Adrenergic Agonists Bind to Adrenergic-Receptor-Like Regions of the Mu Opioid Receptor, Enhancing Morphine and Methionine-Enkephalin Binding: A New Approach to “Biased Opioids”?

 ,
, 
Abstract
:
1. Introduction
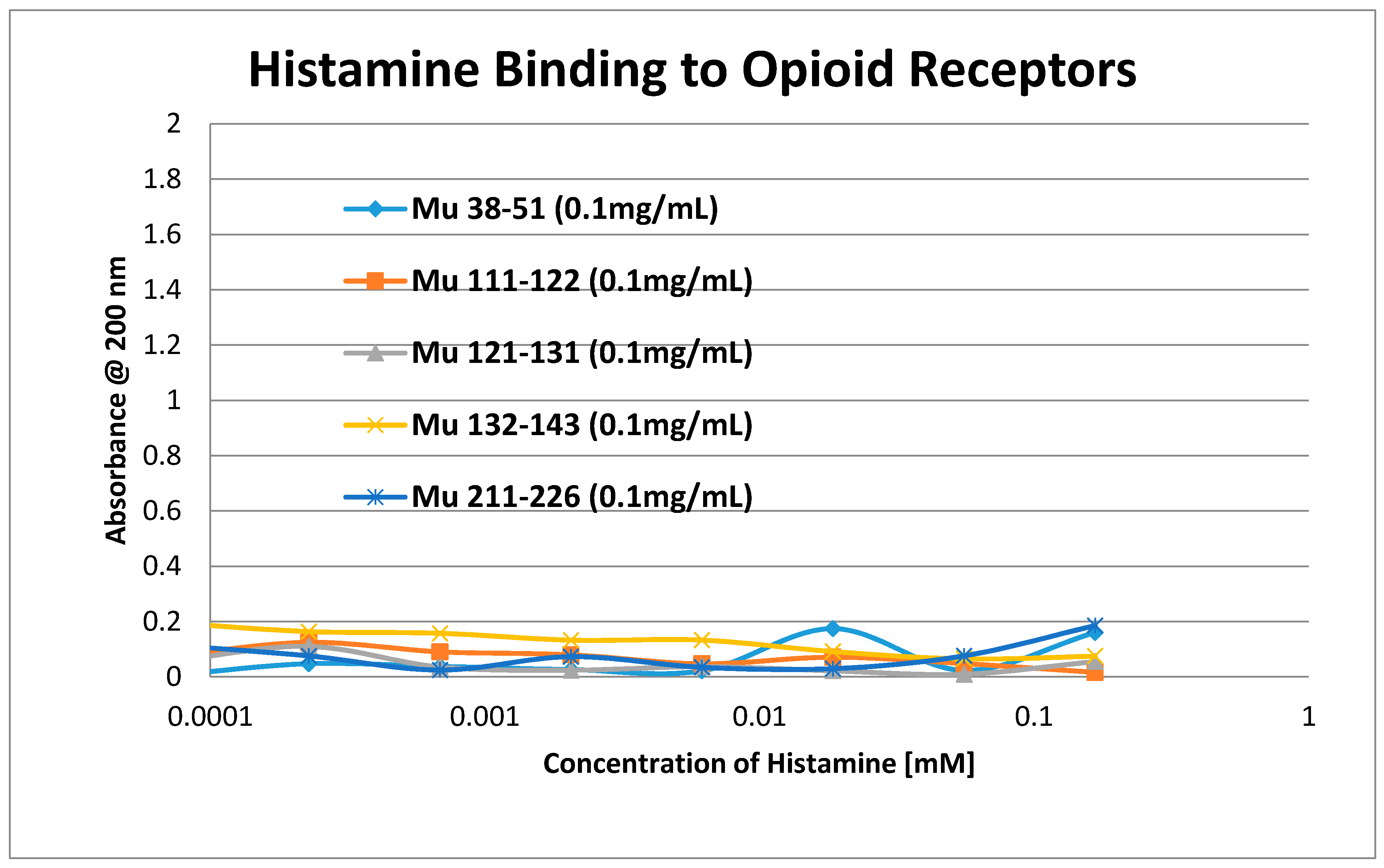
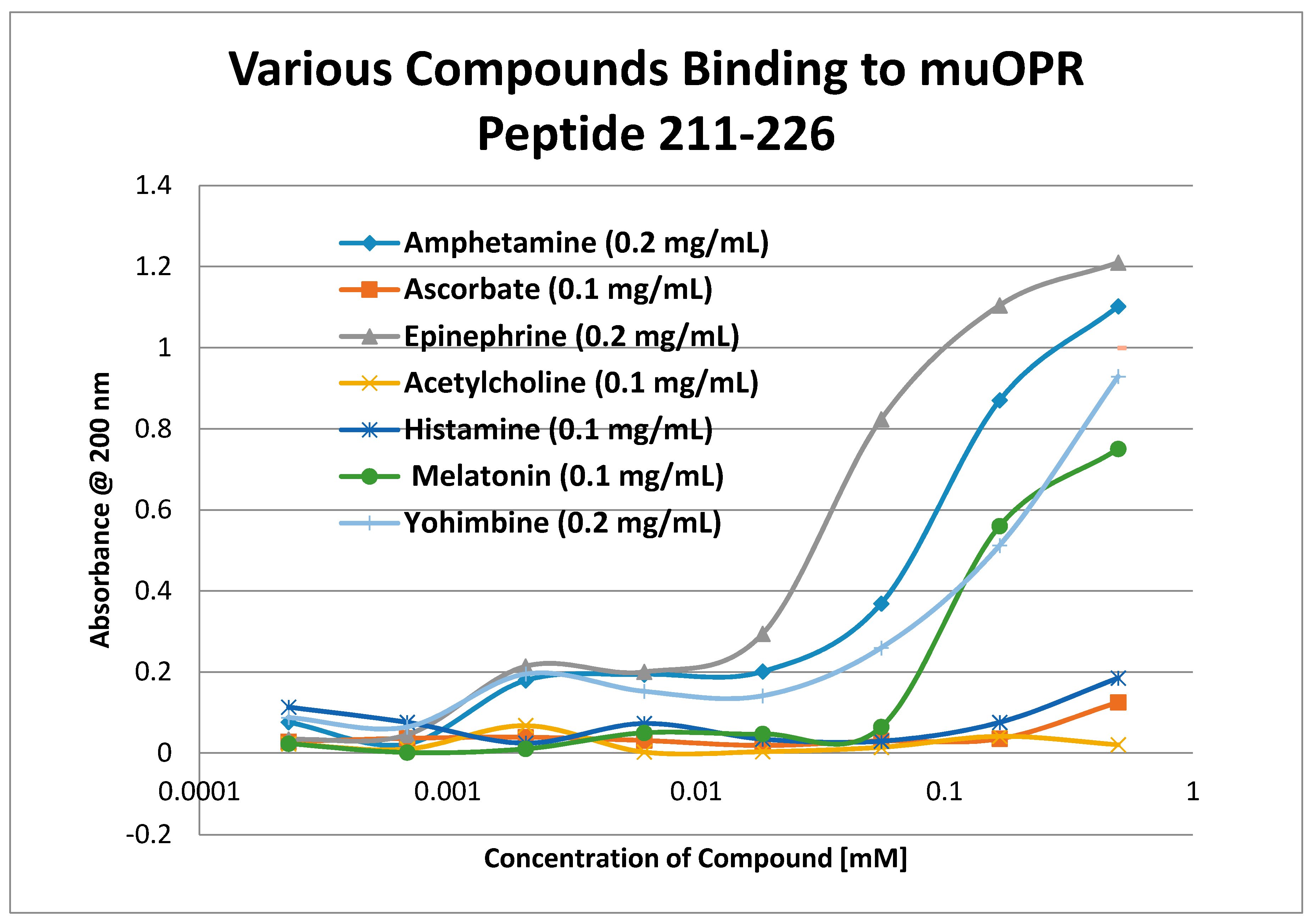
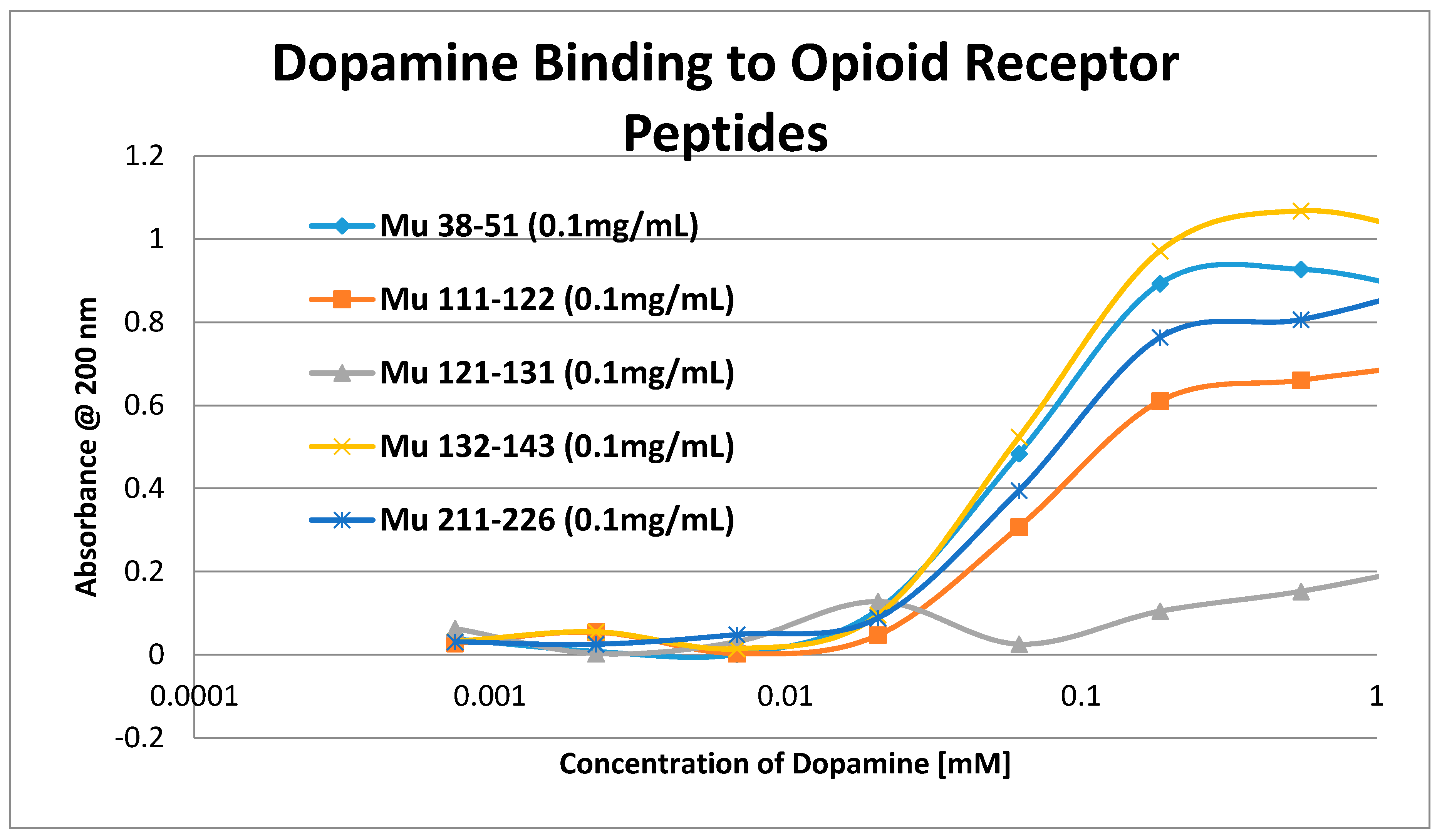
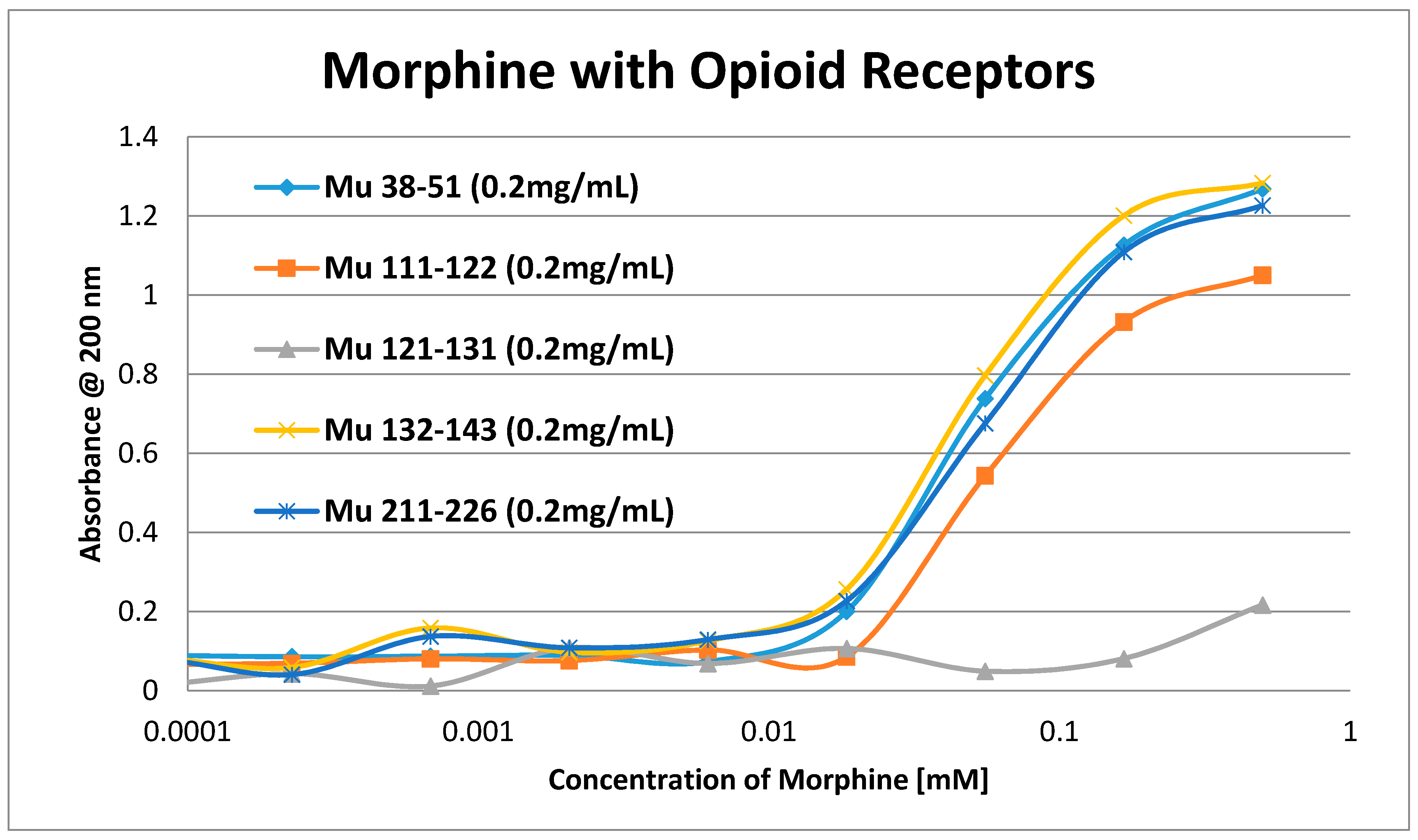
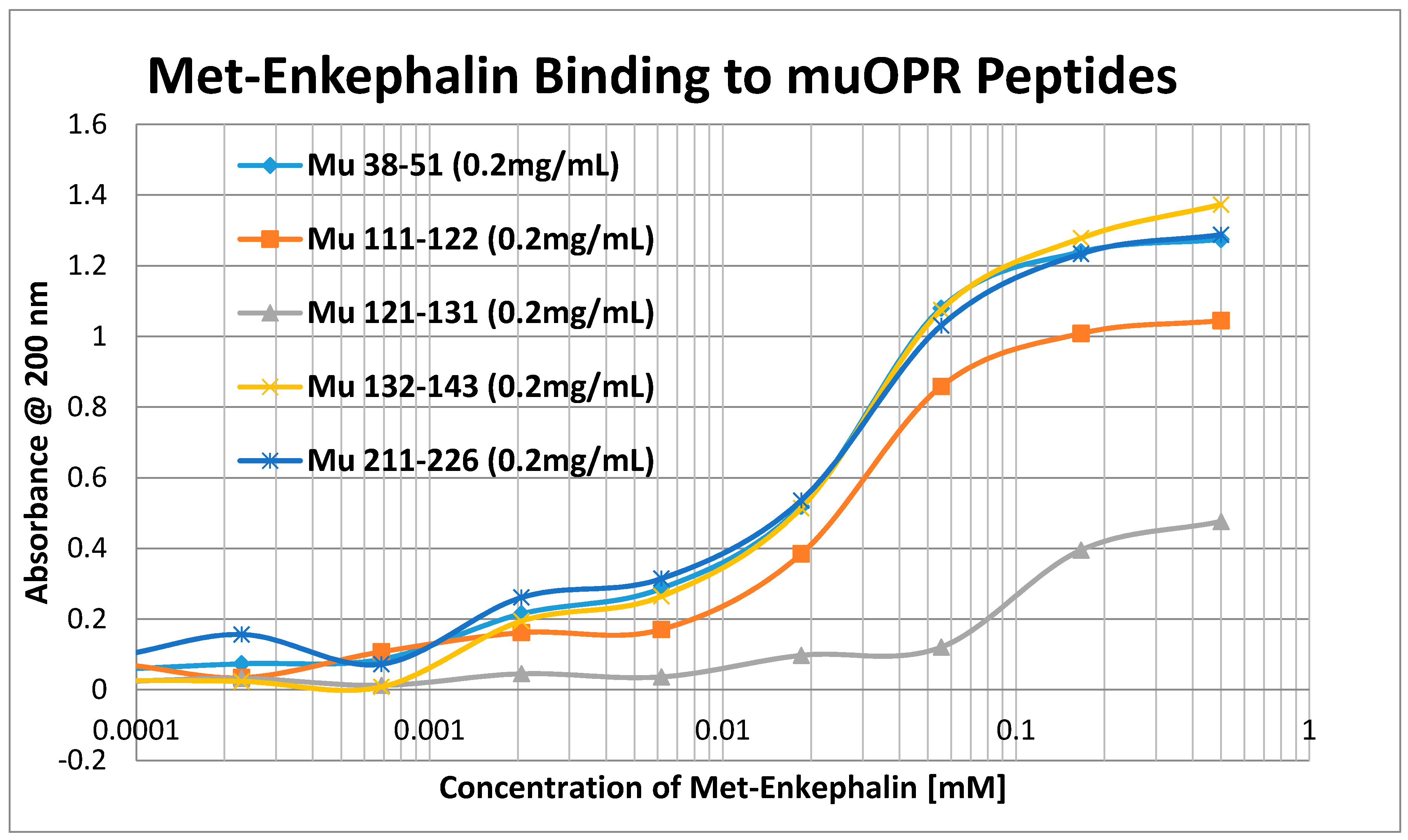
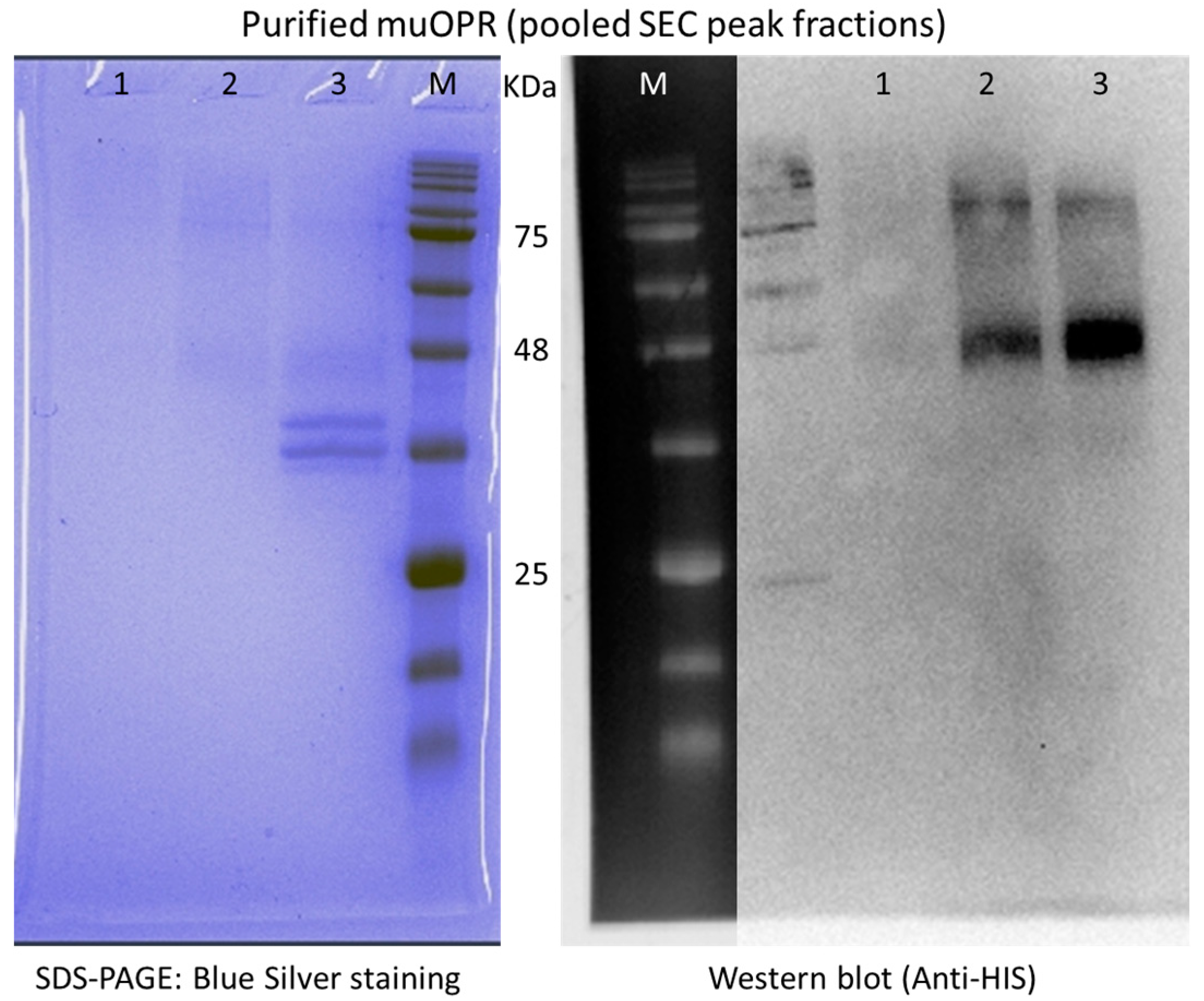
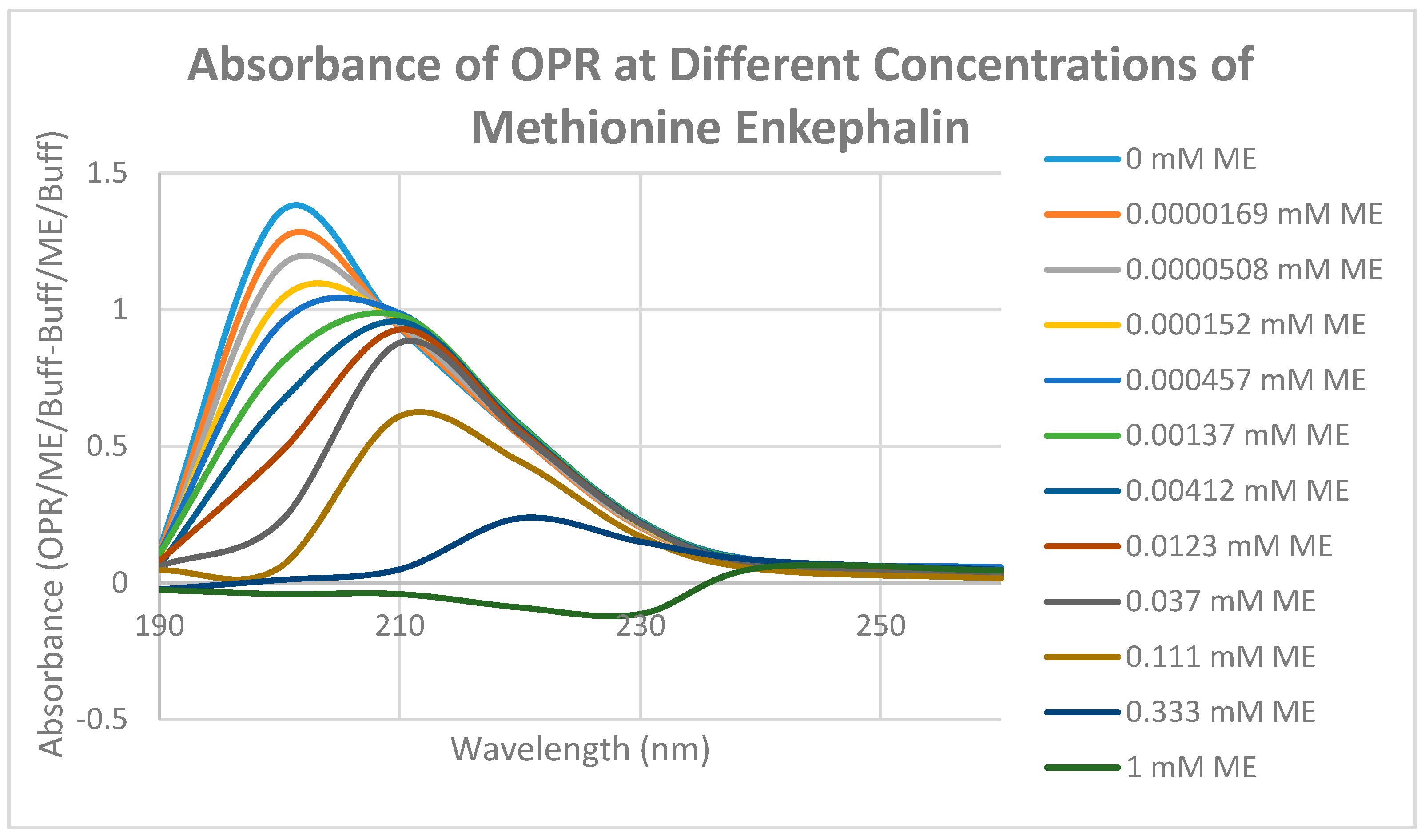
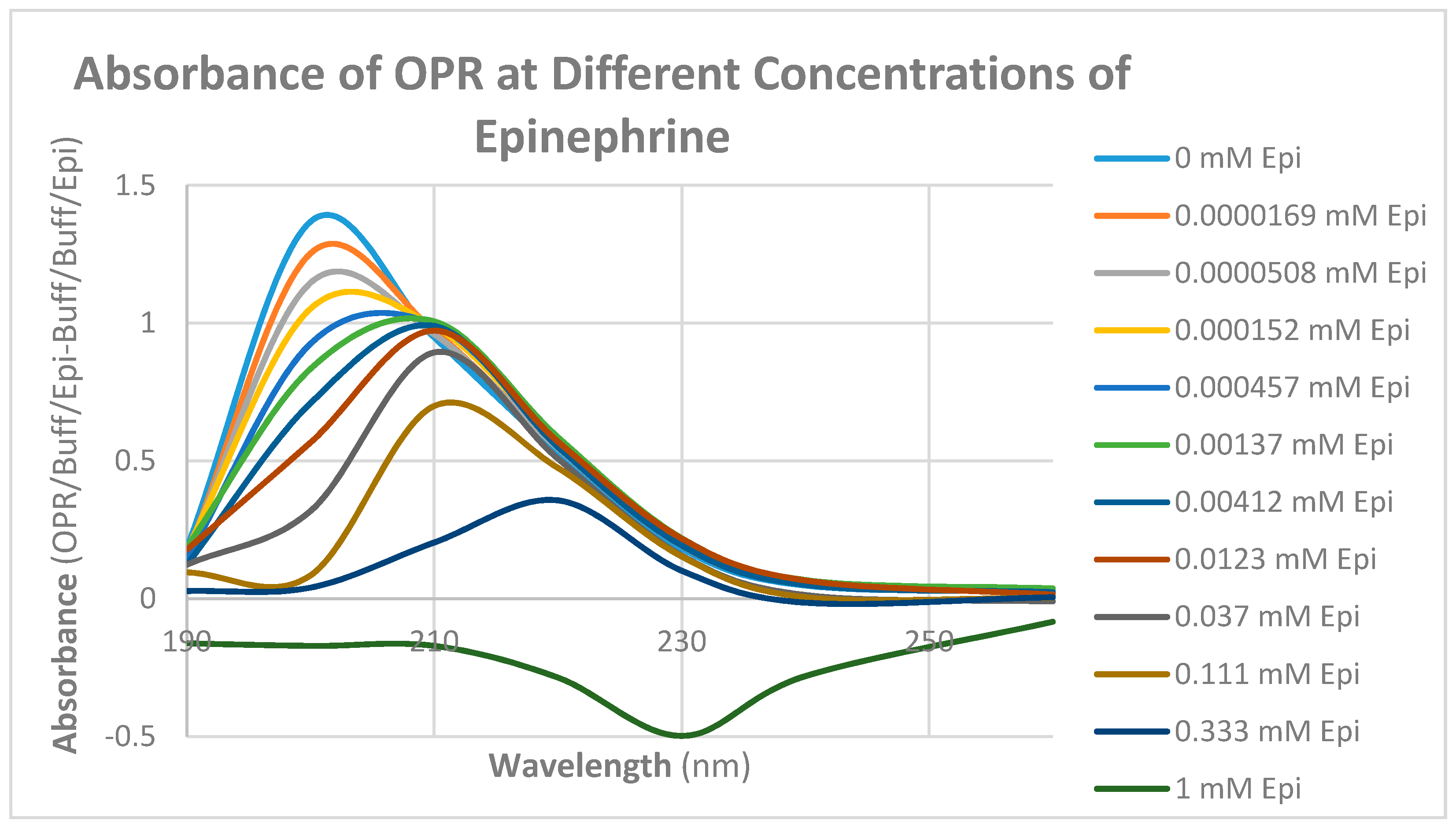
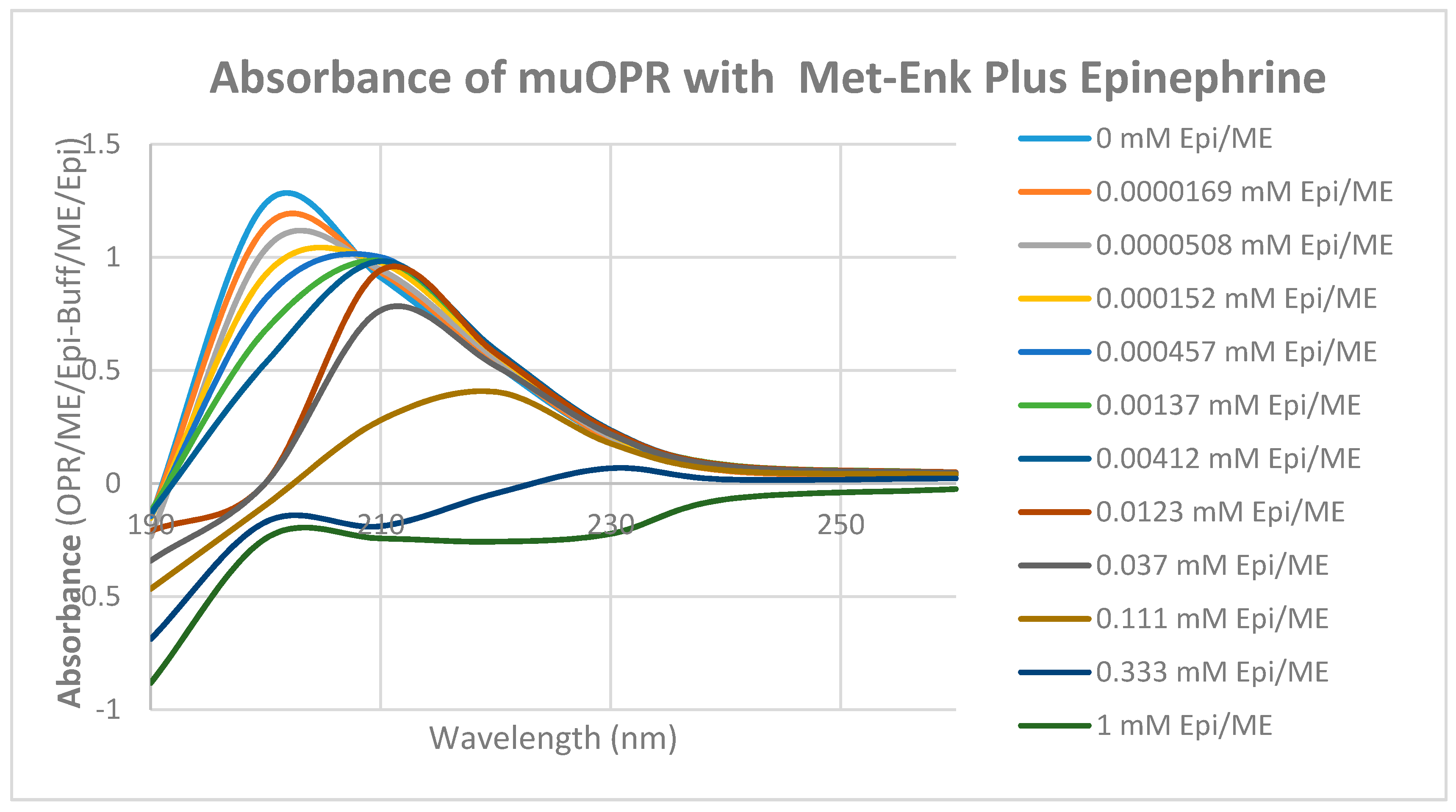
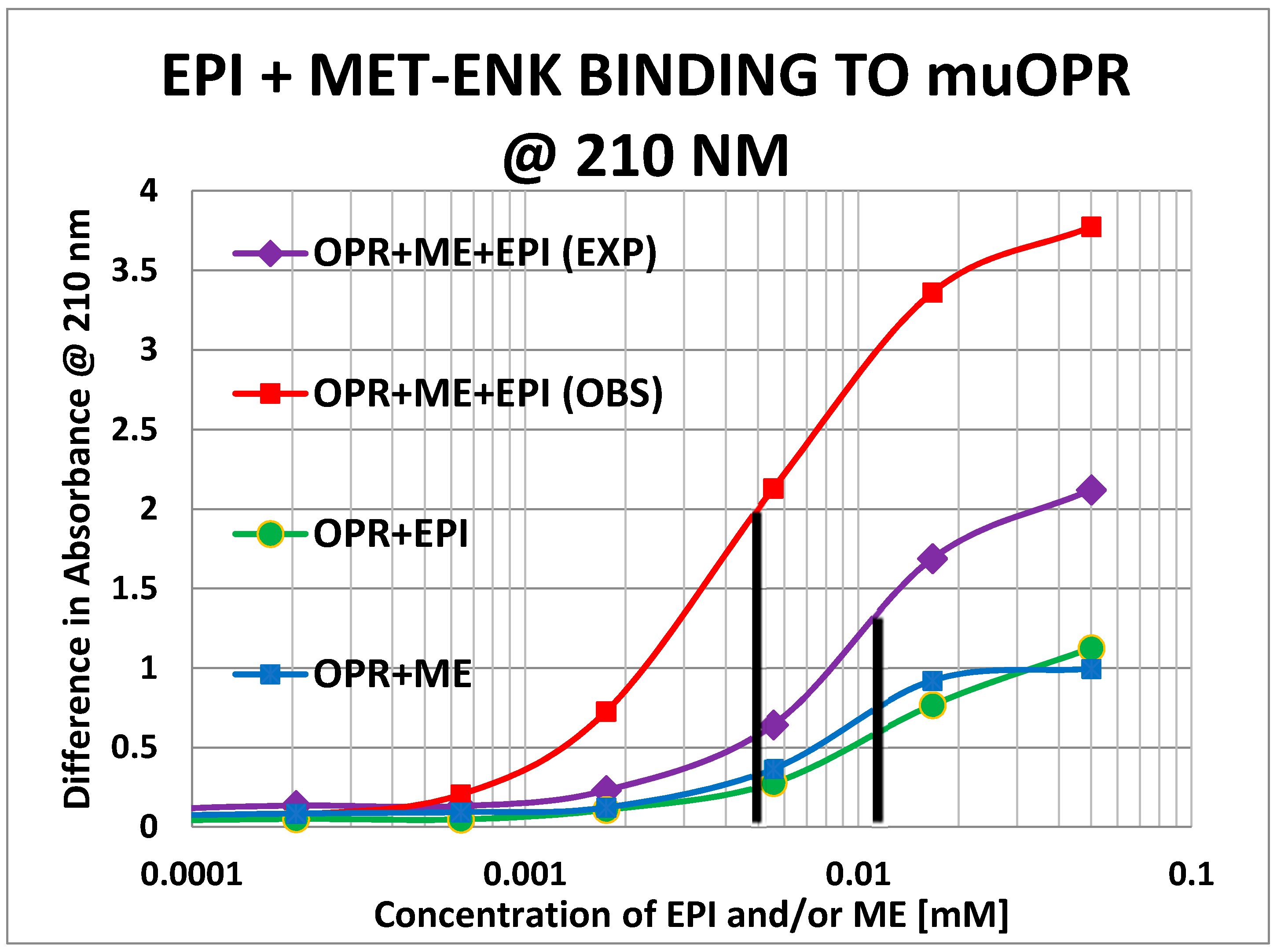
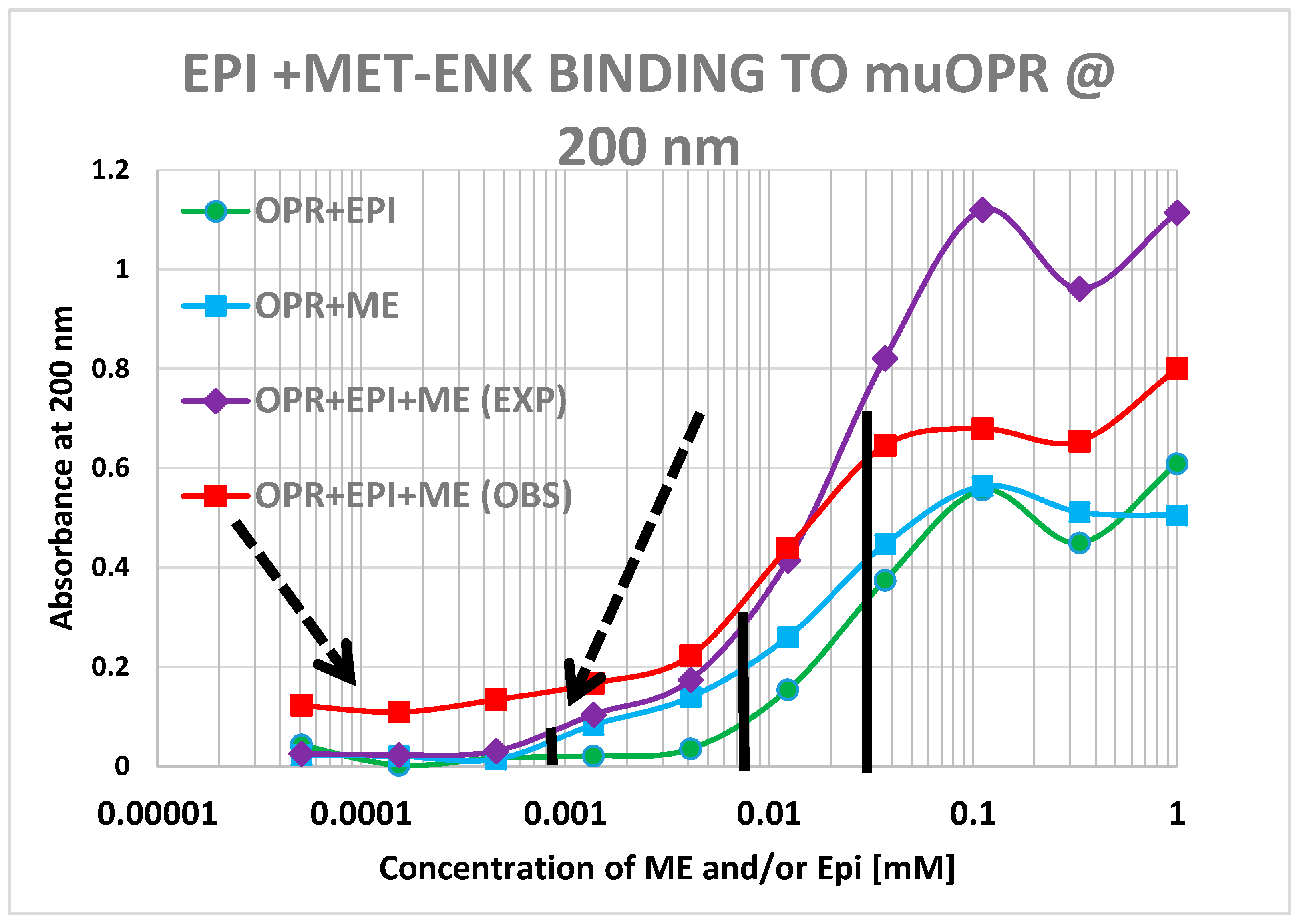
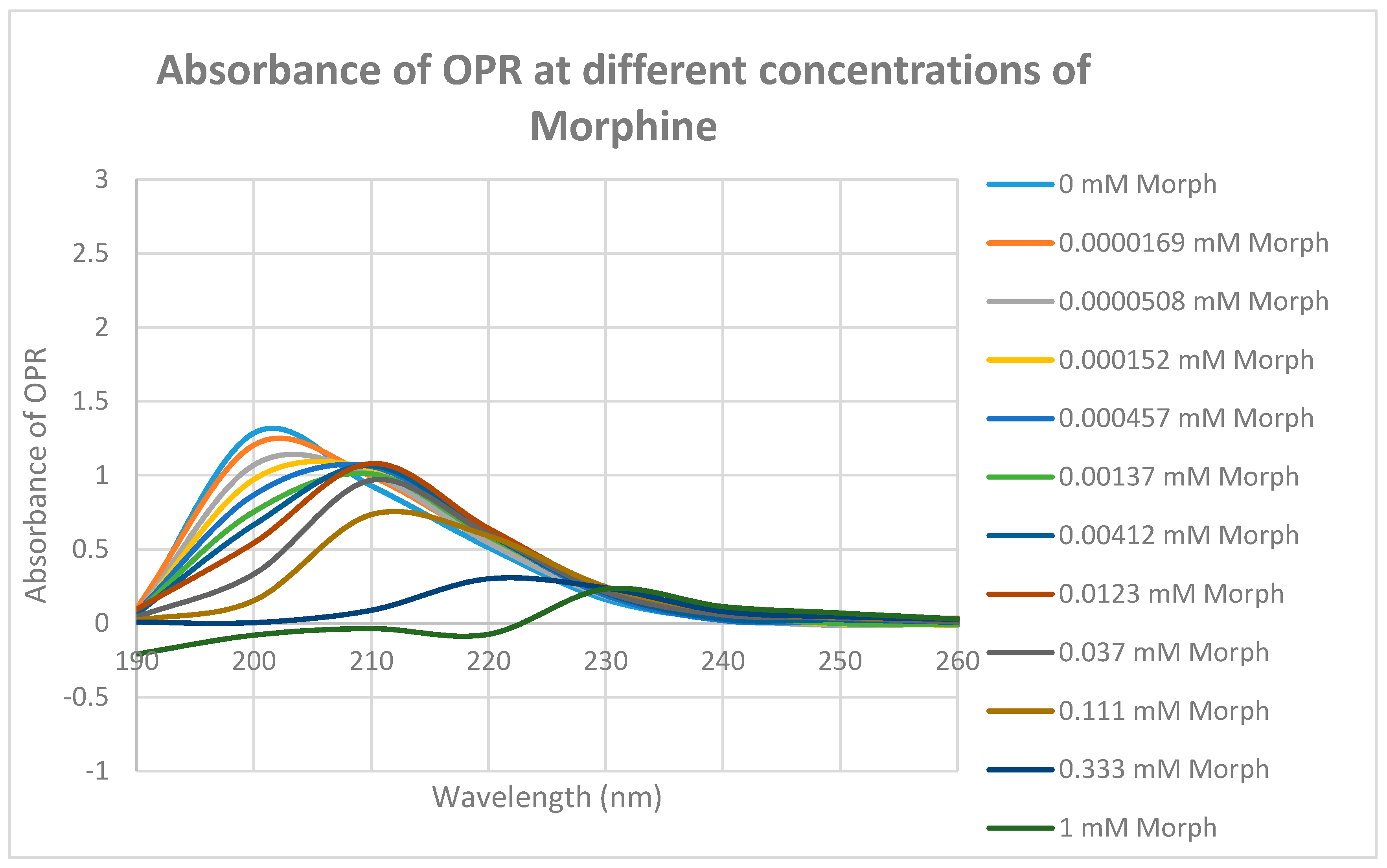
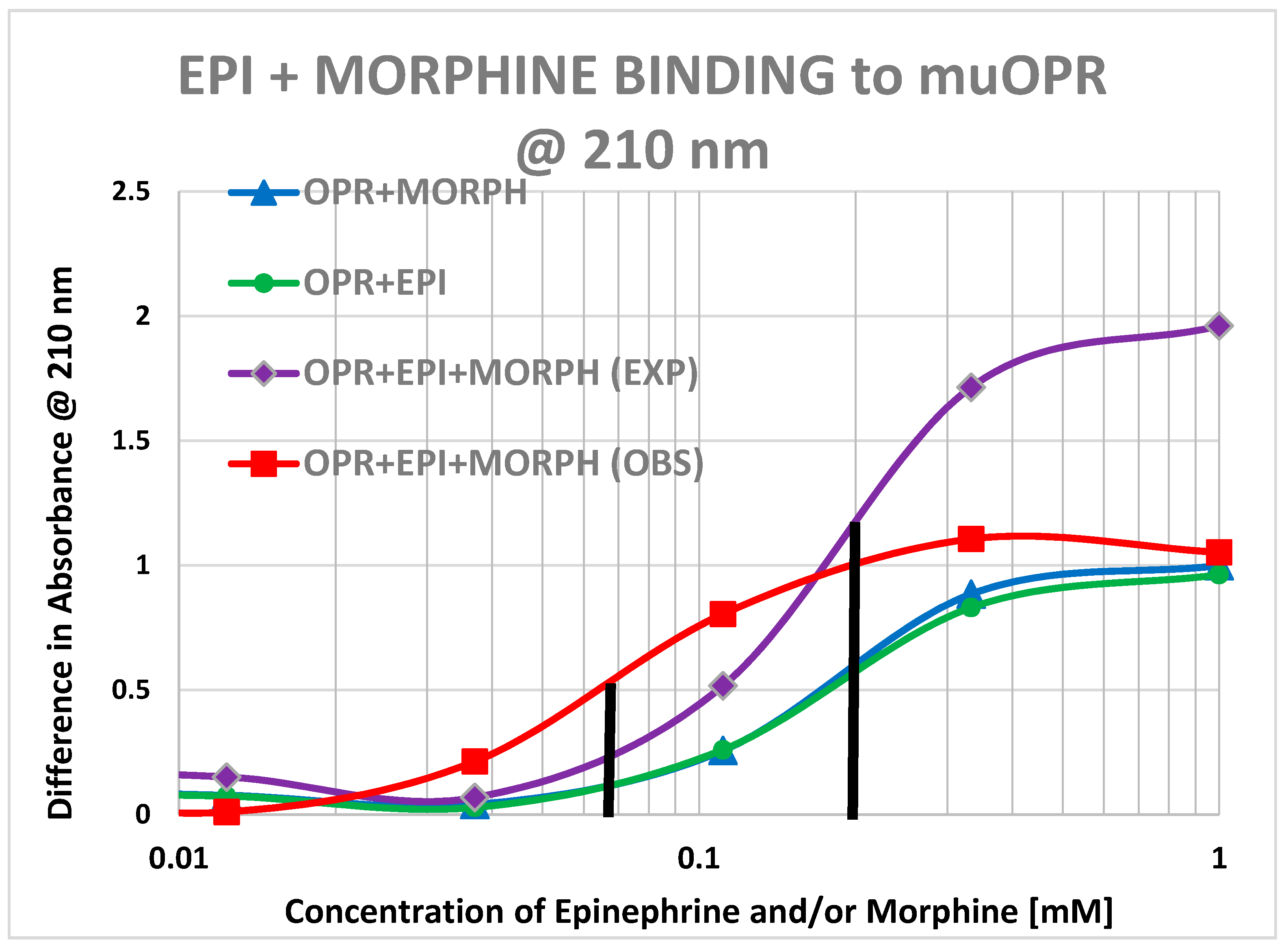
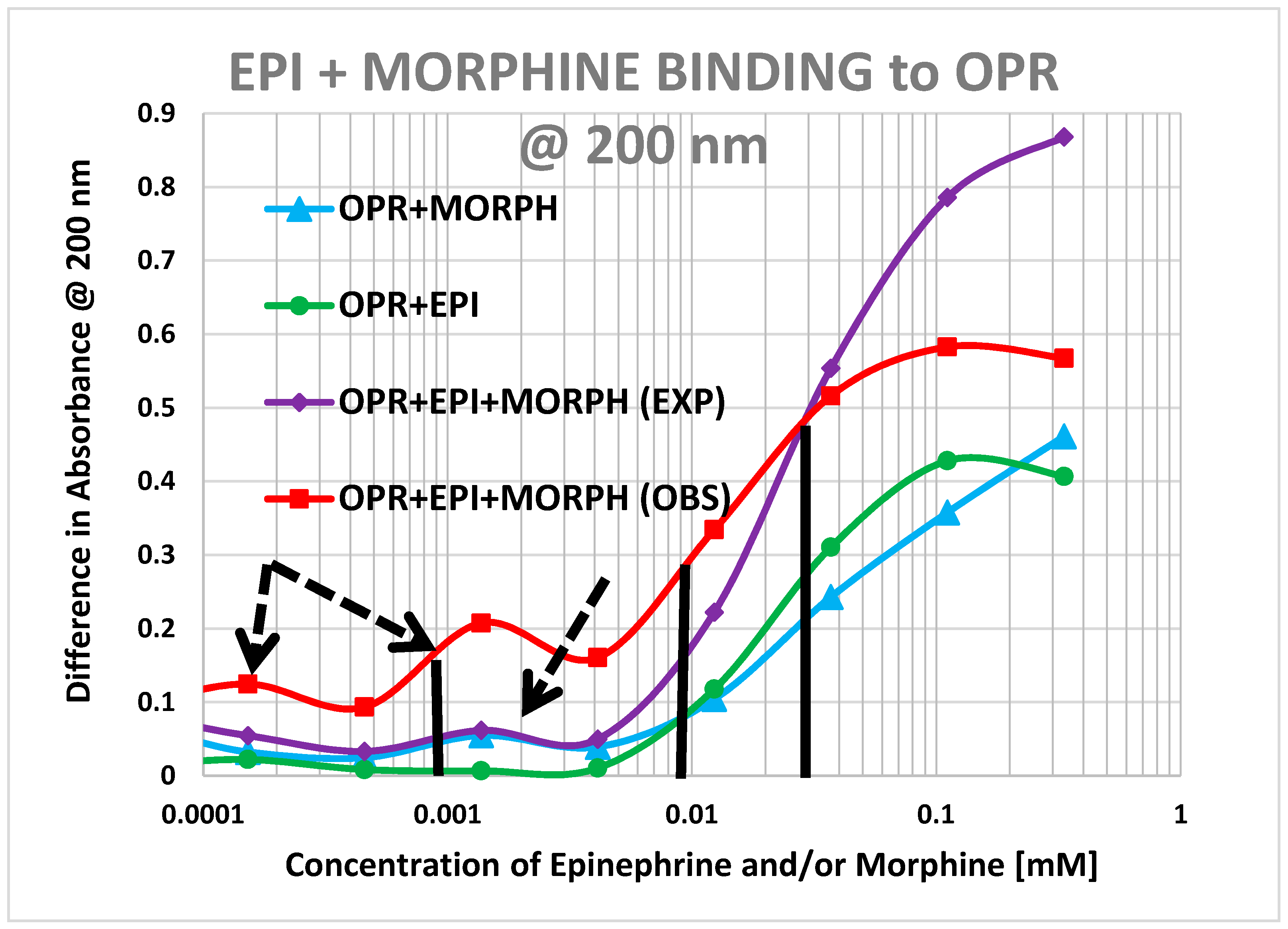
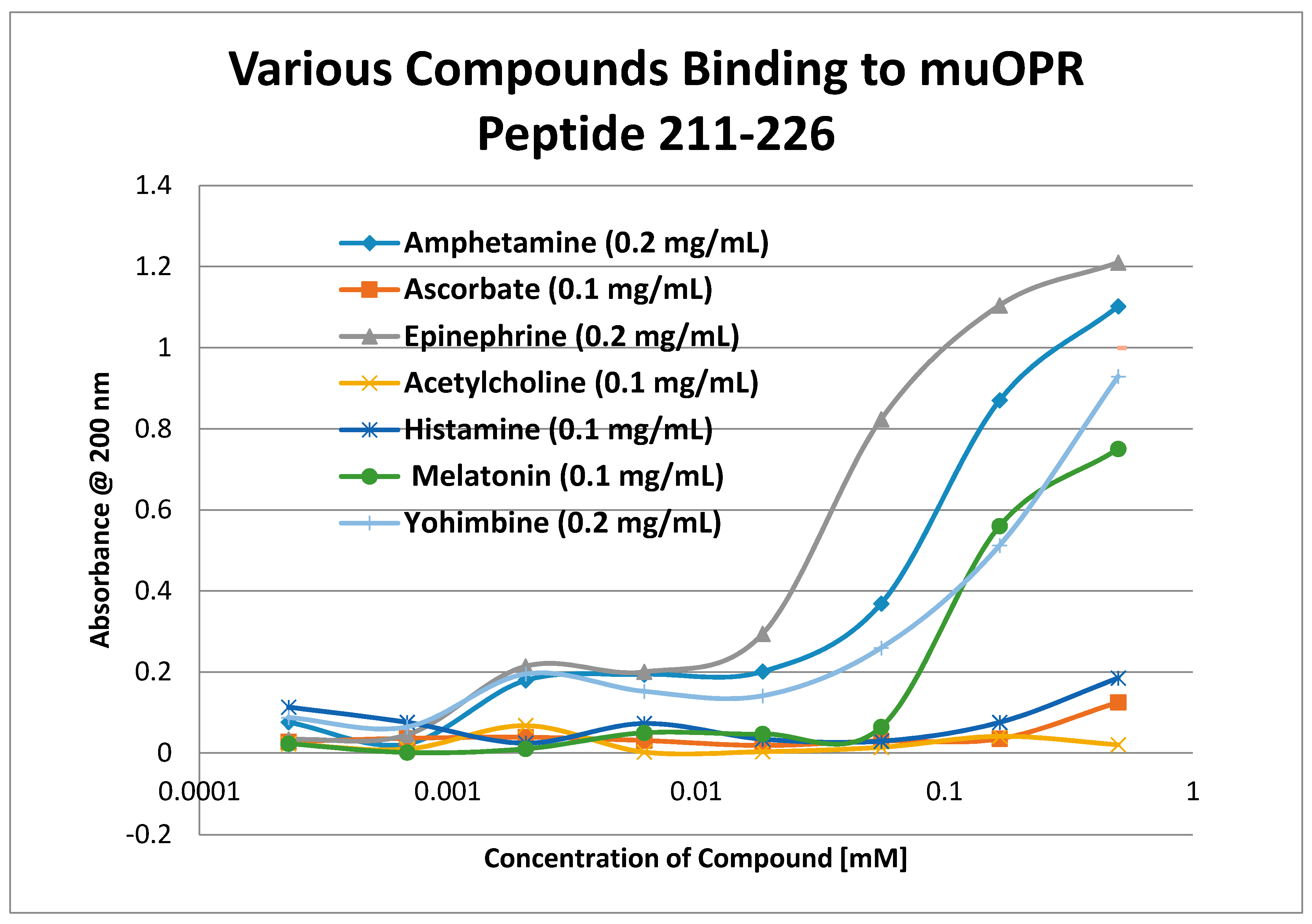
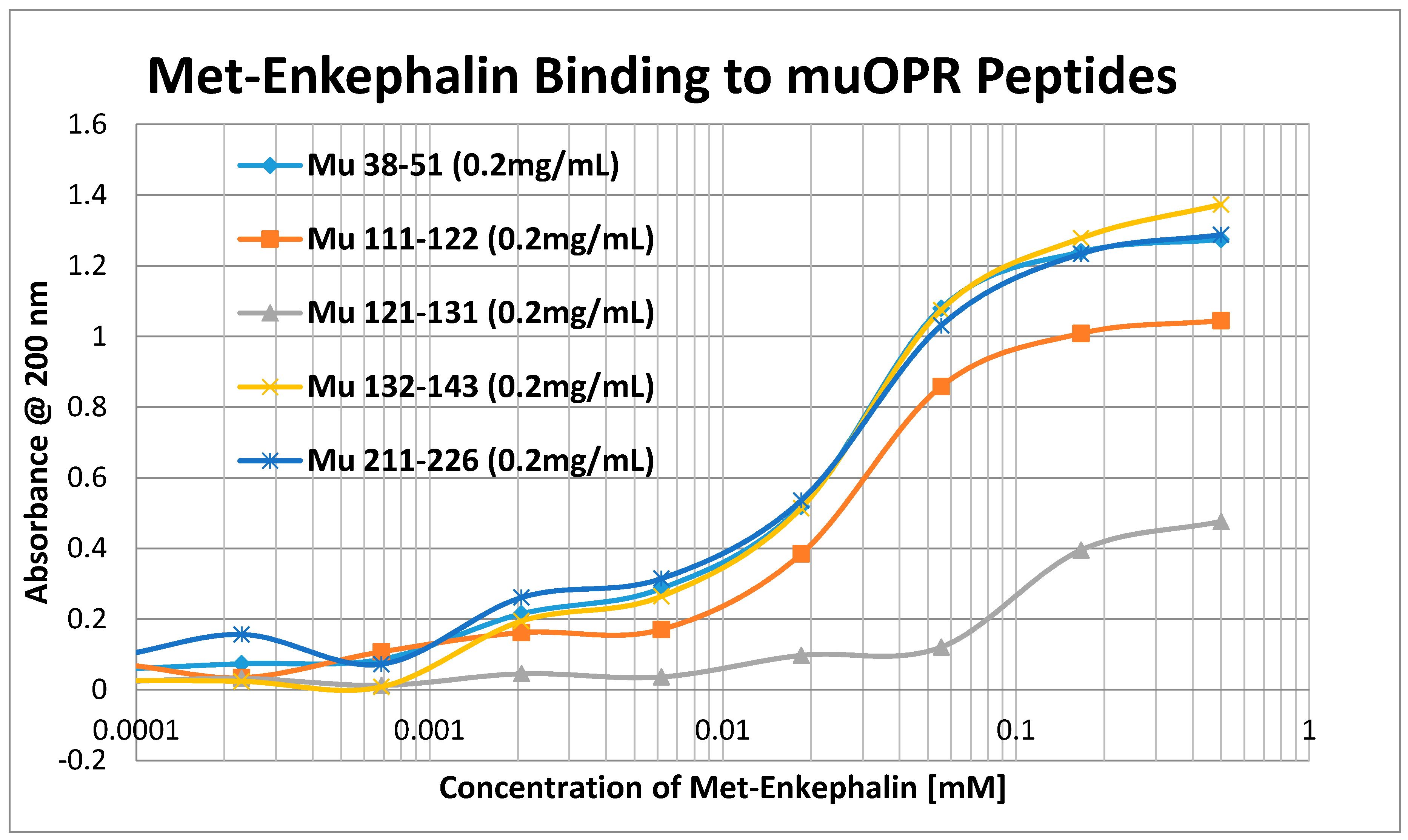
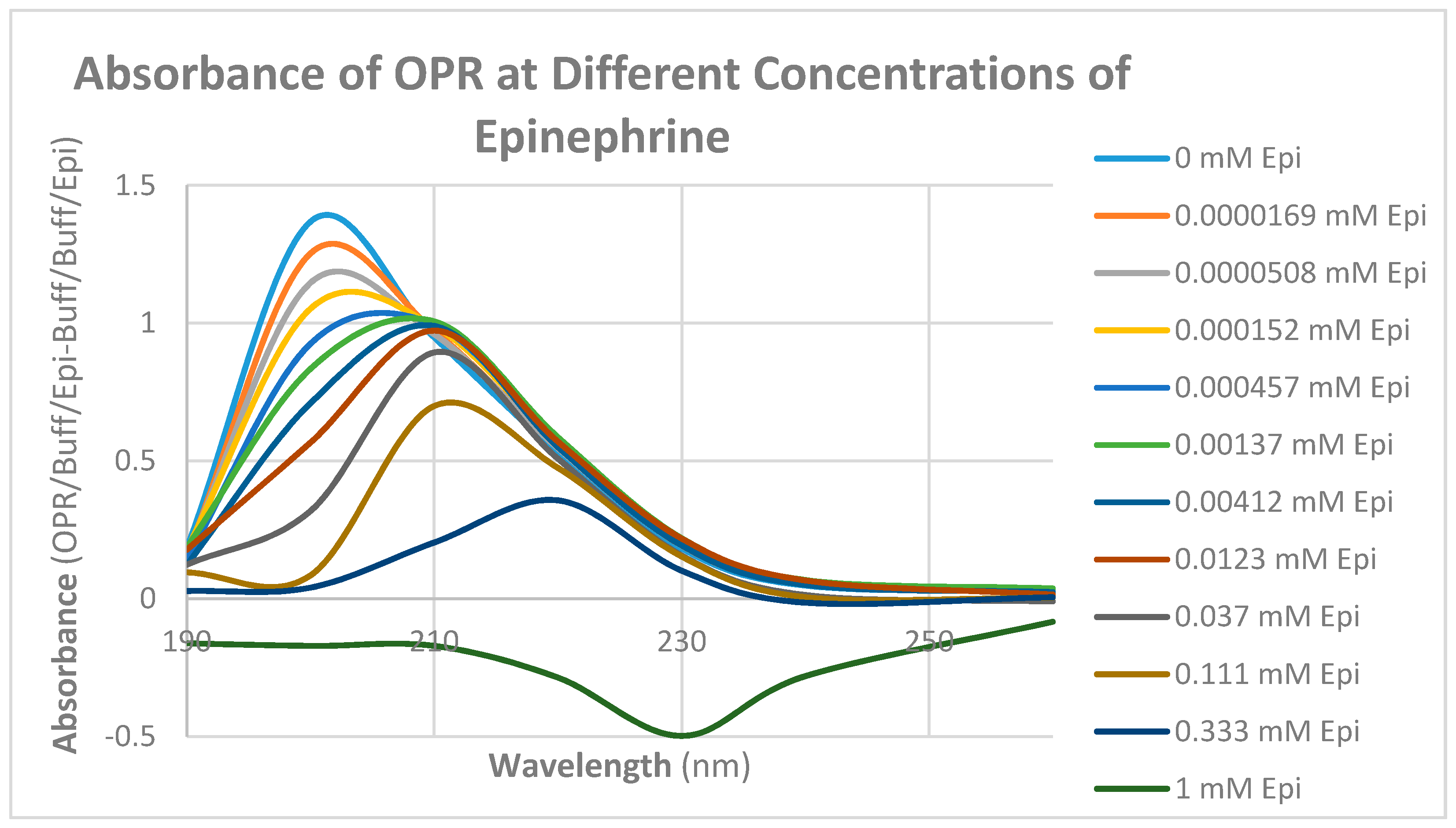
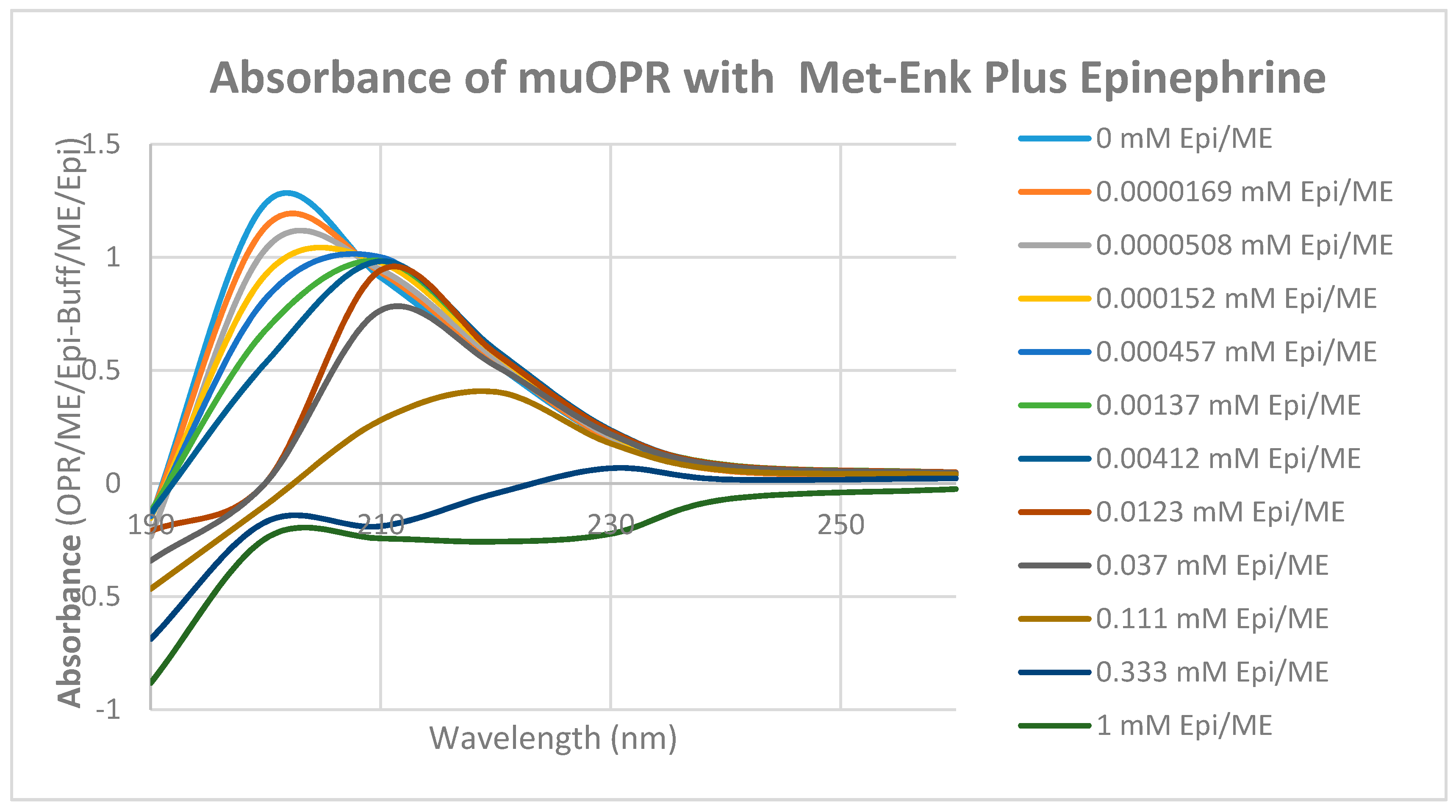
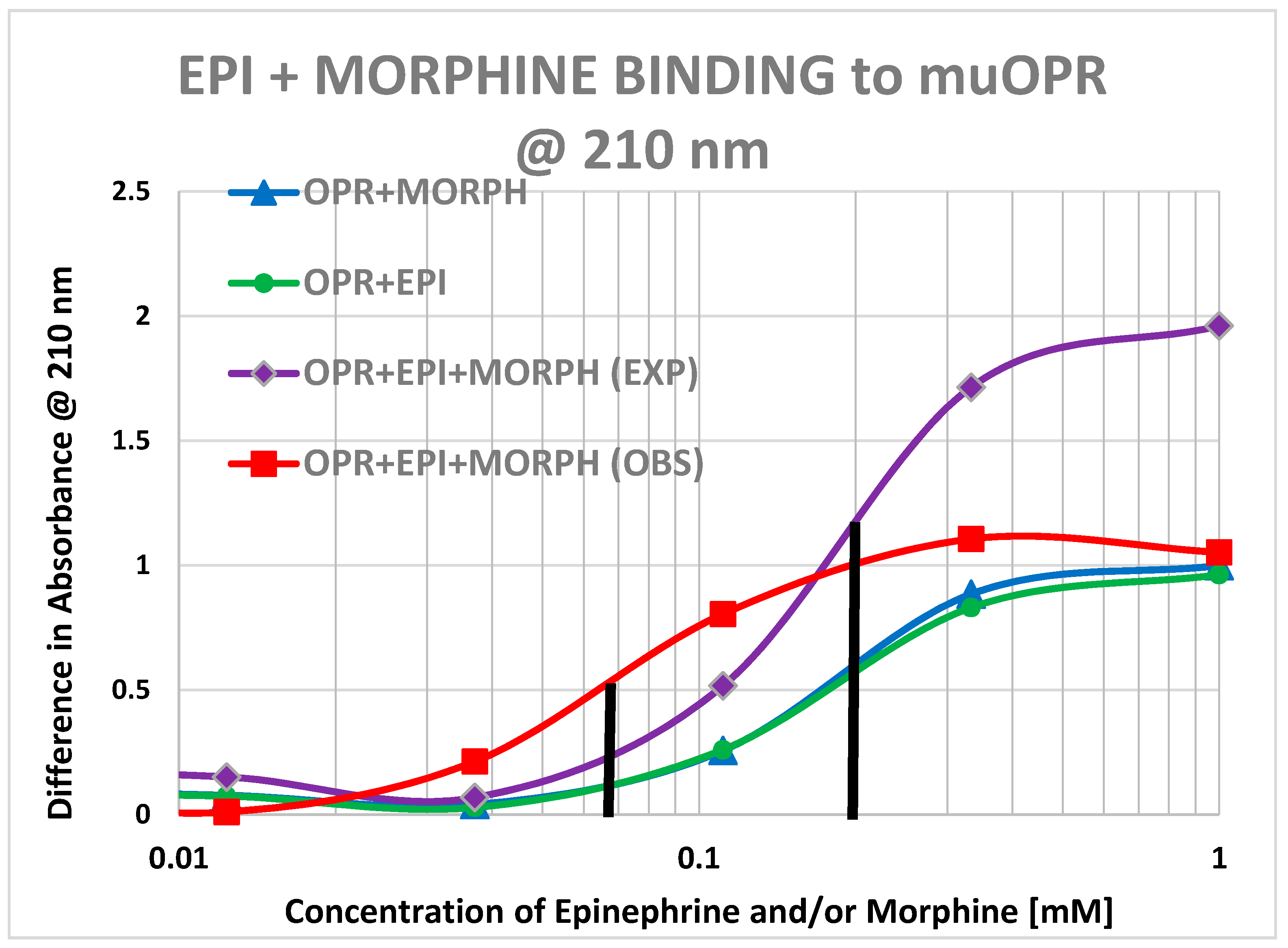
2. Results
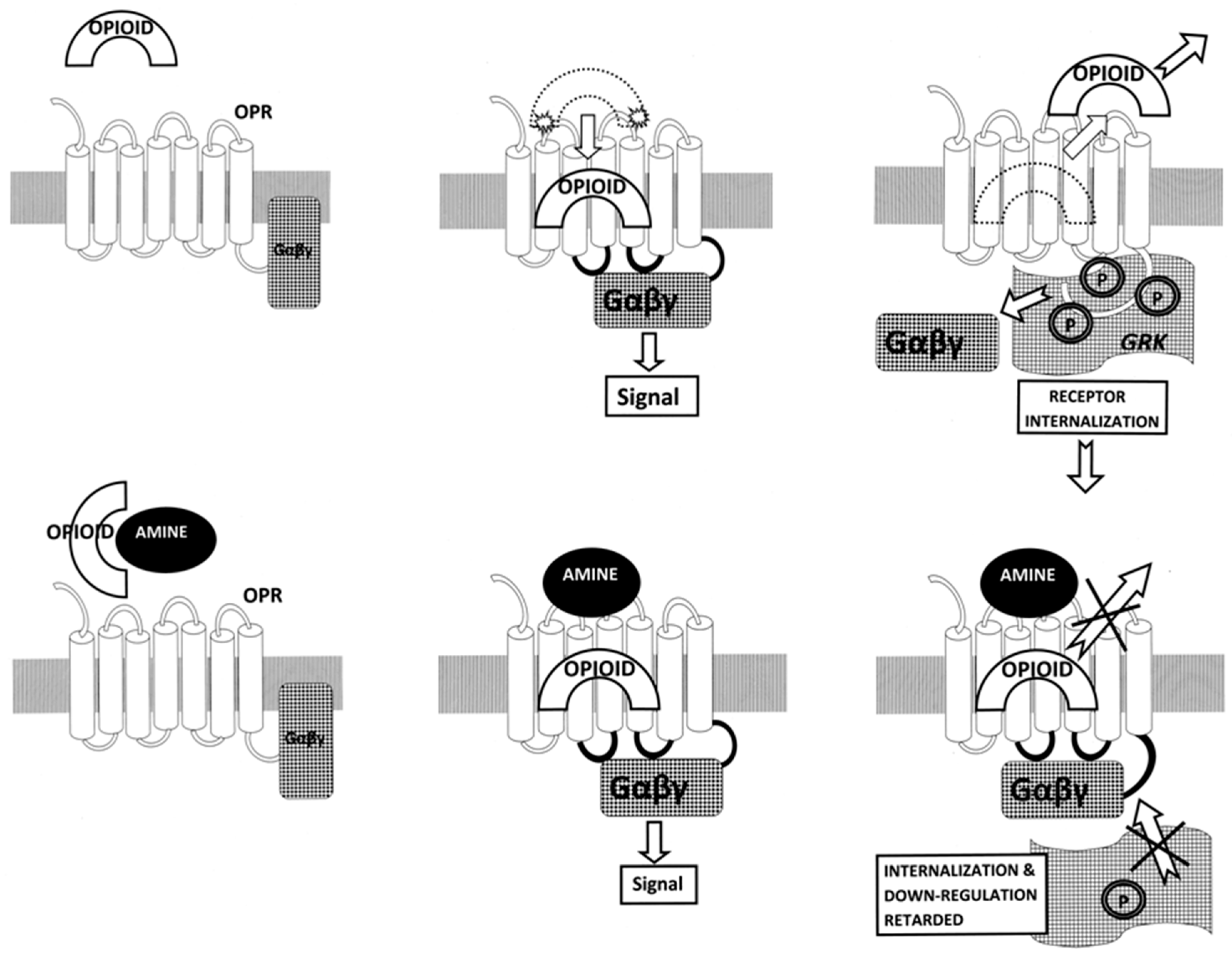
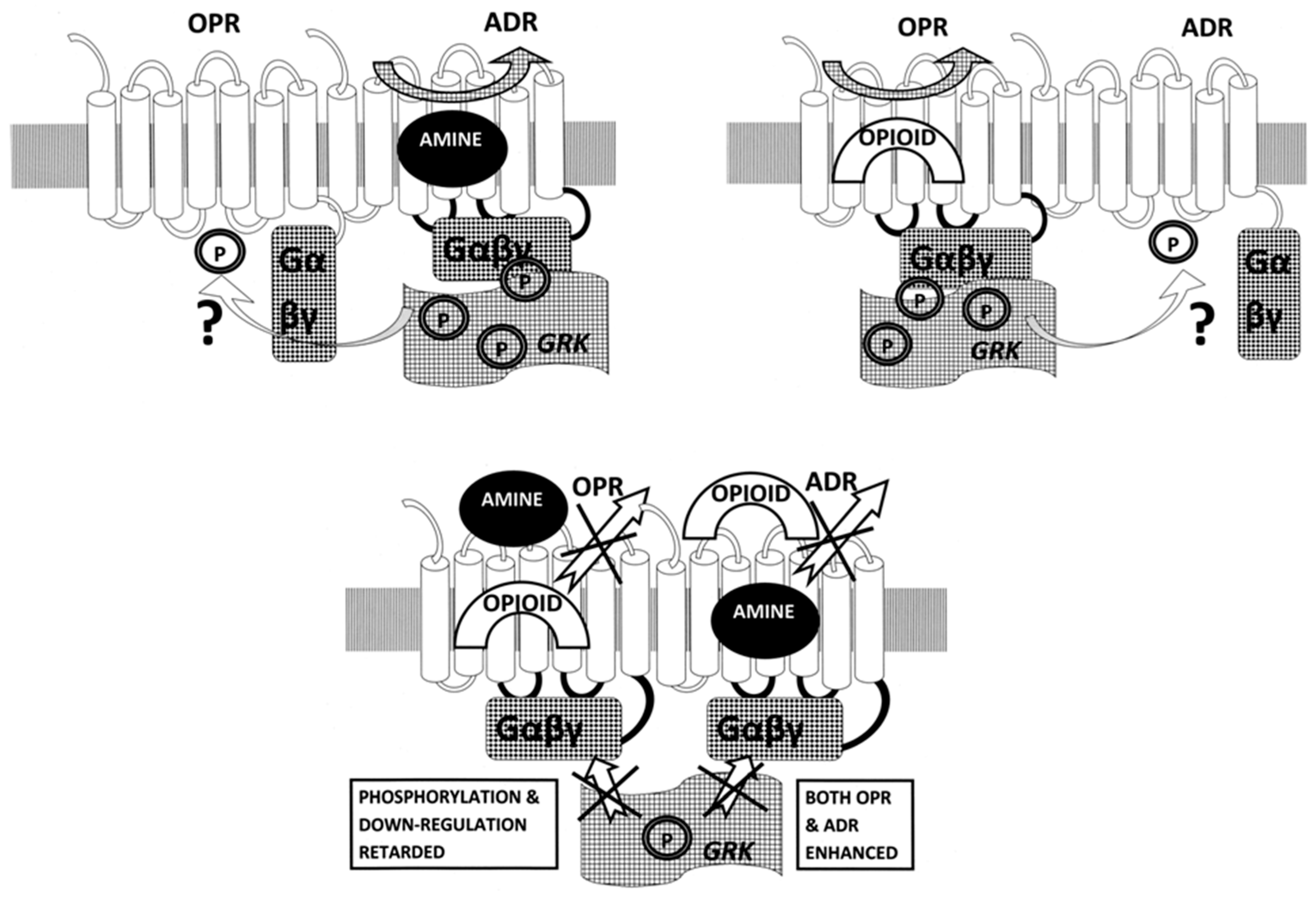
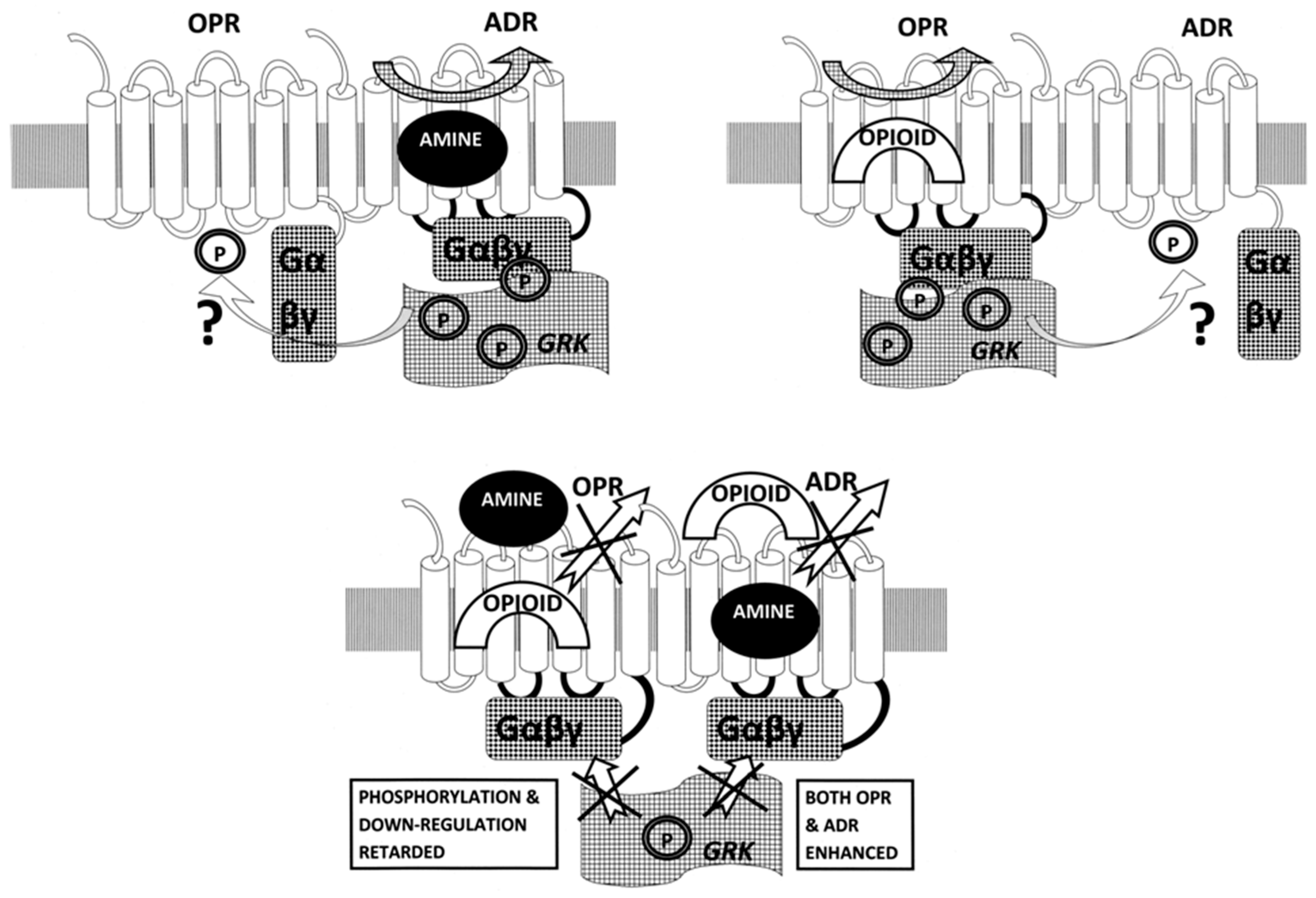
3. Discussion
4. Materials and Methods
4.1. muOPR-ADR Similarity Search
4.2. Opioid Receptor Peptide Synthesis and Preparation
4.3. Opioid Peptide Binding Test Methods
4.4. Human Mu-Opioid Receptor (muOPR) Expression and Purification
4.5. Binding of Epinephrine and Opioids to muOPR Monitored by Ultraviolet Spectroscopy
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Karim, F.; Roerig, S.C. Differential effects of antisense oligodeoxynucleotides directed against Gzα and Goα on antinociception produced by spinal opioid and α2 adrenergic receptor agonists. Pain 2000, 87, 181–191. [Google Scholar] [CrossRef]
- Lawrence, A.J.; Jarrott, B. Neurochemical modulation of cardiovascular control in the nucleus tractus solitarius. Prog. Neurobiol. 1996, 48, 21–53. [Google Scholar] [CrossRef]
- Buccafusco, J.J. Participation of different brain regions in the anti-narcotic withdrawal action of clonidine in the dependent rat. Brain Res. 1990, 513, 8–14. [Google Scholar] [CrossRef]
- Meana, J.J.; Gonzalez-Maeso, J.; Garcia-Sevilla, J.A.; Guimon, J. μ-Opioid receptor and α2-adrenoceptor agonist stimulation of [35S]GTPγS binding to G-proteins in postmortem brains of opioid addicts. Mol. Psychiatry 2000, 5, 308–315. [Google Scholar] [CrossRef] [PubMed]
- Glass, M.J.; Pickel, V.M. α2A-adrenergic receptors are present in μ-opioid receptor containing neurons in rat medial nucleus tractus solitarius. Synapse 2002, 43, 208–218. [Google Scholar] [CrossRef] [PubMed]
- Riedl, M.S.; Schnell, S.A.; Overland, A.C.; Chabot-Doré, A.J.; Taylor, A.M.; Ribeiro-da-Silva, A.; Elde, R.P.; Wilcox, G.L.; Stone, L.S. Coexpression of α 2A-adrenergic and δ-opioid receptors in substance P-containing terminals in rat dorsal horn. J. Comp. Neurol. 2009, 513, 385–398. [Google Scholar] [CrossRef] [PubMed]
- Schultzberg, M.; Hökfelt, T.; Lundberg, J.M.; Terenius, L.; Elfvin, L.G.; Elde, R. Enkephalin-like immunoreactivity in nerve terminals in sympathetic ganglia and adrenal medulla and in adrenal medullary gland cells. Acta Physiol. Scand. 1978, 103, 475–477. [Google Scholar] [CrossRef] [PubMed]
- Charnay, Y.; Leger, L.; Rossier, J.; Jouvet, M.; Dubois, P.M. Evidence for synenkephalin-like immunoreactivity in pontobulbar monoaminergic neurons of the cat. Brain Res. 1985, 335, 160–164. [Google Scholar] [CrossRef]
- Zhuo, H.; Fung, S.J.; Reddy, V.K.; Barnes, C.D. Immunohistochemical evidence for coexistence of methionine-enkephalin and tyrosine hydroxylase in neurons of the locus coeruleus complex projecting to the spinal cord of the cat. J. Chem. Neuroanat. 1992, 5, 1–10. [Google Scholar] [CrossRef]
