Different Lipid Regulation in Ovarian Cancer: Inhibition of the Immune System

 , , and
, , and
Abstract
:1. Introduction
2. Results
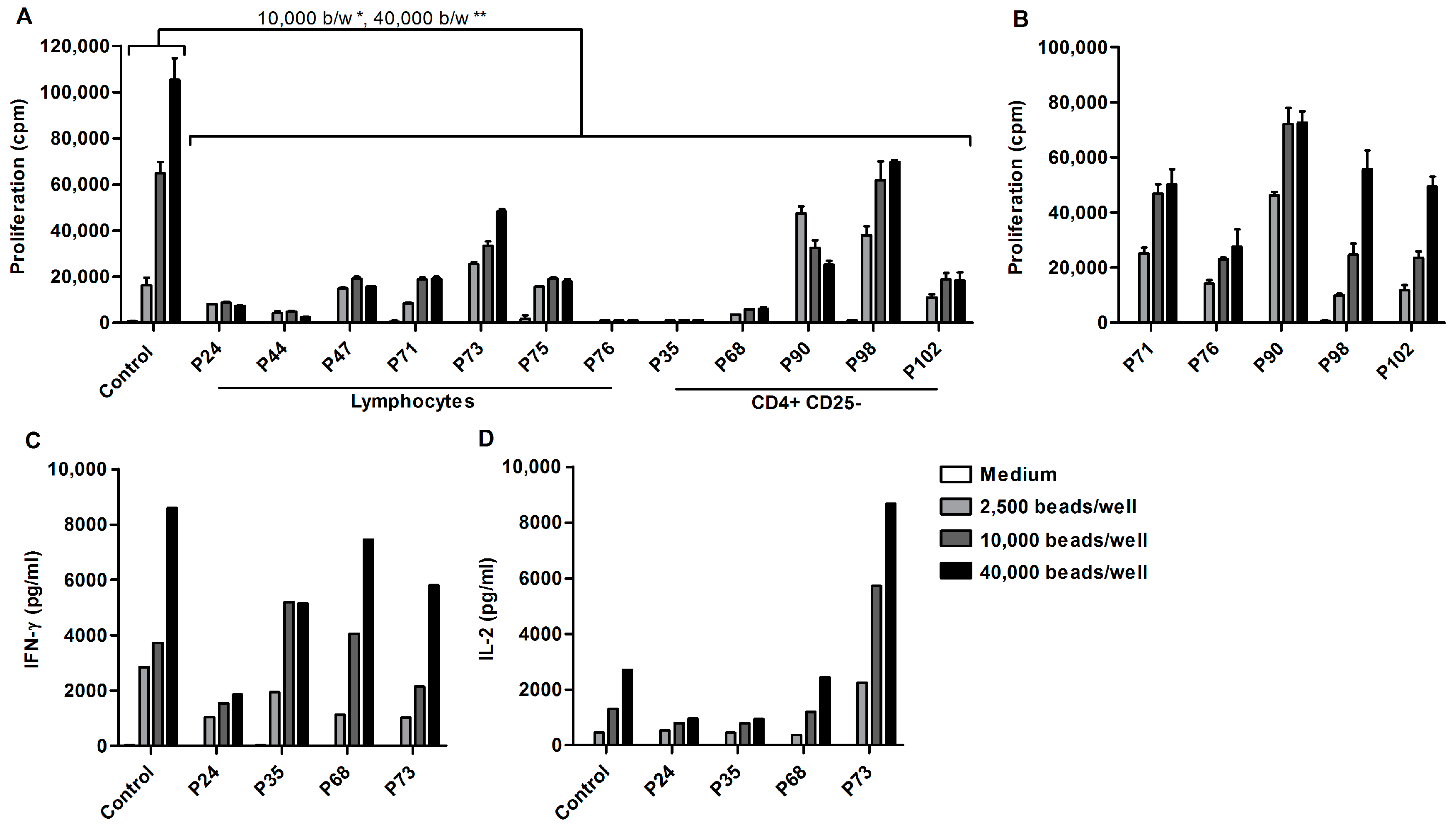
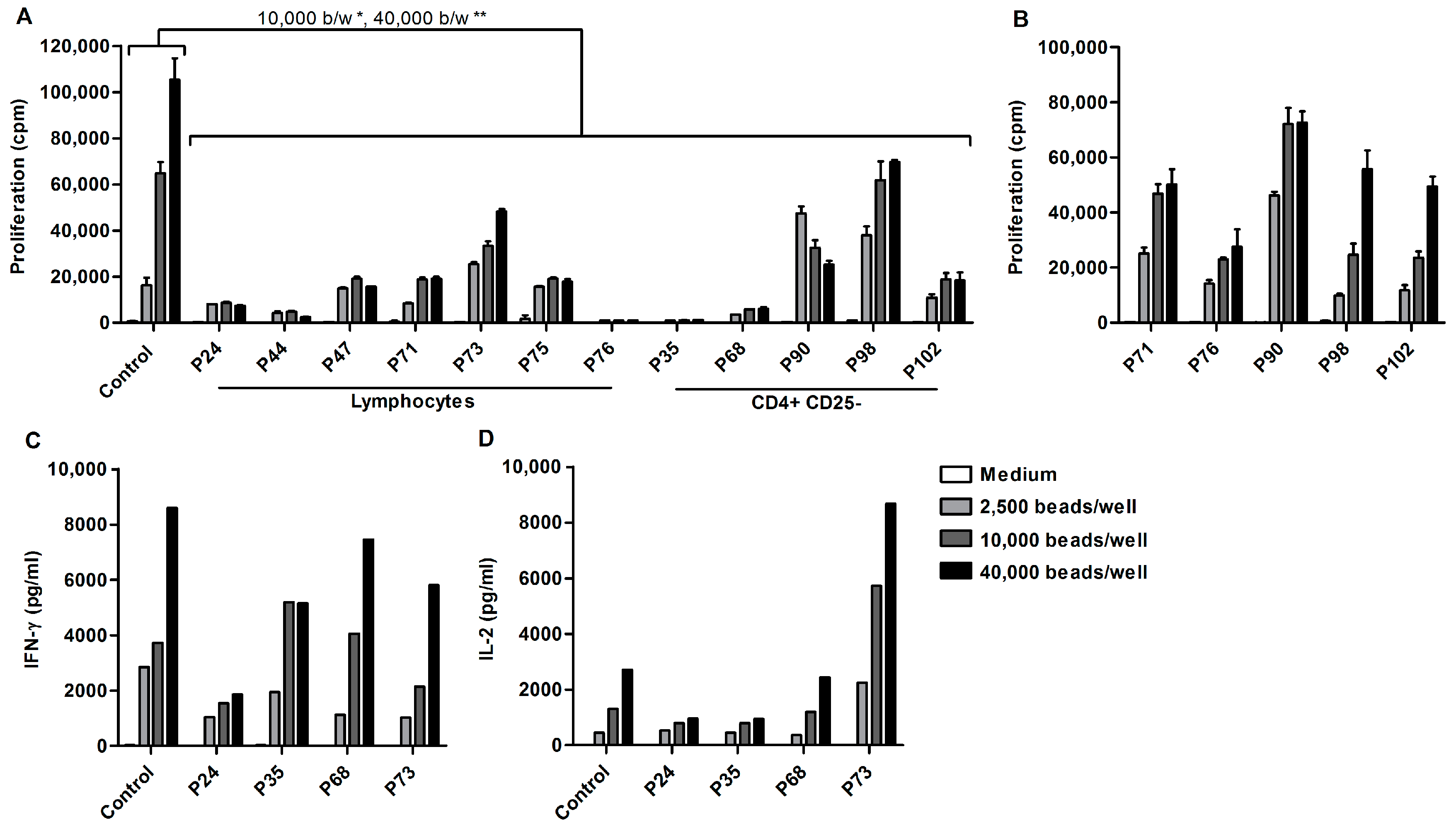
