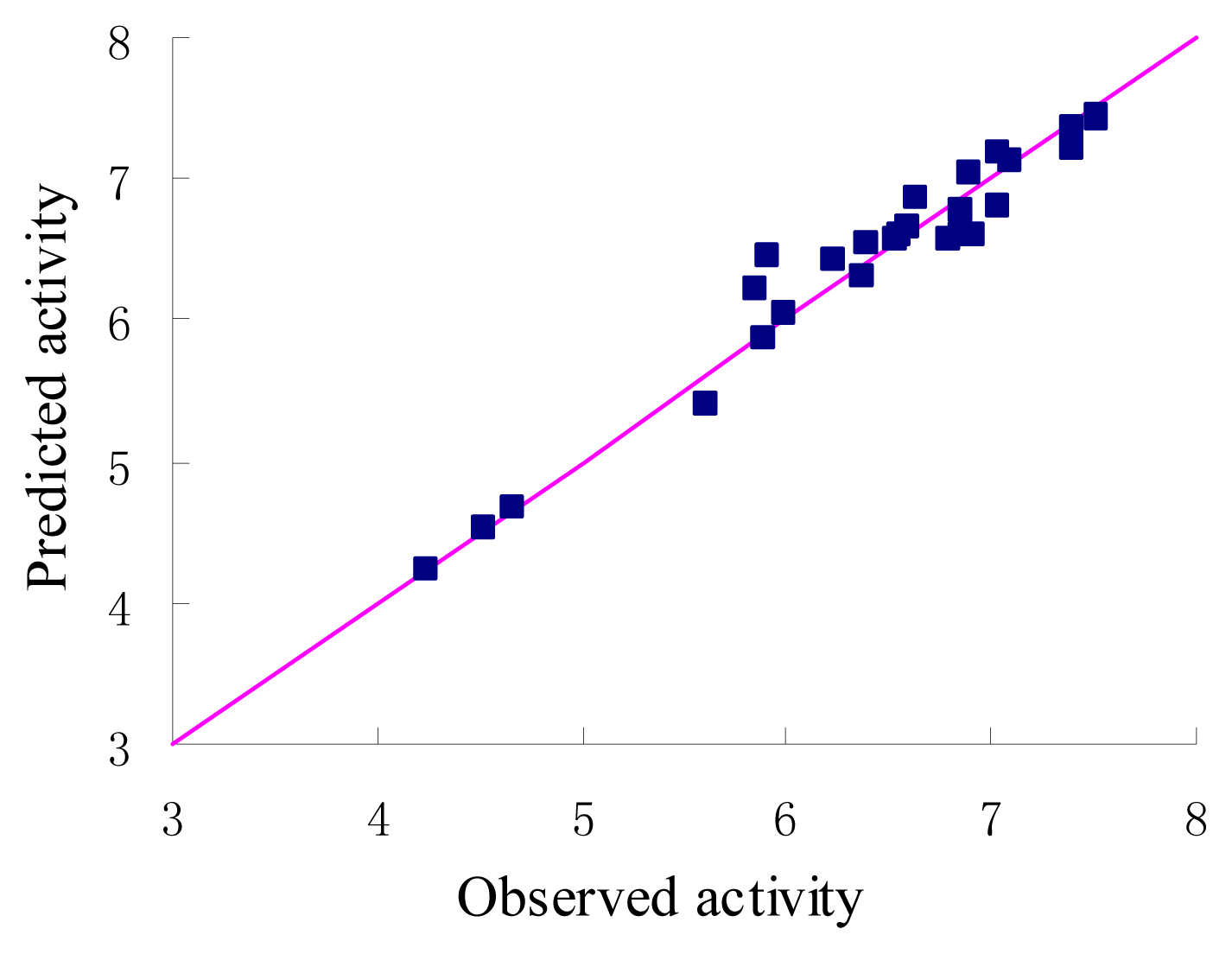
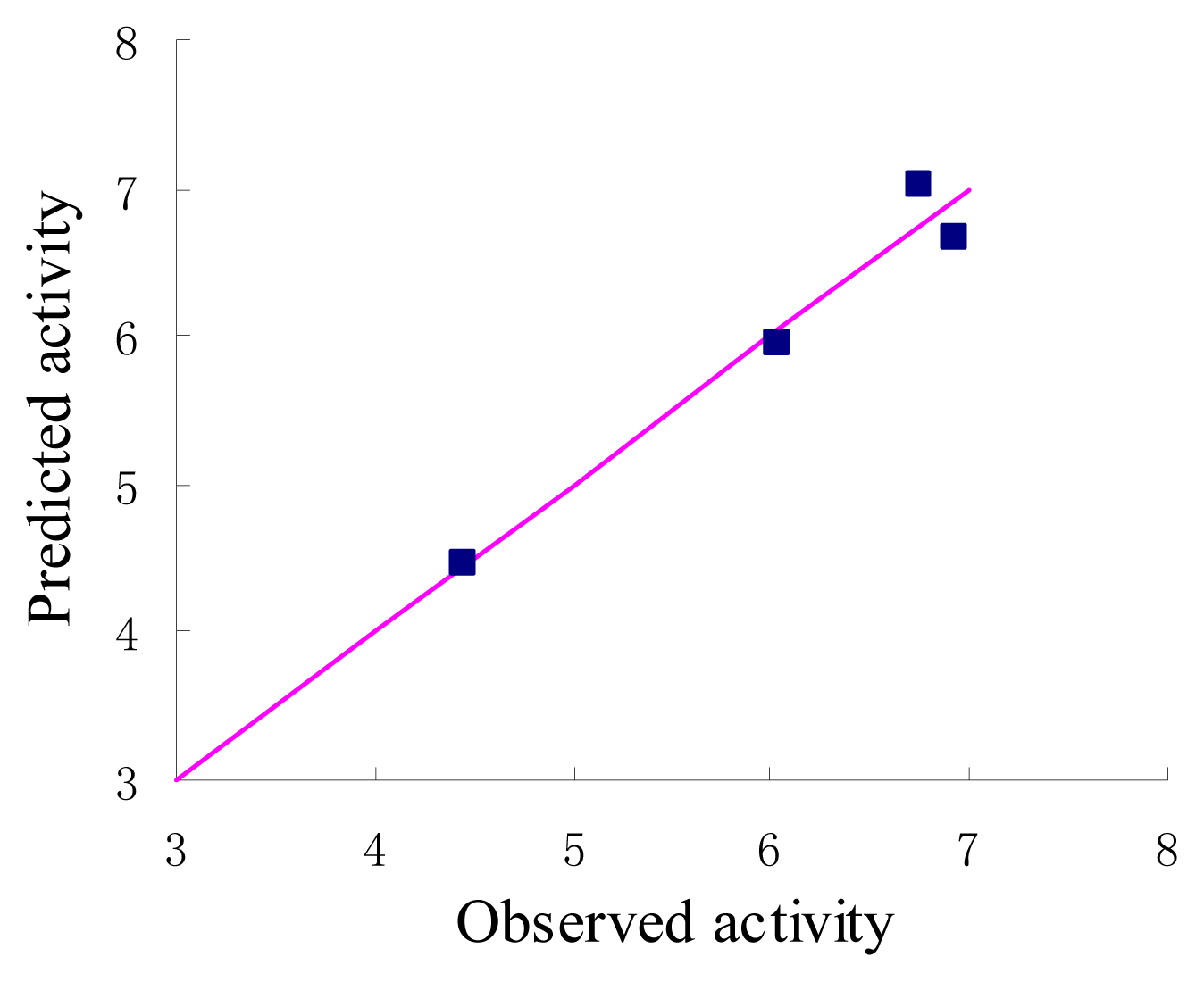
The statistical parameters of CoMFA analysis is summarized in Table 3. The leave-one-out cross-validated PLS analysis of the best model gave rise to a cross-validated value (q2) of 0.741, suggesting that the model is a useful tool for predicting PDE4 inhibitory activity [16]. The correlation coefficient between the calculated and experimental activities, non-cross-validated value (r2) of 0.954 with standard error estimate 0.211. The respective relative contributions of steric and electrostatic fields were 0.915 and 0.085, indicating that steric field is more predominant. Then the condition without electrostatic were studied and the new q2 and r2 values were 0.739 and 0.953 respectively. In fact, the electrostatic contribution was nearly negligible. The actual and predicted values of the training set by the best model are given in Table 1, and the graph of