- Livett, B.G.; Marley, P.D.; Wan, D.C.; Zhou, X.F. Peptide regulation of adrenal medullary function. J. Neural Transm. Suppl. 1990, 29, 77–89. [Google Scholar] [PubMed]
- Stachowiak, M.K.; Goc, A.; Hong, J.S.; Poisner, A.; Jiang, H.K.; Stachowiak, E.K. Regulation of tyrosine hydroxylase gene expression in depolarized non-transformed bovine adrenal medullary cells: Second messenger systems and promoter mechanisms. Brain Res. Mol. Brain Res. 1994, 22, 309–319. [Google Scholar] [CrossRef]
- Carr, D.J.; Gebhardt, B.M.; Paul, D. αAdrenergic and μ-2 opioid receptors are involved in morphine-induced suppression of splenocyte natural killer activity. J. Pharmacol. Exp. Ther. 1993, 264, 1179–1186. [Google Scholar] [PubMed]
- Jordan, B.A.; Trapaidze, N.; Gomes, I.; Nivarthi, R.; Devi, L.A. Oligomerization of opioid receptors with β 2-adrenergic receptors: A role in trafficking and mitogen-activated protein kinase activation. Proc. Natl. Acad. Sci. USA 2001, 98, 343–348. [Google Scholar] [PubMed]
- Jordan, B.A.; Gomes, I.; Rios, C.; Filipovska, J.; Devi, L.A. Functional interactions between mu opioid and α 2A-adrenergic receptors. Mol. Pharmacol. 2003, 64, 1317–1324. [Google Scholar] [CrossRef] [PubMed]
- Rozenfeld, R.; Devi, L.A. Exploring a role for heteromerization in GPCR signaling specificity. Biochem. J. 2011, 433, 11–18. [Google Scholar] [CrossRef] [PubMed]
- Vilardaga, J.-P.; Nikolaev, O.V.; Lorentz, K.; Ferrandon, S.; Zhuang, Z.; Lohse, M.J. Direct inhibition of G protein signaling by cross-conformational switches between a2A-adrenergic and µ-opioid receptors. Nat. Chem. Biol. 2008, 4, 126–131. [Google Scholar] [CrossRef] [PubMed]
- Vilardaga, J.-P.; Agnati, L.F.; Fuxe, K.; Ciruela, F. G-protein-coupled receptor heteromer dynamics. J. Cell Sci. 2010, 123, 4215–4220. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.Q.; Limbird, L.E. Hetero-oligomers of α2A-adrenergic and μ-opioid receptors do not lead to transactivation of G-proteins or altered endocytosis profiles. Biochem. Soc. Trans. 2004, 32, 856–860. [Google Scholar] [CrossRef] [PubMed]
- Fujita, W.; Gomes, I.; Devi, L.A. Revolution in GPCR signalling: Opioid receptor heteromers as novel therapeutic targets: IUPHAR review 10. Br. J. Pharmacol. 2014, 171, 4155–4176. [Google Scholar] [CrossRef] [PubMed]
- Chabot-Doré, A.J.; Millecamps, M.; Naso, L.; Devost, D.; Trieu, P.; Piltonen, M.; Diatchenko, L.; Fairbanks, C.A.; Wilcox, G.L.; Hébert, T.E.; et al. Dual allosteric modulation of opioid antinociceptive potency by α2A-adrenoceptors. Neuropharmacology 2015, 99, 285–300. [Google Scholar] [CrossRef] [PubMed]
- Drouin, C.; Darracq, L.; Trovero, F.; Blanc, G.; Glowinski, J.; Cotecchia, S.; Tassin, J.P. α1b-Adrenergic receptors control locomotor and rewarding effects of psychostimulants and opiates. J. Neurosci. 2002, 22, 2873–2884. [Google Scholar] [PubMed]
- Auclair, A.; Drouin, C.; Cotecchia, S.; Glowinski, J.; Tassin, J.P. 5-HT2A and α1b-adrenergic receptors entirely mediate dopamine release, locomotor response and behavioural sensitization to opiates and psychostimulants. Eur. J. Neurosci. 2004, 20, 3073–3084. [Google Scholar] [CrossRef] [PubMed]
- Kanigel, R. Apprentice to Genius; Macmillan: New York, NY, USA, 1986; pp. 171–201. [Google Scholar]
- Munro, T.A.; Huang, X.-P.; Inglese, C.; Perrone, M.G.; Van’t Veer, A.; Carroll, F.I.; Béguin, C.; Carlezon, W.A., Jr.; Colabufo, N.A.; Cohen, B.M.; et al. Selective κ Opioid Antagonists nor-BNI, GNTI and JDTic Have Low Affinities for Non-Opioid Receptors and Transporters. PLoS ONE 2013, 8, e70701. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lengyel, I.; Toth, F.; Biyashev, D.; Szatmari, I.; Monory, K.; Tomboly, C.; Toth, G.; Benyhe, S.; Borsodi, A. A novel non-opioid binding site for endomorphin-1. J. Physiol. Pharmacol. 2016, 67, 605–616. [Google Scholar] [PubMed]