2.1. Ascites-Derived T Cells Proliferate Poorly upon αCD3/28 Stimulation
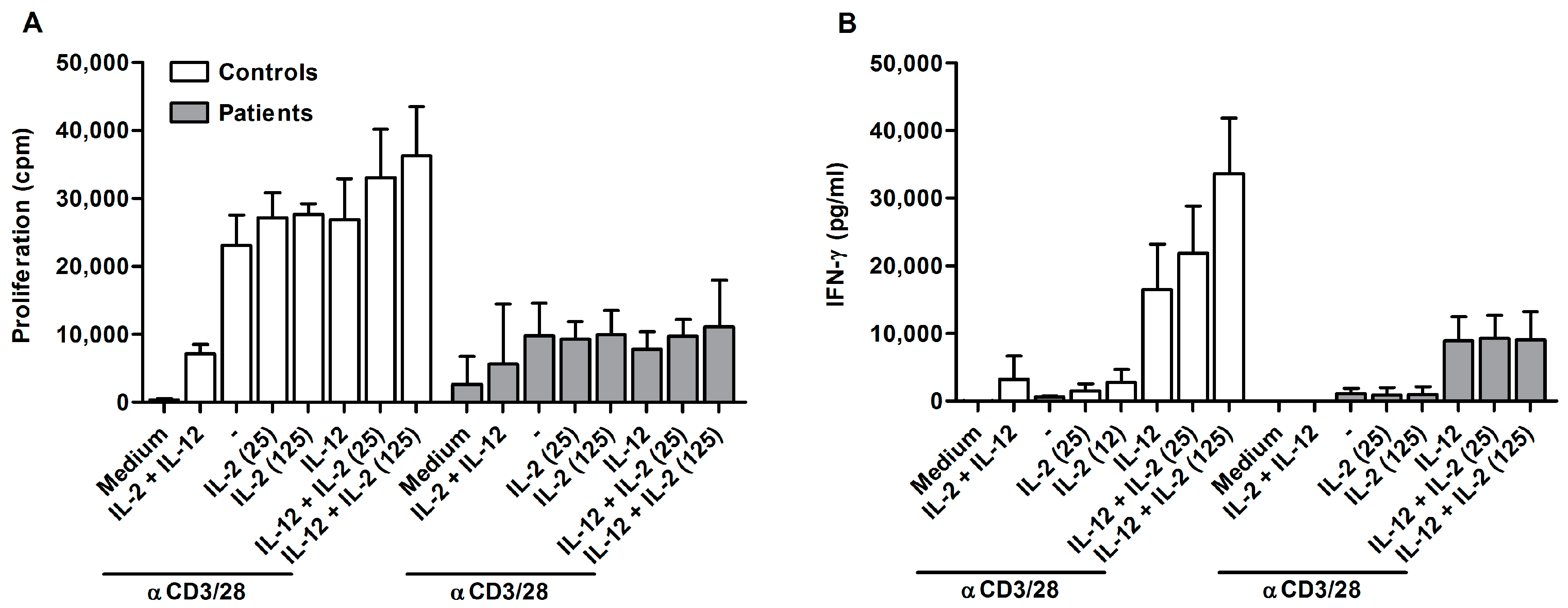