observed activity versus predicted activities of training set molecules from CoMFA analysis is illustrated in Figure 3. The 3D-QSAR model was validated using a test set (in Table 1) of 4 compounds, which were not included in the development of the model. On the basis of the PLS statistics of CoMFA model, this is further validated by the residual values of the test set (Table 1). Figure 4 represents the graph of the actual versus predicted pIC50 values of the test set molecules for CoMFA model.
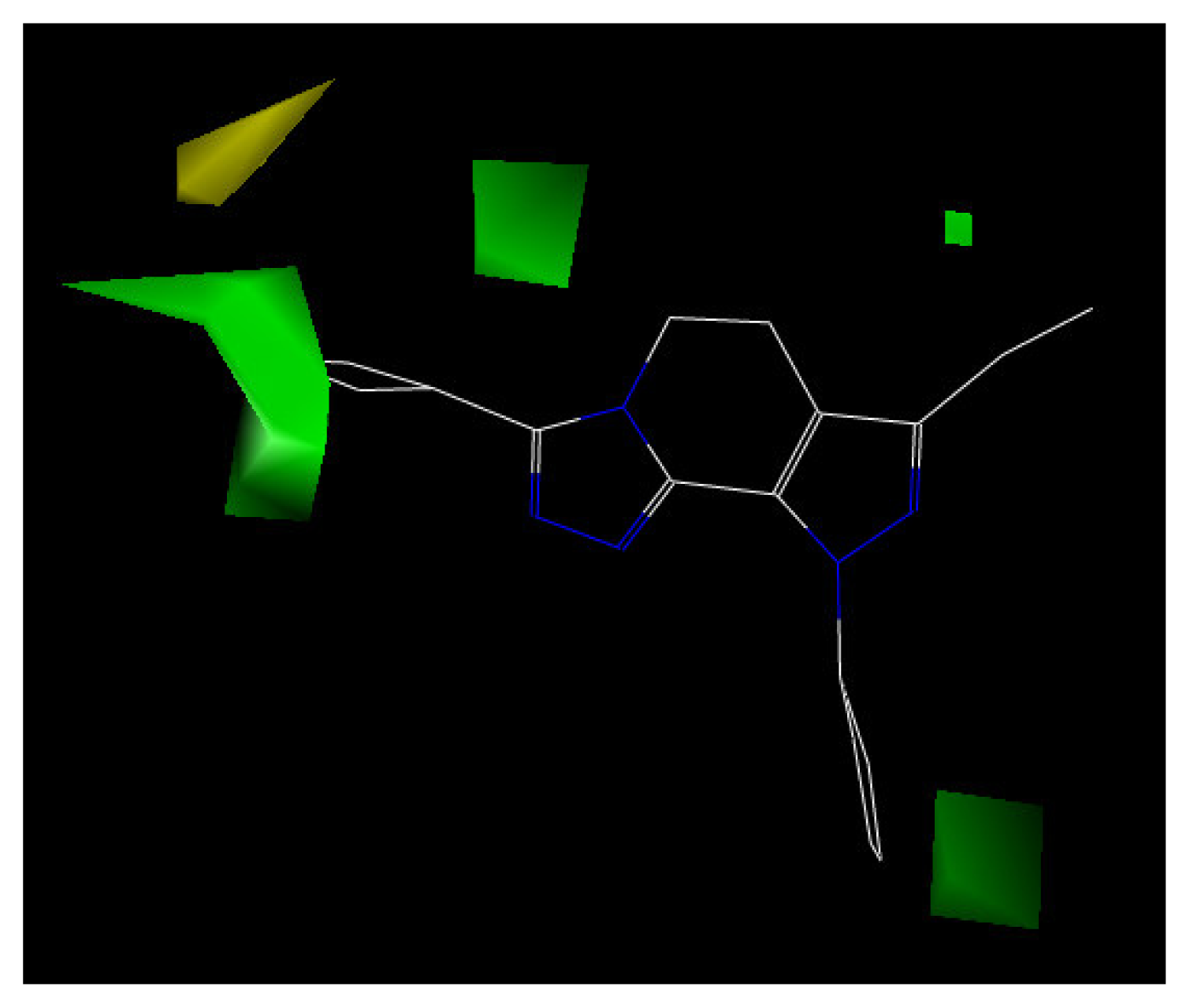
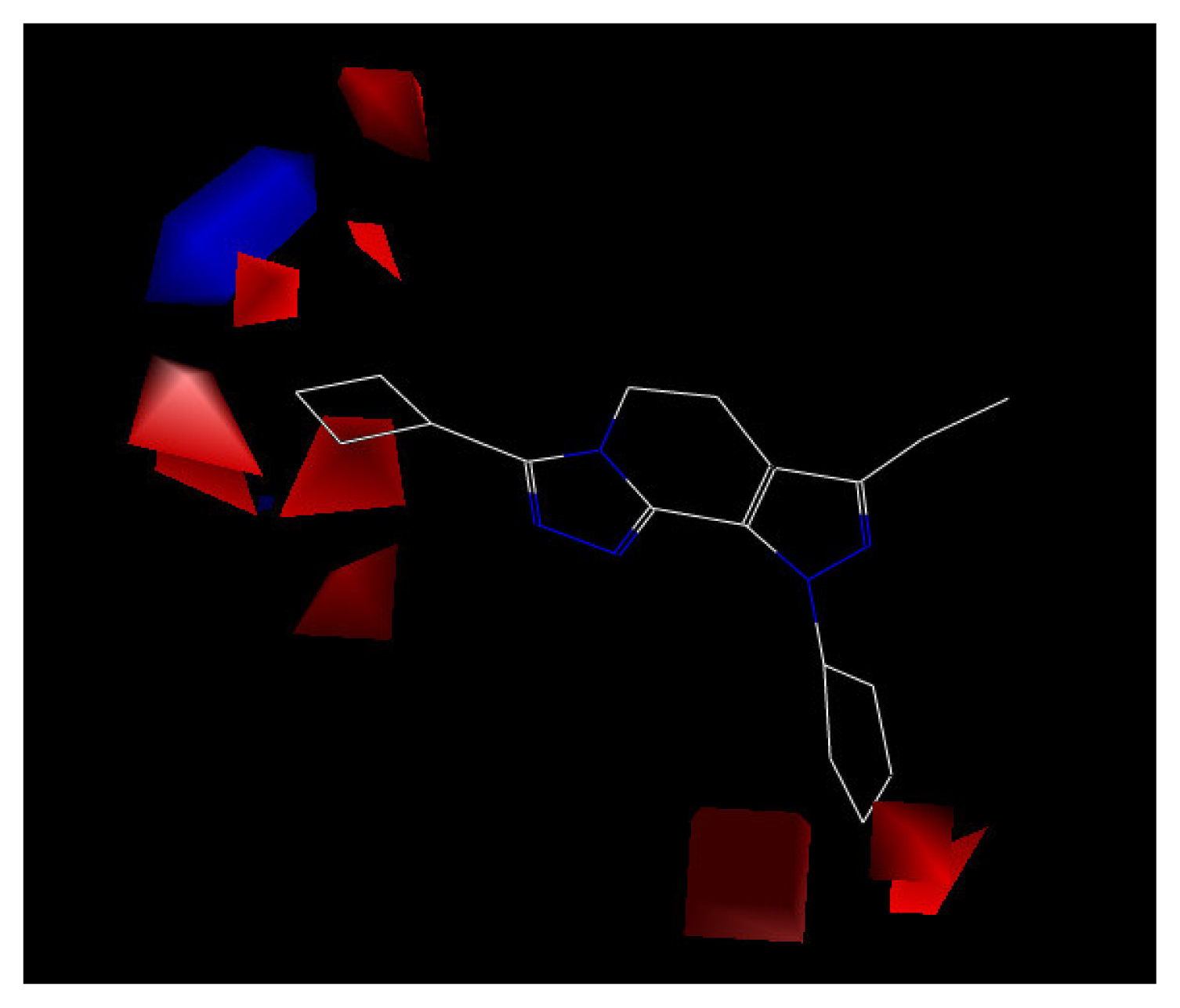
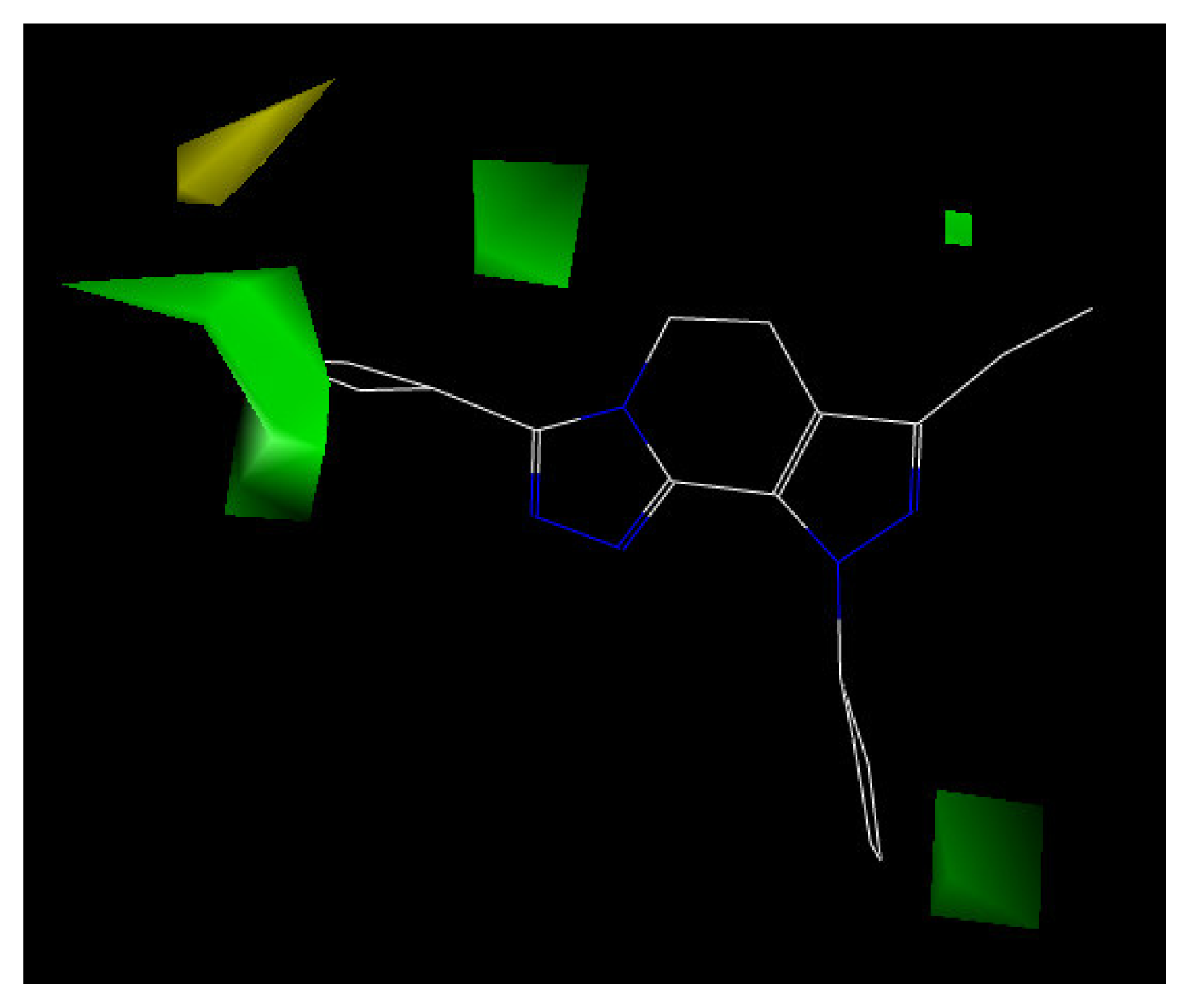
The contour plot representations of the CoMFA results for PDE4 inhibitors are presented in Figures 5 and 6 using compound 24 as reference structure. The green-colored regions indicate areas where steric bulk enhances PDE4 inhibitory activity, while the yellow contours indicate regions where steric bulk is detrimental for the