- He, J.-R.; Molnar, J.; Barraclough, C.A. Morphine amplifies norepinephrine (NE)-induced LH release but blocks NE-stimulated increases in LHRH mRNA levels: Comparison of responses obtained in ovariectomized, estrogen-treated normal and androgen-sterilized rats. Mol. Brain Res. 1993, 20, 71–78. [Google Scholar] [CrossRef]
- Kindman, L.A.; Kates, R.E.; Ginsburg, R. Opioids potentiate contractile response of rabbit myocardium to the β adrenergic agonist isoproterenol. J. Cardiovasc. Pharmacol. 1991, 17, 61–67. [Google Scholar] [CrossRef] [PubMed]
- Lechner, R.B.; Gurll, N.J.; Reynolds, D.G. Naloxone potentiates the cardiovascular effects of catecholamines in canine hemorrhagic shock. Circ. Shock 1985, 16, 347–361. [Google Scholar] [PubMed]
- He, J.-R.; Barraclough, C.A. Morphine but not naloxone enhances luteinizing hormone-releasing hormone neuronal responsiveness to norepinephrine. J. Neuroendocrinol. 1992, 4, 92–99. [Google Scholar] [CrossRef] [PubMed]
- Allgood, S.C.; Gurll, N.J.; Reynolds, D.G. Naloxone requires circulating catecholamines to attenuate the cardiovascular suppression of endotoxic shock. J. Surg. Res. 1988, 44, 73–81. [Google Scholar] [CrossRef]
- Caffrey, J.L.; Hathorne, L.F.; Carter, G.C.; Sinclair, R.J. Naloxone potentiates contractile responses to epinephrine in isolated canine arteries. Circ. Shock 1990, 31, 317–332. [Google Scholar] [PubMed]
- Caffrey, J.L.; Stoll, S.T.; Sinclair, R.J.; Barron, B.A. (+) naloxone enhances vascular contractile responses to added epinephrine. Prog. Clin. Biol. Res. 1990, 328, 375–378. [Google Scholar] [PubMed]
- Lechner, R.B. Naloxone potentiates inotropic but not chronotropic effects of isoproterenol in vitro. Circ. Shock 1993, 39, 226–230. [Google Scholar] [CrossRef] [PubMed]
- Gu, H.; Gaugl, J.F.; Barron, B.A.; Caffrey, J.L. Naloxone enhances cardiac contractile responses to epinephrine without altering epinephrine uptake from plasma. Circ. Shock 1990, 32, 257–271. [Google Scholar] [PubMed]
- McCubbin, J.A.; Surwit, R.S.; Kuhn, C.M.; Cochrane, C.; Feinglos, M.N. Naltrexone potentiates glycemic responses during stress and epinephrine challenge in genetically obese mice. Psychosom. Med. 1989, 51, 441–448. [Google Scholar] [CrossRef] [PubMed]
- Parra, L.; Pérez-Vizcaíno, F.; Alsasua, A.; Martín, M.I.; Tamargo, J. μ- and δ-opioid receptor-mediated contractile effects on rat aortic vascular smooth muscle. Eur. J. Pharmacol. 1995, 277, 99–105. [Google Scholar] [CrossRef]
- Lee, C.H.; Berkowitz, B.A. Stereoselective and calcium-dependent contractile effects of narcotic antagonist analgesics in the vascular smooth muscle of the rat. J. Pharmacol. Exp. Ther. 1976, 198, 347–356. [Google Scholar] [PubMed]
- Lee, C.H.; Berkowitz, B.A. Calcium antagonist activity of methadone, l-acetylmethadol and l-pentazocine in the rat aortic strip. J. Pharmacol. Exp. Ther. 1977, 202, 646–653. [Google Scholar] [PubMed]
- Deyo, S.N.; Swift, R.M.; Miller, R.J. Morphine and endorphins modulate dopamine turnover in rat median eminence. Proc. Natl. Acad. Sci. USA 1979, 76, 3006–3009. [Google Scholar] [CrossRef] [PubMed]
- Deyo, S.N.; Swift, R.M.; Miller, R.J.; Fang, V.S. Development of tolerance to the prolactin-releasing action of morphine and its modulation by hypothalamic dopamine. Endocrinology 1980, 106, 1469–1474. [Google Scholar] [CrossRef] [PubMed]
- Tagaya, E.; Tamaoki, J.; Chiyotani, A.; Konno, K. Stimulation of opioid mu-receptors potentiates β adrenoceptor-mediated relaxation of canine airway smooth muscle. J. Pharmacol. Exp. Ther. 1995, 275, 1288–1292. [Google Scholar] [PubMed]
- Root-Bernstein, R.S.; Dillon, P.F. Fostering adventure research. A case study of the discovery that ascorbic acid enhances adrenergic drug activity. Drug Dev. Res. 2002, 57, 58–74. [Google Scholar] [CrossRef]