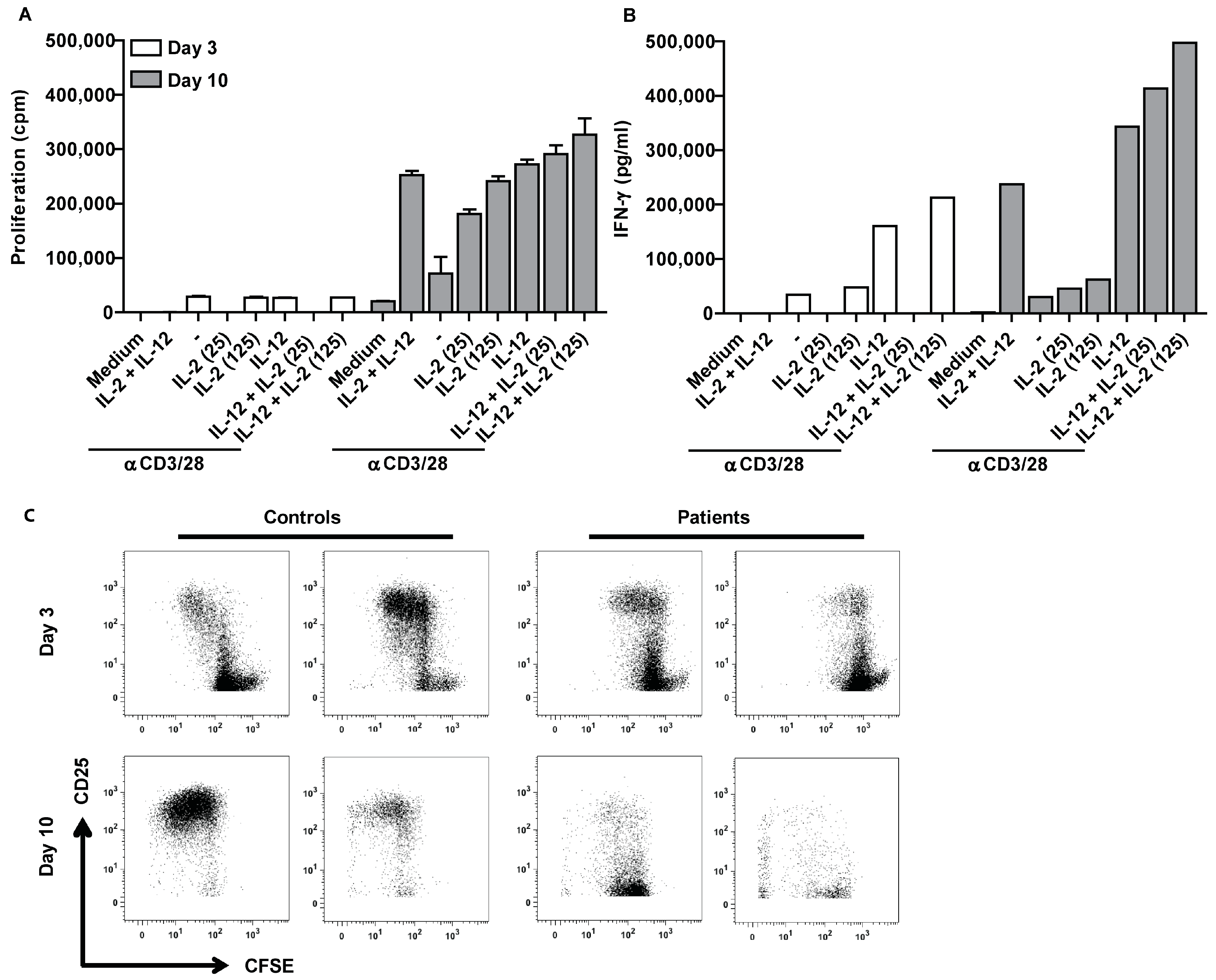
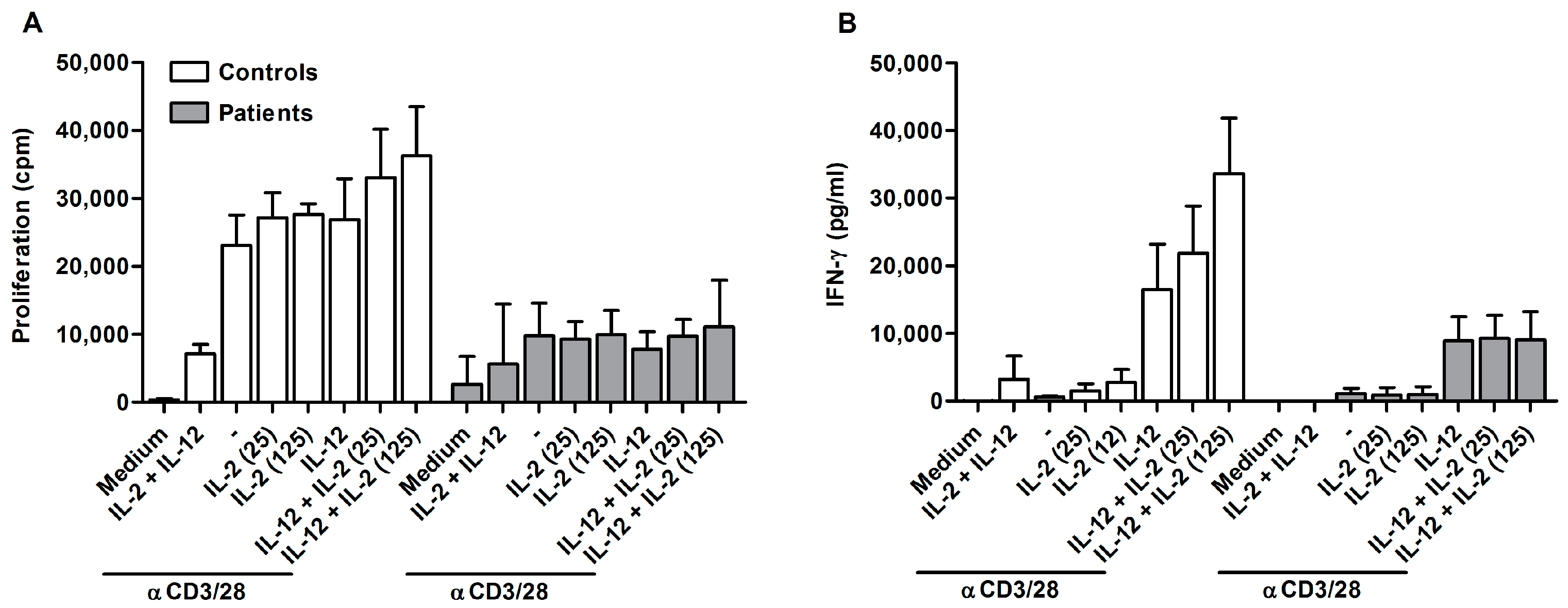
2.2. Ascites-Derived CD4+CD25- T