biological activity. Blue-colored regions show areas where electropositive charged groups enhance PDE4 inhibitory activity, while red regions represent where electronegative charged groups improve the activity.
The electrostatic contour map displayed in Figure 5 shows a region of red polyhedral space, indicating that the electron-rich groups are beneficial to the activity. Compounds 4 and 7 have higher inhibition activity than compound 10 because the chlorine atom is placed within the electron-rich charge red contour. Additionally, a blue polyhedron in the top left corner of Figure 5, indicates that electron-rich substituents will reduce the biological activity. Compound 11, with a strong electron withdrawing substituent, 4-trifluoromethylphenyl, can not fit into the electropositively charged blue contour. Thereby, compound 11 has lower activity than other compounds in the model.
The steric contour map is displayed in Figure 6. The plot shows the cyclobutyl group of molecule 24 placed between two big green polyhedrons. If a substituent, such as 3-pentyl group, is attached on molecule 28, it occupies the green contour and enhances the activity. In compounds 25 and 29, where the substituent group R is cyclopentyl and 1-methylcyclohex-1-yl, respectively, these fit into the green contour, and the activity of these compounds are higher. Compounds 20, 21 and 22, with methyl, ethyl and propyl substituents, show less activity than compound 28. Additionally, the contour plot shows a yellow polyhedron in the top left corner of Figure 6. If a bulky substituent occupies the yellow contour this will depress the biological activity. Compounds 8, 9 and 11, with 4-methoxyphenyl, 4-chlorophenyl and 4-trifluoromethylphenyl substituents, respectively, are very close to the yellow contour which represents the unfavored steric region. Consequently, these compounds almost have no activity.