- Root-Bernstein, R.S.; Dillon, P.F. A common molecular motif characterizes extracellular allosteric enhancers of GPCR aminergic receptors and suggests their mechanism of action. Curr. Med. Chem. 2014, 21, 3673–3686. [Google Scholar] [CrossRef] [PubMed]
- Root-Bernstein, R.; Fewins, J.; Dillon, P.F. Tartaric Acid Enhances Adrenergic Receptor Activity: Test of a General Theory of Extracellular Aminergic GPCR Enhancer Discovery. Curr. Drug Discov. Technol. 2014, 11, 293–307. [Google Scholar] [CrossRef] [PubMed]
- Abel, S.; Harris, S.C. Morphine-benzedrine analgesia in obstetrics. Fed Proc. 1947, 6, 67. [Google Scholar] [PubMed]
- Milosevic, M.P. Effect of adrenaline on the analgesic response of mice to morphine and related drugs. Arch. Int. Pharmacodyn. Ther. 1955, 104, 50–56. [Google Scholar] [PubMed]
- Goyagi, T.; Nishikawa, T. The addition of epinephrine enhances postoperative analgesia by intrathecal morphine. Anesth. Analg. 1995, 81, 508–513. [Google Scholar] [PubMed]
- Goyagi, T.; Nishikawa, T. Oral clonidine premedication enhances the quality of postoperative analgesia by intrathecal morphine. Anesth. Analg. 1996, 82, 1192–1196. [Google Scholar] [PubMed]
- Sasson, S.; Unterwald, E.M.; Kornetsky, C. Potentiation of morphine analgesia by d-amphetamine. Psychopharmacology 1986, 90, 163–165. [Google Scholar] [CrossRef] [PubMed]
- Izenwasser, S.; Kornetsky, C. Potentiation of morphine analgesia by d-amphetamine is mediated by norepinephrine and not dopamine. Pain 1988, 33, 363–368. [Google Scholar] [CrossRef]
- Huang, K.S.; Tseng, C.H.; Cheung, K.S.; Hui, Y.L.; Tan, P.P. Influence of epinephrine as an adjuvant to epidural morphine for postoperative analgesia. Ma Zui Xue Za Zhi 1993, 31, 245–248. (In Chinese) [Google Scholar] [PubMed]
- Sierralta, F.; Naquira, D.; Pinardi, G.; Miranda, H.F. α-Adrenoceptor and opioid receptor modulation of clonidine-induced antinociception. Br. J. Pharmacol. 1996, 119, 551–554. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.W.; Seah, Y.S.; Chung, K.T.; Liu, M.D. Postoperative pain relief in primigravida caesarean section patients--combination of intrathecal morphine and epinephrine. Acta Anaesthesiol. Sin. 1999, 37, 111–114. [Google Scholar] [PubMed]
- Gulati, A.; Bhalla, S.; Matwyshyn, G.; Zhang, Z.; Andurkar, S.V. Determination of adrenergic and imidazoline receptor involvement in augmentation of morphine and oxycodone analgesia by clonidine and BMS182874. Pharmacology 2009, 83, 45–58. [Google Scholar] [CrossRef] [PubMed]
- Fairbanks, C.A.; Posthumus, I.J.; Kitto, K.F.; Stone, L.S.; Wilcox, G.L. Moxonidine, a selective imidazoline/α2 adrenergic receptor agonist, synergizes with morphine and deltorphin II to inhibit substance P-induced behavior in mice. Pain 2000, 84, 13–20. [Google Scholar] [CrossRef]
- Gupta, S.; Raval, D.; Patel, M.; Patel, N.; Shah, N. Addition of epidural Clonidine enhances postoperative analgesia: A double-blind study in total knee- replacement surgeries. Anesth. Essays Res. 2010, 4, 70–74. [Google Scholar] [CrossRef] [PubMed]
- Engelman, E.; Marsala, C. Efficacy of adding clonidine to intrathecal morphine in acute postoperative pain: Meta-analysis. Br. J. Anaesth. 2013, 110, 21–27. [Google Scholar] [CrossRef] [PubMed]
- Katz, D.; Hamburger, J.; Gutman, D.; Wang, R.; Lin, H.M.; Marotta, M.; Zahn, J.; Beilin, Y. The effect of adding subarachnoid epinephrine to hyperbaric bupivacaine and morphine for repeat cesarean delivery: A double-blind prospective randomized control trial. Anesth. Analg. 2017. [Google Scholar] [CrossRef] [PubMed]
- Ito, S.; Mori, T.; Namiki, M.; Suzuki, T.; Sawaguchi, T. Complicated interaction between psychostimulants and morphine in expression of phenotype of behavior in the dopaminergic system of BALB/c mice. J. Pharmacol. Sci. 2007, 105, 326–333. [Google Scholar] [CrossRef] [PubMed]