Cells Are Unresponsive to IL-2
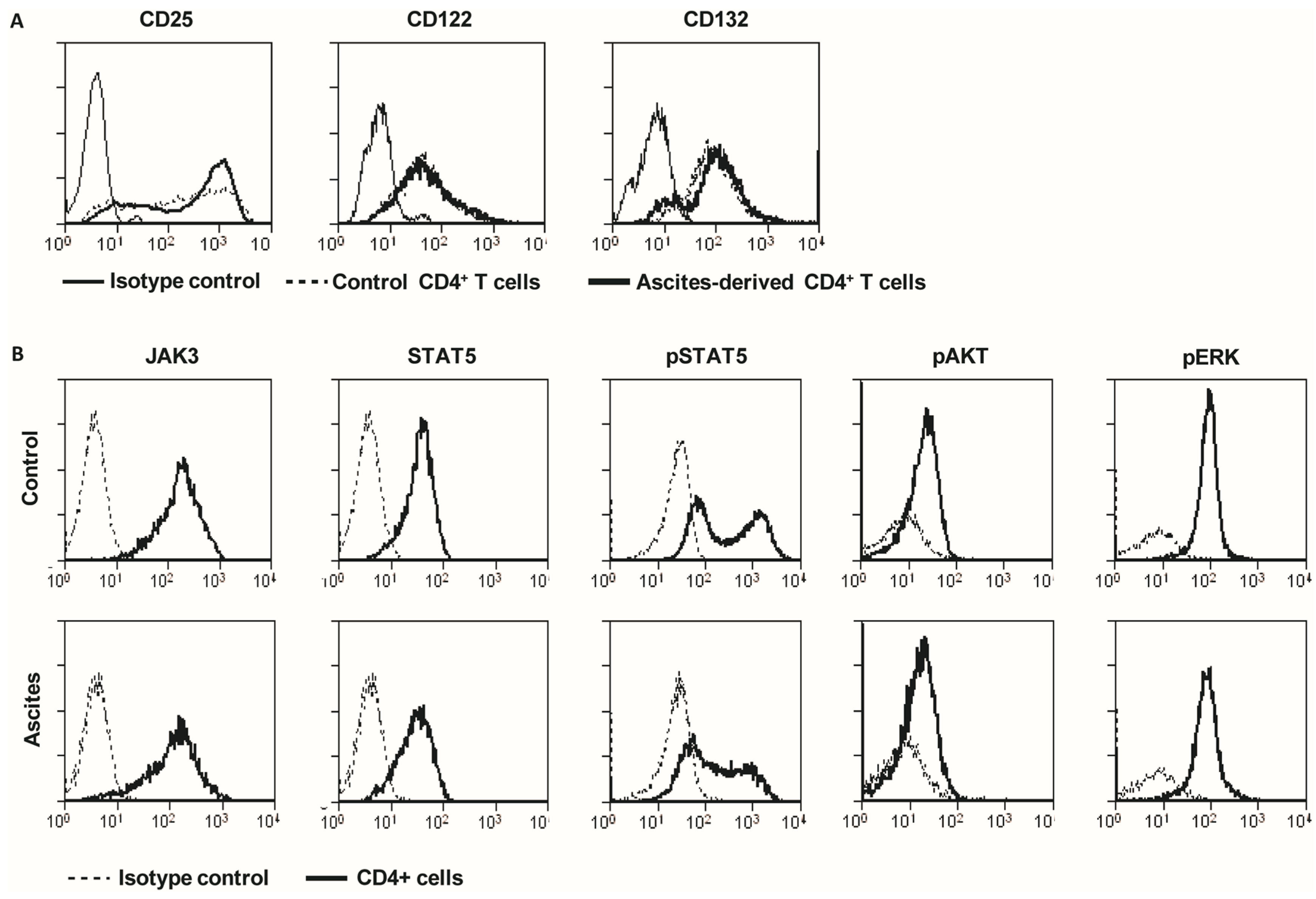
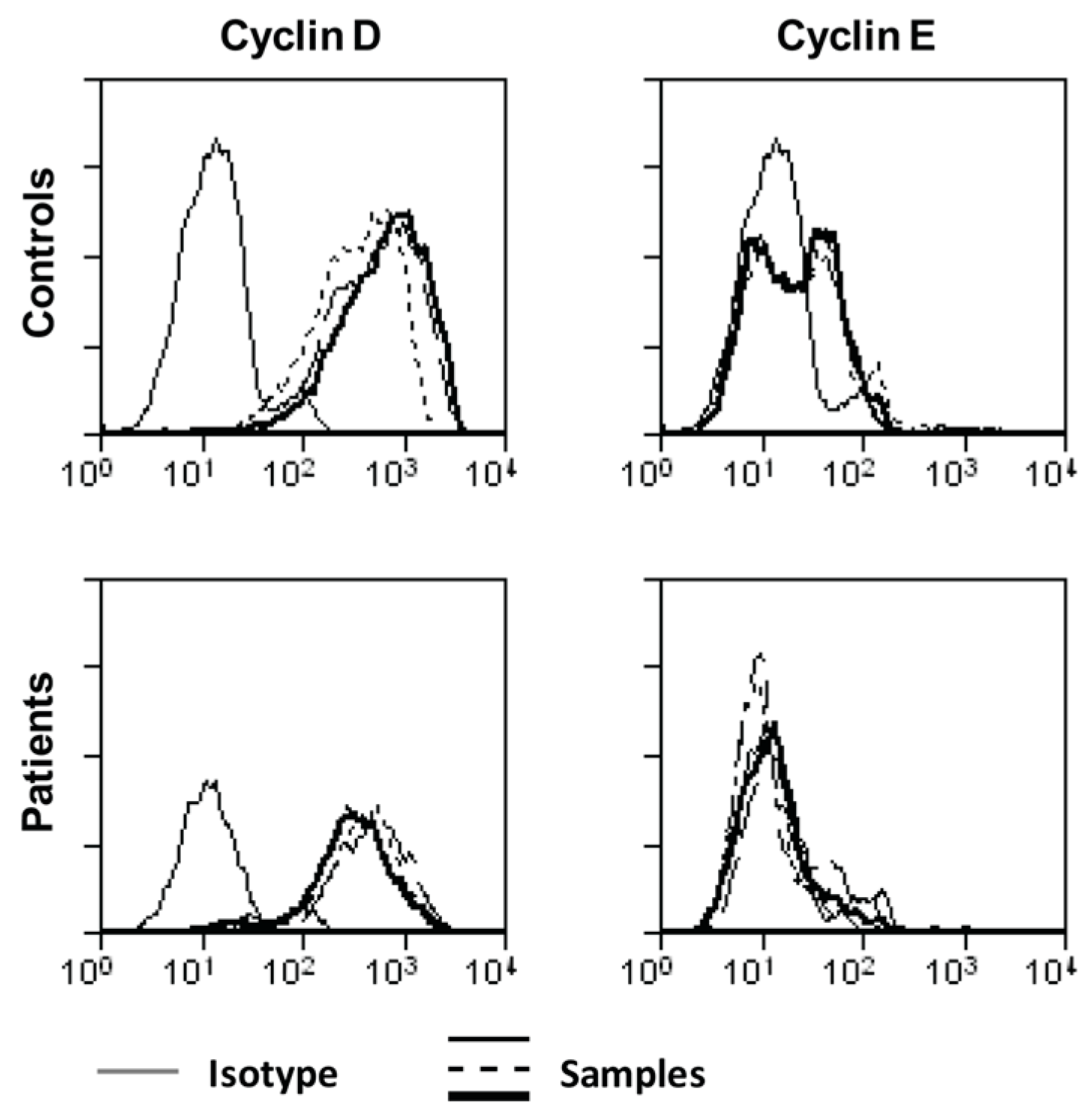