4. Conclusions
3D QSAR studies were performed using CoMFA on a series of derivatives with inhibitory activity on phosphodiesterase-4. A satisfactory CoMFA model was obtained with LOO cross-validation q2 and non-cross-validated r2 values of 0.741 and 0.954 respectively. The developed model also possesses promising predictive ability as discerned by the testing on the external test set, and should be useful to elucidate the relationship between compound structures and biological activities and to facilitate design of more potent phosphodiesterase-4 inhibitors. For example, use of a p-trifluoromethyl substituent in a cyclobutyl maybe increase the biological activity, since a fluorine atom at that position participates in the electron-rich charge red contour and the trifluoromethyl will fit into the green contour. Similarly, other p-substituents on the cycylobutyl moiety might show even greater inhibitory power than a simple cyclobutyl.
Figure 1.
Molecule 24.
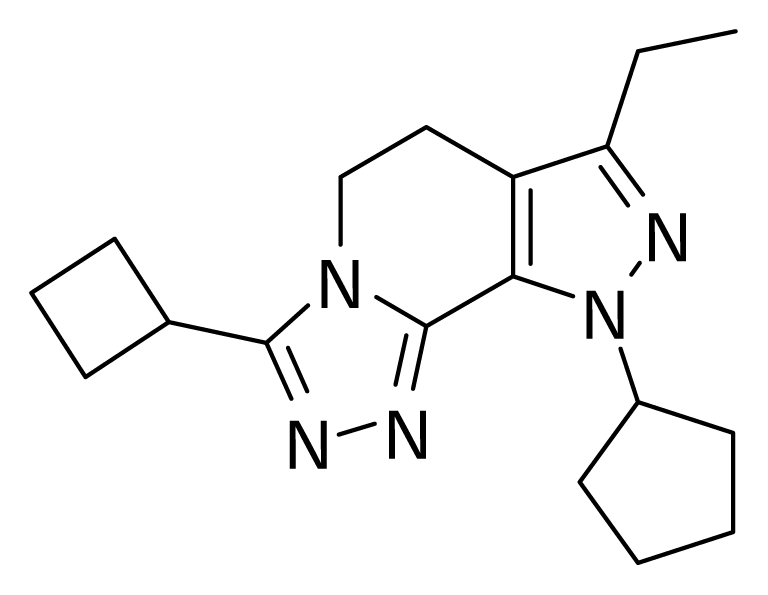
Figure 2.
Alignment of the compounds used in the training set of 3D-QSAR analysis.

Figure 3.
Graph of observed versus predicted activities of the training set.
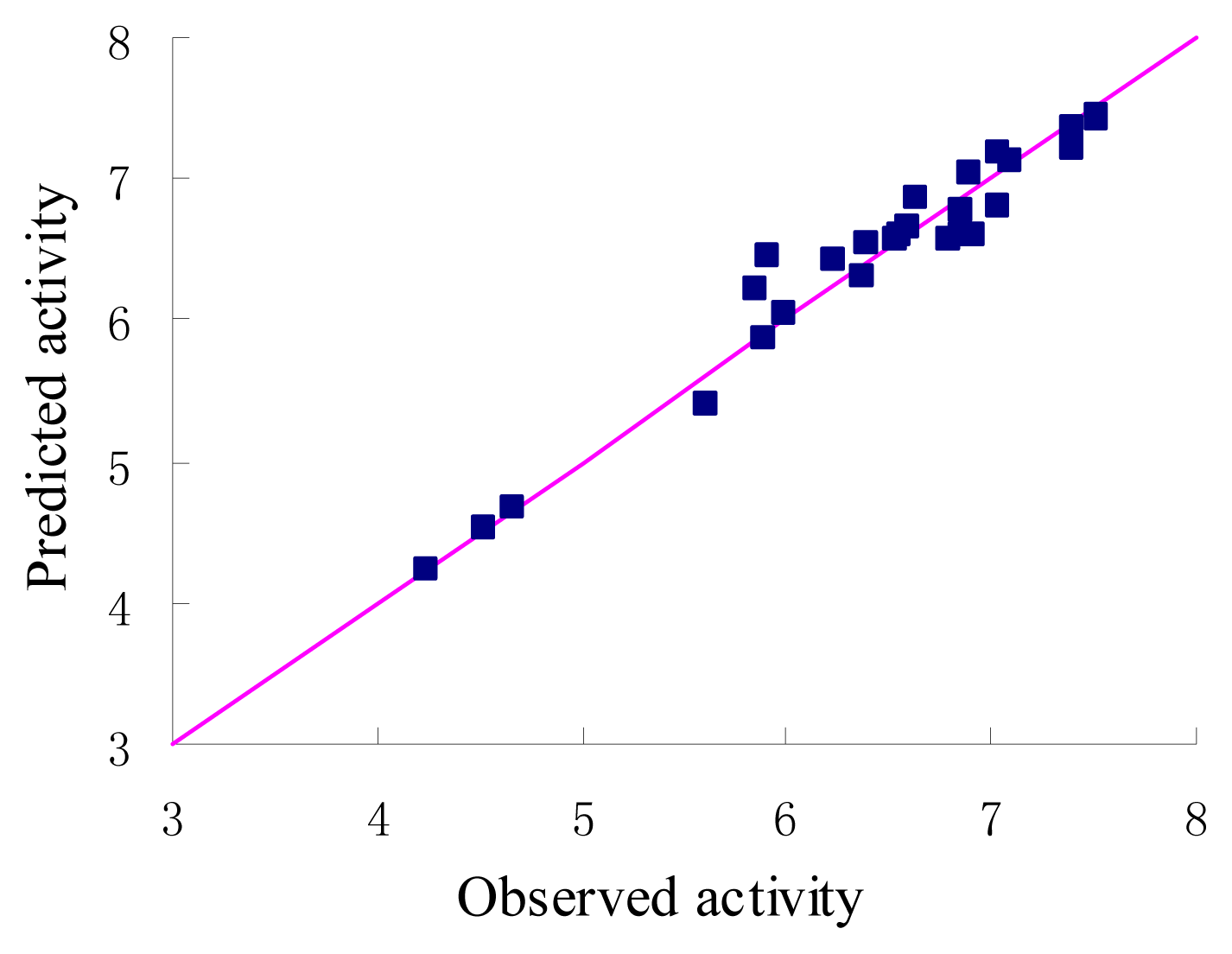
Figure 4.