- Mori, T.; Ito, S.; Narita, M.; Suzuki, T.; Sawaguchi, T. Combined effects of psychostimulants and morphine on locomotor activity in mice. J. Pharmacol. Sci. 2004, 96, 450–458. [Google Scholar] [CrossRef] [PubMed]
- Trujillo, K.A.; Smith, M.L.; Guaderrama, M.M. Powerful Behavioral Interactions between Methamphetamine and Morphine. Pharmacol. Biochem. Behav. 2011, 99, 451–458. [Google Scholar] [CrossRef] [PubMed]
- Mori, T.; Ito, S.; Kita, T.; Narita, M.; Suzuki, T.; Sawaguchi, T. Effects of mu-, delta- and kappa-opioid receptor agonists on methamphetamine-induced self-injurious behavior in mice. Eur. J. Pharmacol. 2006, 532, 81–87. [Google Scholar] [CrossRef] [PubMed]
- Hubner, C.B.; Bain, G.T.; Kornetsky, C. The combined effects of morphine and d-amphetamine on the threshold for brain stimulation reward. Pharmacol. Biochem. Behav. 1987, 28, 311–315. [Google Scholar] [CrossRef]
- Schaefer, G.J.; Michael, R.P. Interactions of naloxone with morphine, amphetamine and phencyclidine on fixed interval responding for intracranial self-stimulation in rats. Psychopharmacology 1990, 102, 263–268. [Google Scholar] [CrossRef] [PubMed]
- Stone, L.S.; Wilcox, G.L. α-2-Adrenergic and opioid receptor additivity in rat locus coeruleus neurons. Neurosci. Lett. 2004, 361, 265–268. [Google Scholar] [CrossRef] [PubMed]
- Lipman, J.J.; Tolchard, S. Separate and Combined Effects of Morphine and Amphetamine on Quantitative Electroencephalogram and Regional Cerebral Glucose Uptake in a Rat Model. Neuropsychobiology 1990, 23, 89–98. [Google Scholar] [CrossRef] [PubMed]
- Sarto, G. Modulation of the effect of morphine on the isolated guinea pig intestine by noradrenaline and serotonin. Boll. Soc. Ital. Biol. Sper. 1981, 57, 394–400. [Google Scholar] [PubMed]
- Zhang, H.X.; Tang, J.; Li, S.J.; Han, J.S. Inhibitory effect of morphine on guinea pig ileum contraction was modified by four monoamine blockers. Zhongguo Yao Li Xue Bao 1983, 4, 92–95. [Google Scholar] [PubMed]
- Gintzler, A.R.; Musacchio, J.M. Interaction between serotonin and morphine in the guinea-pig ileum. J. Pharmacol. Exp. Ther. 1974, 189, 484–492. [Google Scholar] [PubMed]
- Dillon, P.F.; Root-Bernstein, R.; Robinson, N.E.; Abraham, W.M.; Berney, C. Receptor-mediated enhancement of β adrenergic drug activity by ascorbate in vitro and in vivo. PLoS ONE 2010, 5, e15130. [Google Scholar] [CrossRef] [PubMed]
- Root-Bernstein, R.S. Catecholamines bind to enkephalins, morphiceptin and morphine. Brain Res. Bull. 1987, 18, 509–532. [Google Scholar] [CrossRef]
- Dillon, P.F.; Root-Bernstein, R.S.; Sears, P.R.; Olson, L.K. Natural electrophoresis of norepinephrine and ascorbic acid. Biophys. J. 2000, 79, 370–376. [Google Scholar] [CrossRef]
- Dillon, P.F.; Root-Bernstein, R.S.; Lieder, C.M. Antioxidant-independent ascorbate enhancement of catecholamine-induced contractions of vascular smooth muscle. Am. J. Physiol. Heart Circ. Physiol. 2004, 286, H2353–H2360. [Google Scholar] [CrossRef] [PubMed]
- Surath, S.; Banerjee, K.; Rupley, J.A. Binding of oligosaccharides to lysozyme temperature and pH dependence of the interaction. J. Biol. Chem. 1973, 248, 2117–2124. [Google Scholar]
- Matsumoto, I.; Jinbo, A.; Kitigaki, H.; Golovtchenko-Matsumoto, B.; Seno, N. Detection of lectin-sugar interaction by ultraviolet difference spectroscopy. J. Biochem. 1980, 88, 1093–1096. [Google Scholar] [CrossRef] [PubMed]
- Harris, W.R.; Pecoraro, V.L. Thermodynamic binding constants for gallium transferring. Biochemistry 1983, 22, 292–299. [Google Scholar] [CrossRef] [PubMed]
- Khaled, M.A.; Mullins, D.W.; Swindle, M.; Lacey, J.C., Jr. Complexes of polyadenylic acid and the methyl esters of amino acids. Orig. Life 1983, 13, 87–96. [Google Scholar] [CrossRef] [PubMed]