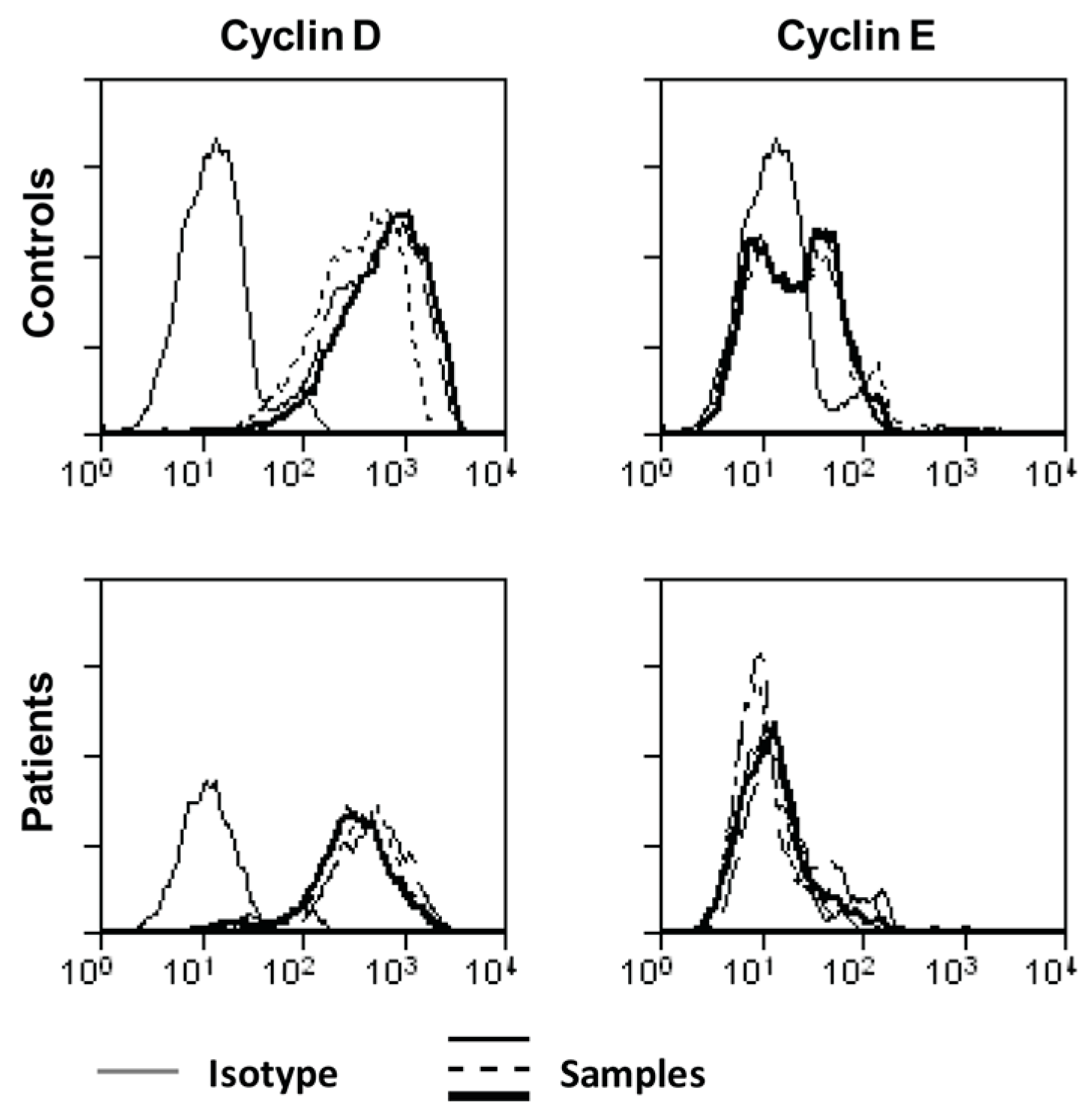
2.3. Ascites-Derived CD4+CD25- T Cells Have a Normal Expression