Graph of observed versus predicted activities of the test set.
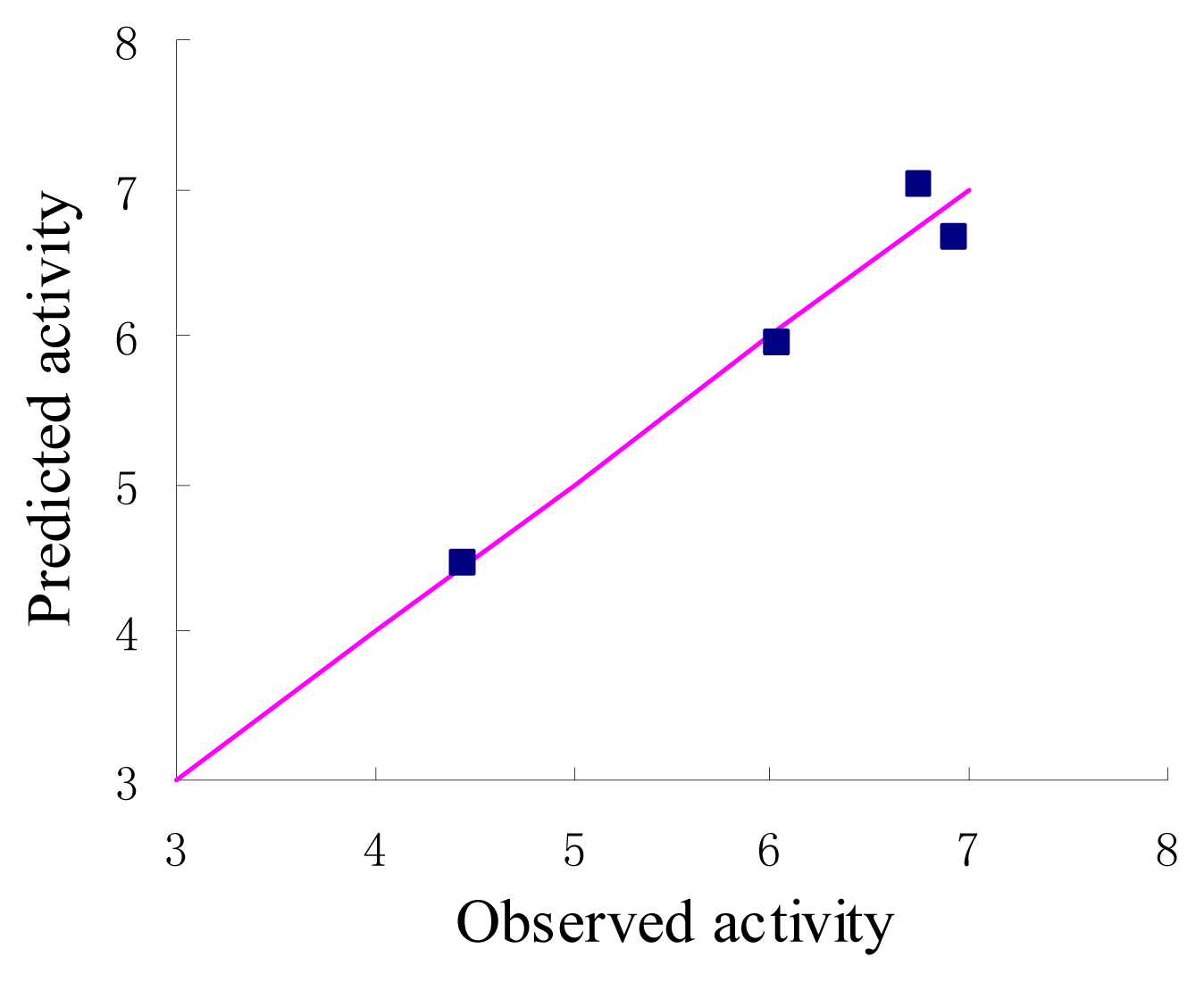
Figure 5.
CoMFA contour map of the electrostatic field with compound 24.