- Root-Bernstein, R.S.; Dobbelstein, C. Insulin binds to glucagon forming a complex that is hyper-antigenic and inducing complementary antibodies having an idiotype-antiidiotype relationship. Autoimmunity 2001, 33, 153–169. [Google Scholar] [CrossRef] [PubMed]
- Root-Bernstein, R.; Podufaly, A.; Dillon, P.F. Estradiol binds to insulin and insulin receptor decreasing insulin binding in vitro. Front. Endocrinol. 2014, 5, 118. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Kubicek, J.; Labahn, J. Expression and purification of functional human mu opioid receptor from E.coli. PLoS ONE 2013, 8, e56500. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Magnusdottir, A.; Johansson, I.; Dahlgren, L.G.; Nordlund, P.; Berglund, H. Enabling IMAC purification of low abundance recombinant proteins from E. coli lysates. Nat. Methods 2009, 6, 477–478. [Google Scholar] [CrossRef] [PubMed]
- Wolf, S.; Grünewald, S. Sequence, Structure and Ligand Binding Evolution of Rhodopsin-Like G Protein-Coupled Receptors: A Crystal Structure-Based Phylogenetic Analysis. PLoS ONE 2015, 10, e0123533. [Google Scholar] [CrossRef] [PubMed]
- Onogi, T.; Minami, M.; Katao, Y.; Nakagawa, T.; Aoki, Y.; Toya, T.; Katsumata, S.; Satoh, M. DAMGO, a mu-opioid receptor selective agonist, distinguishes between mu- and delta-opioid receptors around their first extracellular loops. FEBS Lett. 1995, 357, 93–97. [Google Scholar] [CrossRef]
- Monroe, P.J.; Perschke, S.E.; Crisp, T.; Smith, D.J. Evaluation of the interactions of serotonergic and adrenergic drugs with mu, delta, and kappa opioid binding sites. Neurosci. Lett. 1991, 133, 229–232. [Google Scholar] [CrossRef]
- Manglik, A.; Kruse, A.C.; Kobilka, T.S.; Thian, F.S.; Mathiesen, J.M.; Sunahara, R.K.; Pardo, L.; Weis, W.I.; Kobilka, B.K.; Granier, S. Crystal structure of the µ-opioid receptor bound to a morphinan antagonist. Nature 2012, 485, 321–326. [Google Scholar] [CrossRef] [PubMed]
- Wilkinson, M.; Jacobson, W.; Wilkinson, D.A. The noradrenergic neurotoxins DSP4 and xylamine bind to opiate receptors. Brain Res. Bull. 1985, 14, 493–495. [Google Scholar] [CrossRef]
- Jacobson, W.; Wilkinson, M.; Gibson, C.J. Direct effects of the adrenergic neurotoxin DSP4 on central opiate receptors: Implications for neuroendocrine studies. Exp. Brain Res. 1985, 59, 570–574. [Google Scholar] [CrossRef] [PubMed]
- Ventura, C.; Bastagli, L.; Bernardi, P.; Caldarera, C.M.; Guarnieri, C. Opioid receptors in rat cardiac sarcolemma: Effect of phenylephrine and isoproterenol. Biochim. Biophys. Acta 1989, 987, 69–74. [Google Scholar] [CrossRef]
- Chabot-Doré, A.J.; Millecamps, M.; Stone, L.S. The delta-opioid receptor is sufficient, but not necessary, for spinal opioid-adrenergic analgesic synergy. J. Pharmacol. Exp. Ther. 2013, 347, 773–780. [Google Scholar] [CrossRef] [PubMed]
- Chabot-Doré, A.J.; Schuster, D.J.; Stone, L.S.; Wilcox, G.L. Analgesic synergy between opioid and α2-adrenoceptors. Br. J. Pharmacol. 2015, 172, 388–402. [Google Scholar] [CrossRef] [PubMed]
- Qin, F.; Yan, C.; Patel, R.; Liu, W.; Dong, E. Vitamins C and E attenuate apoptosis, β-adrenergic receptor desensitization, and sarcoplasmic reticular Ca2+ ATPase downregulation after myocardial infarction. Free Radic. Biol. Med. 2006, 40, 1827–1842. [Google Scholar] [CrossRef] [PubMed]
- Mak, S.; Newton, G.E. Vitamin C augments the inotropic response to dobutamine in humans with normal left ventricular function. Circulation 2001, 103, 826–830. [Google Scholar] [CrossRef] [PubMed]
- Shinke, T.; Shite, J.; Takaoka, H.; Hata, K.; Inoue, N.; Yoshikawa, R.; Matsumoto, H.; Masai, H.; Watanabe, S.; Ozawa, T.; et al. Vitamin C restores the contractile response to dobutamine and improves myocardial efficiency in patients with heart failure after anterior myocardial infarction. Am. Heart J. 2007, 154. [Google Scholar] [CrossRef] [PubMed]
- Monahan, K.D.; Eskurza, I.; Seals, D.R. Ascorbic acid increases cardiovagal baroreflex sensitivity in healthy older men. Am. J. Physiol. Heart Circ. Physiol. 2004, 286, H2113–H2117. [Google Scholar] [CrossRef] [PubMed]
- Jablonski, K.L.; Seals, D.R.; Eskurza, I.; Monahan, K.D.; Donato, A.J. High-dose ascorbic acid infusion abolishes chronic vasoconstriction and restores resting leg blood flow in healthy older men. J. Appl. Physiol. 2007, 103, 1715–1721. [Google Scholar] [CrossRef] [PubMed]
- Park, W.K.; Chang, C.H.; Chae, J.E.; Kim, M.H.; Cho, Y.L.; Ahn, D.S. Phosphodiesterase inhibition by naloxone augments the inotropic actions of β-adrenergic stimulation. Acta Anaesthesiol. Scand. 2009, 53, 1043–1051. [Google Scholar] [CrossRef] [PubMed]
- Milne, B.; Sutak, M.; Cahill, C.M.; Jhamandas, K. Low doses of α 2-adrenoceptor antagonists augment spinal morphine analgesia and inhibit development of acute and chronic tolerance. Br. J. Pharmacol. 2008, 155, 1264–1278. [Google Scholar] [CrossRef] [PubMed]
- Satarian, L.; Javan, M.; Fathollahi, Y. Epinephrine inhibits analgesic tolerance to intrathecal administrated morphine and increases the expression of calcium-calmodulin-dependent protein kinase IIα. Neurosci. Lett. 2008, 430, 213–217. [Google Scholar] [CrossRef] [PubMed]
- Heimans, R.L. Catecholamines and the actions of morphine on the guinea-pig ileum. Arch. Int. Pharmacodyn. Ther. 1975, 216, 11–18. [Google Scholar]
- Ferri, S.; Reina, R.; Santagostino, A. Dopamine and the depressant action of morphine on stimulated guinea-pig ileum. Br. J. Pharmacol. 1977, 59, 25–28. [Google Scholar] [CrossRef] [PubMed]
- Goldstein, A.; Schulz, R. Morphine-tolerant longitudinal muscle strip from guinea-pig ileum. Br. J. Pharmacol. 1973, 48, 655–666. [Google Scholar] [CrossRef] [PubMed]
- Garzón, J.; Sánchez-Blázquez, P. Administration of myr+-Gi2α subunits prevents acute tolerance (tachyphylaxis) to mu-opioid effects in mice. Neuropharmacology 2001, 40, 560–569. [Google Scholar] [CrossRef]
- Wadman, M. “Biased” opioids could yield safer pain relief. Science 2017, 358, 847–848. [Google Scholar] [CrossRef] [PubMed]
- Root-Bernstein, R.S.; Dillon, P.F.; Hollingsworth, R. A tethered ascorbate-norepinephrine compound, 4-UT, displays long-acting adrenergic activity on rabbit aortic smooth muscle. Drug Res. Dev. 2008, 69, 242–250. [Google Scholar] [CrossRef]
- Berque-Bestel, I.; Lezoualc’h, F.; Jockers, R. Bivalent ligands as specific pharmacological tools for G protein-coupled receptor dimers. Curr. Drug Discov. Technol. 2008, 5, 312–318. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Miller, W. A time-efficient, linear-space local similarity algorithm. Adv. Appl. Math. 1991, 12, 337–357. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| MuOPR 38–51 | DGNLSDPCGPNRTD |
| MuOPR 111–122 | NLALADALATST |
| MuOPR 121–131 | TLPFQSVNYL |
| MuOPR 132–143 | MGTWPFGTILCK |
| MuOPR 211–226 | KYRQGSIDCTLTFSHP |
| B2AR 93–100 | HILMKMWT |
| B2AR 97–106 | KMWTFGN |
| B2AR 103–113 | NFWCEFTSID |
| B2AR 175–188 | RATHQEAINCYANE |
| D1DR 89–100 | FWPFGSFCNWV |
| D1DR 175–188 | ATSLAETINCDS |
| Hist Rec 77–87 | GAVVMPMNILYL |
| Hist Rec 89–98 | LMSKWSLGRP |
| Hist Rec 96–107 | RPLCLFWLSMD |
| Hist Rec 105–115 | SMDYVASTASI |
| Hist Rec 166–172 | NHFMQQT |
| Hist Rec 177–183 | RDKCETD |
| Insulin Rec 105–113 | NLTVIRGSR |
| Insulin Rec 157–166 | TIDWSRILDS |
| Kd (µM) @ 200 nm | Morph | Nalox | MENK | Epi | NorEpi | Amph | DOP | L-DOPA | Salb | Prop |
| Mu 38–51 | 35 | 0.5/35 | 0.15/55 | 1.2/35 | 1.4/45 | 1.25/90 | 60 | 50 | 33 | 250 |
| Mu 111–122 | 50 | 0.5/38 | 0.33/80 | 1.25/40 | 1.3/40 | 1.3/100 | 65 | 60 | 25 | 300 |