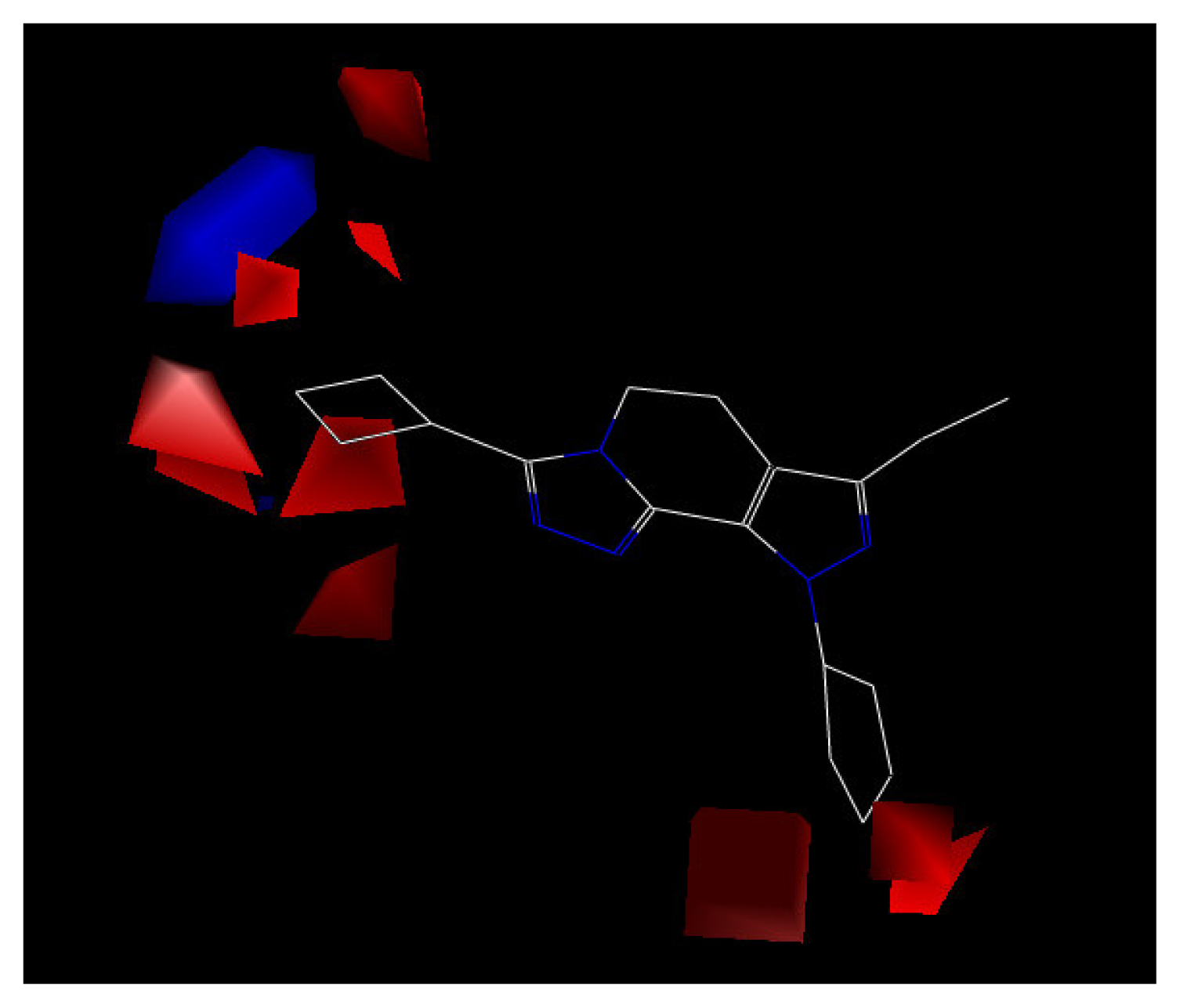
Figure 6.
CoMFA contour maps of steric field with compound 24.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | ||||
|---|---|---|---|---|
| Compound | R | pIC50 (Observed) | pIC50 (Predicted) | Residual |
| 1△ | phenyl | 6.921 | 6.657 | 0.264 |
| 2 | 2-methoxyphenyl | 6.237 | 6.413 | −0.176 |
| 3 | 2-methylphenyl | 6.796 | 6.564 | 0.232 |
| 4 | 2-chlorophernyl | 6.538 | 6.559 | −0.021 |
| 5 | 2-trifluoromethylphenyl | 6.367 | 6.296 | 0.071 |
| 6 | 3-methoxyphenyl | 5.886 | 5.863 | 0.023 |
| 7 | 3-chlorophenyl | 5.854 | 6.223 | −0.369 |
| 8 | 4-methoxyphenyl | 4.523 | 4.527 | −0.004 |
| 9 | 4-methylphenyl | 4.658 | 4.664 | −0.006 |
| 10△ | 4-chlorophernyl | 4.444 | 4.471 | −0.027 |
| 11 | 4-trifluoromethylphenyl | 4.244 | 4.24 | 0.004 |
| 12 | 2-pyridyl | 6.585 | 6.659 | −0.074 |
| 13 | 3-pyridyl | 5.921 | 6.441 | −0.520 |
| 14 | 4-pyridyl | 7.046 | 6.812 | 0.234 |
| 15 | 2-furanyl | 6.553 | 6.582 | −0.029 |
| 16 | 2-thienyl | 6.854 | 6.591 | 0.263 |
| 17 | 3-chloro-4-methylthien-2-yl | 6.000 | 6.049 | −0.049 |
| 18 | benzyl | 6.398 | 6.551 | −0.153 |
| 19 | 3-thenyl | 6.921 | 6.595 | 0.326 |
| 20△ | methyl | 6.041 | 5.948 | 0.093 |
| 21 | ethyl | 6.886 | 7.021 | −0.135 |
| 22△ | propyl | 6.745 | 7.009 | −0.264 |
| 23 | butyl | 7.398 | 7.217 | 0.181 |
| 24 | cyclobutyl | 7.523 | 7.417 | 0.106 |
| 25 | cyclopentyl | 7.046 | 7.185 | −0.139 |
| 26 | cyclohexyl | 6.854 | 6.759 | 0.095 |
| 27 | 4-tetrahydropyranyl | 6.638 | 6.867 | −0.229 |
| 28 | 3-pentyl | 7.523 | 7.448 | 0.075 |
| 29 | 1-methylcyclohex-1-yl | 7.398 | 7.365 | 0.033 |
| 30 | tert-butyl | 7.097 | 7.132 | −0.035 |
| 31 | Bicycle[2.2.2]octanyl | 5.620 | 5.385 | 0.235 |
△The molecule was included in test set
| grid spacing (Å) | 1.0 | 1.5 | 2.0 | 2.5 | 3.0 |
|---|---|---|---|---|---|
| q2 | 0.335 | 0.297 | 0.741 | 0.295 | 0.223 |
| r2 | 0.540 | 0.524 | 0.954 | 0.566 | 0.345 |
| standard error | 0.630 | 0.641 | 0.211 | 0.623 | 0.738 |
| PLS statistics | CoMFA |
|---|---|
| q2 (leave-one out cross-validated predicted power of model) | 0.741 |
| r2 (correlation coefficient squared of PLS analysis) | 0.954 |
| N (optimum number of components obtained from cross-validated PLS analysis and the same used in final non cross-validated analysis) | 5 |
| Standard error of estimate (SEE) | 0.211 |
| F-test value (F-value) | 103.397 |
| Steric field contribution from CoMFA | 0.915 |
| Electrostatic field contribution from CoMFA | 0.085 |
References and Notes
- Mallia, P.; Contoli, M.; Caramori, G.; Pandit, A.; Johnston, S.L.; Papi, A. Exacerbations of Asthma and Chronic Obstructive Pulmonary Disease (COPD): Focus on Virus Induced Exacerbations. Curr. Pharm. Des 2007, 13, 73–97. [Google Scholar]
- Liam, G.H.; John, T.L.; Lorcan, P.A.M. Inflammation in Chronic Obstructive Pulmonary Disease: Implications for New Treatment Strategies. Curr. Med. Chem 2007, 14, 187–796. [Google Scholar]
- Houslay, M.D.; Schafer, P.; Zhang, K.Y. Keynote review: phosphodiesterase-4 as a therapeutic target. Drug. Discov. Today 2005, 10, 1503–1519. [Google Scholar]