| Mu 121–131 | 900 | >1000 | 3.5/90 | >1000 | >1000 | >1000 | >1000 | >1000 | >1000 | >1000 |
| Mu 132–143 | 35 | 0.5/42 | 0.4 /70 | 1.4/35 | 1.35/40 | 1.1/85 | 60 | 50 | 35 | 150 |
| Mu 211–226 | 30 | 1.0/45 | 1.0/65 | 1.2/40 | 1.3/45 | 1.2/90 | 65 | 60 | 33 | 250 |
| B2AR 93–100 | 8 | 5 | ||||||||
| B2AR 97–106 | 1 | 6 | ||||||||
| B2AR 103–113 | 27 | 3 | ||||||||
| B2AR 175–188 | 50 | 40 | ||||||||
| D1DR 89–100 | 20 | 5 | ||||||||
| D1DR 175–188 | 10 | 20 | ||||||||
| HIST 77–87 | 70 | 30 | ||||||||
| HIST 89–98 | 5 | 4 | ||||||||
| HIST 96–107 | 15 | 10 | ||||||||
| HIST 105–115 | 50 | 40 | ||||||||
| HIST 166–172 | 0.6/30 | 130 | ||||||||
| HIST 177–183 | 30 | 150 | ||||||||
| INSR 105–113 | 10 | 20 | ||||||||
| INSR 157–166 | 60 | 200 | ||||||||
| Kd (µM) @ 200 nm | Yoh | Phen | 5HT | Mel | ACh | Hist | EDTA | ASC | RIBO | GLUC |
| Mu 38–51 | 300 | >1000 | 100 | 100 | >1000 | >1000 | 900 | >1000 | 50 | >1000 |
| Mu 111–122 | 400 | >1000 | 100 | 150 | >1000 | >1000 | 950 | >1000 | >1000 | >1000 |
| Mu 121–131 | >1000 | >1000 | 350 | 900 | >1000 | >1000 | 40 | >1000 | >1000 | >1000 |
| Mu 132–143 | 250 | >1000 | 100 | 100 | >1000 | >1000 | 700 | >1000 | 100 | >1000 |
| Mu 211–226 | 3.3/250 | >1000 | 90 | 100 | >1000 | >1000 | 900 | >1000 | >1000 | >1000 |
| B2AR 93–100 | 300 | 60 | 400 | >1000 | ||||||
| B2AR 97–106 | 50 | 60 | 600 | >1000 | ||||||
| B2AR 103–113 | >1000 | 150 | >1000 | |||||||
| B2AR 175–188 | 350 | 300 | 500 | >1000 | ||||||
| D1DR 89–100 | 900 | 300 | 600 | >1000 | ||||||
| D1DR 175–188 | 50 | 300 | 300 | >1000 | ||||||
| HIST 77–87 | 60 | 300 | >1000 | >1000 | ||||||
| HIST 89–98 | 20 | 30 | 400 | >1000 | ||||||
| HIST 96–107 | 90 | 40 | 500 | >1000 | ||||||
| HIST 105–115 | 60 | 300 | 450 | >1000 | ||||||
| HIST 166–172 | 250 | 350 | >1000 | |||||||
| HIST 177–183 | 20 | 40 | >1000 | |||||||
| INSR 105–113 | >1000 | >1000 | 500 | >1000 | ||||||
| INSR 157–166 | >1000 | >1000 | >1000 |
| Compound | Kd @ 200 nm (μM) | Kd @ 210 nm (μM) |
|---|---|---|
| Acetylcholine | >1000 | >1000 |
| Histamine | >1000 | >1000 |
| Ascorbic Acid (Vitamin C) | >1000 | >1000 |
| Epinephrine | 30 | 20 |
| Met-Enkephalin | 15/0.8 | 12 |
| Met-Enkephalin + Epinephrine | 4/<0.01? | 5 |
| Morphine | 60/0.9 | 20 |
| Morphine + Epinephrine | 9/0.9/<0.01? | 6 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Root-Bernstein, R.; Turke, M.; Subhramanyam, U.K.T.; Churchill, B.; Labahn, J. Adrenergic Agonists Bind to Adrenergic-Receptor-Like Regions of the Mu Opioid Receptor, Enhancing Morphine and Methionine-Enkephalin Binding: A New Approach to “Biased Opioids”? Int. J. Mol. Sci. 2018, 19, 272. https://doi.org/10.3390/ijms19010272
Root-Bernstein R, Turke M, Subhramanyam UKT, Churchill B, Labahn J. Adrenergic Agonists Bind to Adrenergic-Receptor-Like Regions of the Mu Opioid Receptor, Enhancing Morphine and Methionine-Enkephalin Binding: A New Approach to “Biased Opioids”? International Journal of Molecular Sciences. 2018; 19(1):272. https://doi.org/10.3390/ijms19010272
Chicago/Turabian StyleRoot-Bernstein, Robert, Miah Turke, Udaya K. Tiruttani Subhramanyam, Beth Churchill, and Joerg Labahn. 2018. "Adrenergic Agonists Bind to Adrenergic-Receptor-Like Regions of the Mu Opioid Receptor, Enhancing Morphine and Methionine-Enkephalin Binding: A New Approach to “Biased Opioids”?" International Journal of Molecular Sciences 19, no. 1: 272. https://doi.org/10.3390/ijms19010272
APA StyleRoot-Bernstein, R., Turke, M., Subhramanyam, U. K. T., Churchill, B., & Labahn, J. (2018). Adrenergic Agonists Bind to Adrenergic-Receptor-Like Regions of the Mu Opioid Receptor, Enhancing Morphine and Methionine-Enkephalin Binding: A New Approach to “Biased Opioids”? International Journal of Molecular Sciences, 19(1), 272. https://doi.org/10.3390/ijms19010272