- Huang, Z; Mancini, J.A. Phosphodiesterase 4 inhibitors for the treatment of asthma and COPD. Curr. Med. Chem. 2006, 13, 3253–3262. [Google Scholar]
- Barnes, P.J.; Stockley, R.A. COPD: current therapeutic interventions and future approaches. Eur. Respir. J 2005, 25, 1084–1106. [Google Scholar]
- Burnouf, C.; Pruniaux, M.P. Recent advances in PDE4 inhibitors as immunoregulators and anti-inflammatory drugs. Curr. Pharm. Des 2002, 8, 1255–1296. [Google Scholar]
- Claus, k.; Martin, F. Phosphodiesterase-4 inhibitors as a novel approach for the treatment of respiratory disease: cilomilast. Expert. Opin. Investig. Drugs 2007, 16, 109–124. [Google Scholar]
- Duplantier, A.J.; Bachert, E.L.; Cheng, J.B.; Cohan, V.L.; Jenkinson, T.H.; Kraus, K.G.; Mckechney, M.W.; Pillar, J.D.; Watson, J.W. SAR of a Series of 5,6-Dihydro-(9H)-pyrazolo [3,4-c]-1,2,4-triazolo[4,3-α] pytidines as Potent Inhibitons of Human Eosinophil Phosphodiesterase. J. Med. Chem 2007, 50, 344–349. [Google Scholar]
- Lipworth, B.J. Phosphodiesterase-4 inhibitors for asthma and chronic obstructive pulmonary disease. Lancet 2005, 365, 167–175. [Google Scholar]
- Oding, J.O. Inhibitors of PDE4: a review of recent patent literature. Expert. Opin. Ther. Patents 2005, 15, 773–787. [Google Scholar]
- Huang, M.; Yang, D.Y.; Shang, Z.; Zou, J.; Yu, Q. 3D-QSAR Studies on 4-Hydroxyphenylpyruvate Dioxygenase Inhibitors by Comparative Molecular Field Analysis (CoMFA). Bioorg. Med. Chem. Lett 2002, 12, 2271–2275. [Google Scholar]
- Ozlem, T.A.; Betul, T.G.; Yildiz, I.; Esin, A.S.; Ismail, Y. 3D-QSAR analysis on benzazole derivatives as eukaryotic topoisomerase II inhibitors by using comparative molecular field analysis method. Bioorg. Med. Chem 2005, 13, 6354–6359. [Google Scholar]
- Sybyl 7.0; Tripos Inc: St. Louis, USA.
- Hokuldsson, A. PLS regression methods. J. Chemometrics 1988, 2, 211–228. [Google Scholar]
- Cramer, R.D.; Patterson, D.E.; Bunce, J.D. Comparative Molecular Field Analysis (CoMFA). 1. Effect of Shape on Binding of Steroids to Carrier Proteins. J. Am. Chem. Soc 1998, 110, 5959–5967. [Google Scholar]
- Xu, M.; Zhang, A.Q.; Han, S.K.; Wang, L.S. Studies of 3D-quantitative structure-activity relationships on a set of nitroaromatic compounds: CoMFA, advanced CoMFA and CoMSIA. Chemsphere 2002, 48, 707–715. [Google Scholar]
© 2007 by MDPI ( http://www.mdpi.org) Reproduction is permitted for noncommercial purposes.
Share and Cite
MDPI and ACS Style
Yang, Z.; Sun, P. 3D-QSAR Study of Potent Inhibitors of Phosphodiesterase-4 Using a CoMFA Approach. Int. J. Mol. Sci. 2007, 8, 714-722. https://doi.org/10.3390/i8070714
AMA Style
Yang Z, Sun P. 3D-QSAR Study of Potent Inhibitors of Phosphodiesterase-4 Using a CoMFA Approach. International Journal of Molecular Sciences. 2007; 8(7):714-722. https://doi.org/10.3390/i8070714
Chicago/Turabian StyleYang, Zhaoqi, and Pinghua Sun. 2007. "3D-QSAR Study of Potent Inhibitors of Phosphodiesterase-4 Using a CoMFA Approach" International Journal of Molecular Sciences 8, no. 7: 714-722. https://doi.org/10.3390/i8070714