of IL-2R and Its Downstream Signaling Components
2.4. Acellular Fraction of Ascites Inhibits Proliferation of Control PBLs
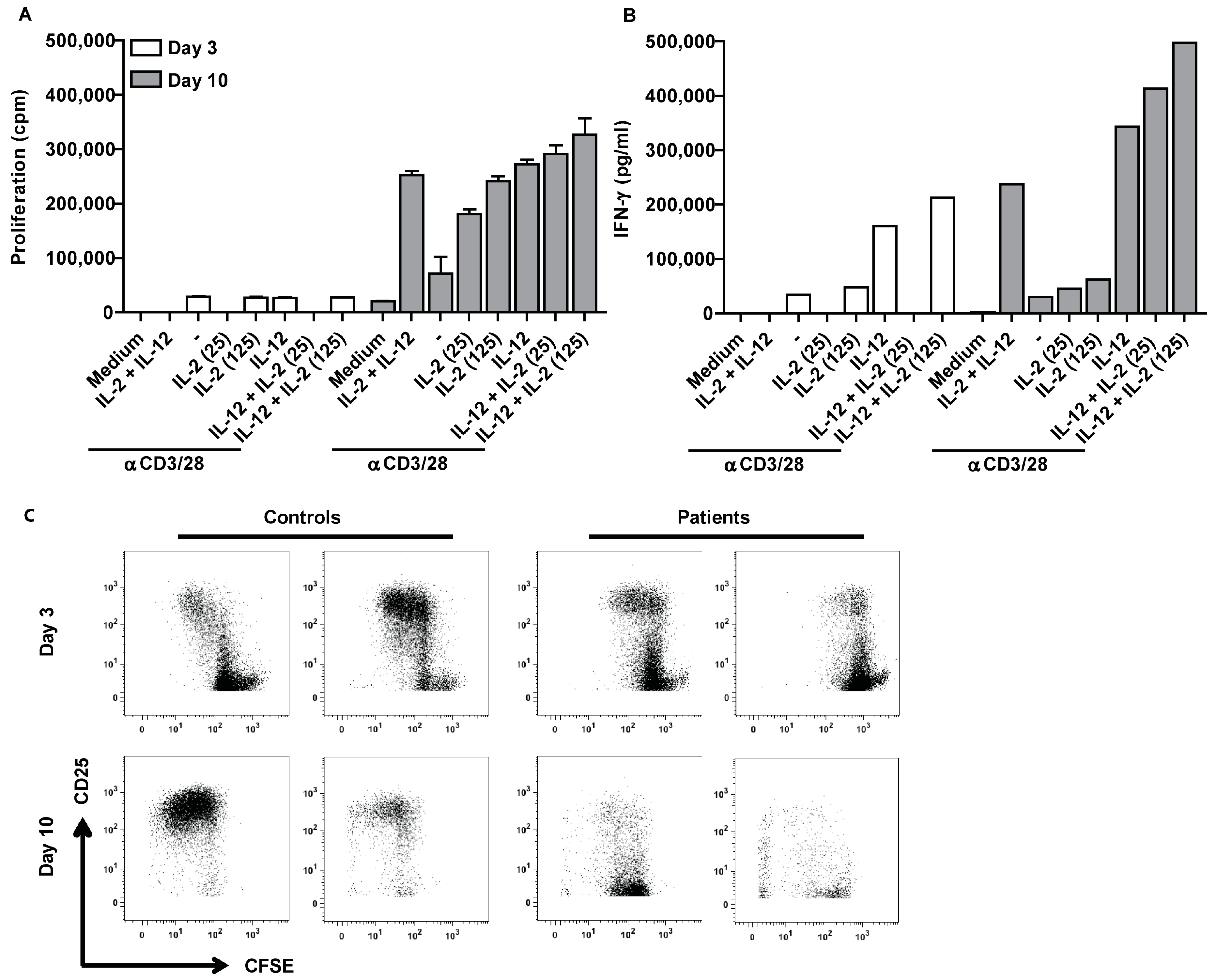
2.5. Suppression of Ascites-Derived T Cells Is Reversible
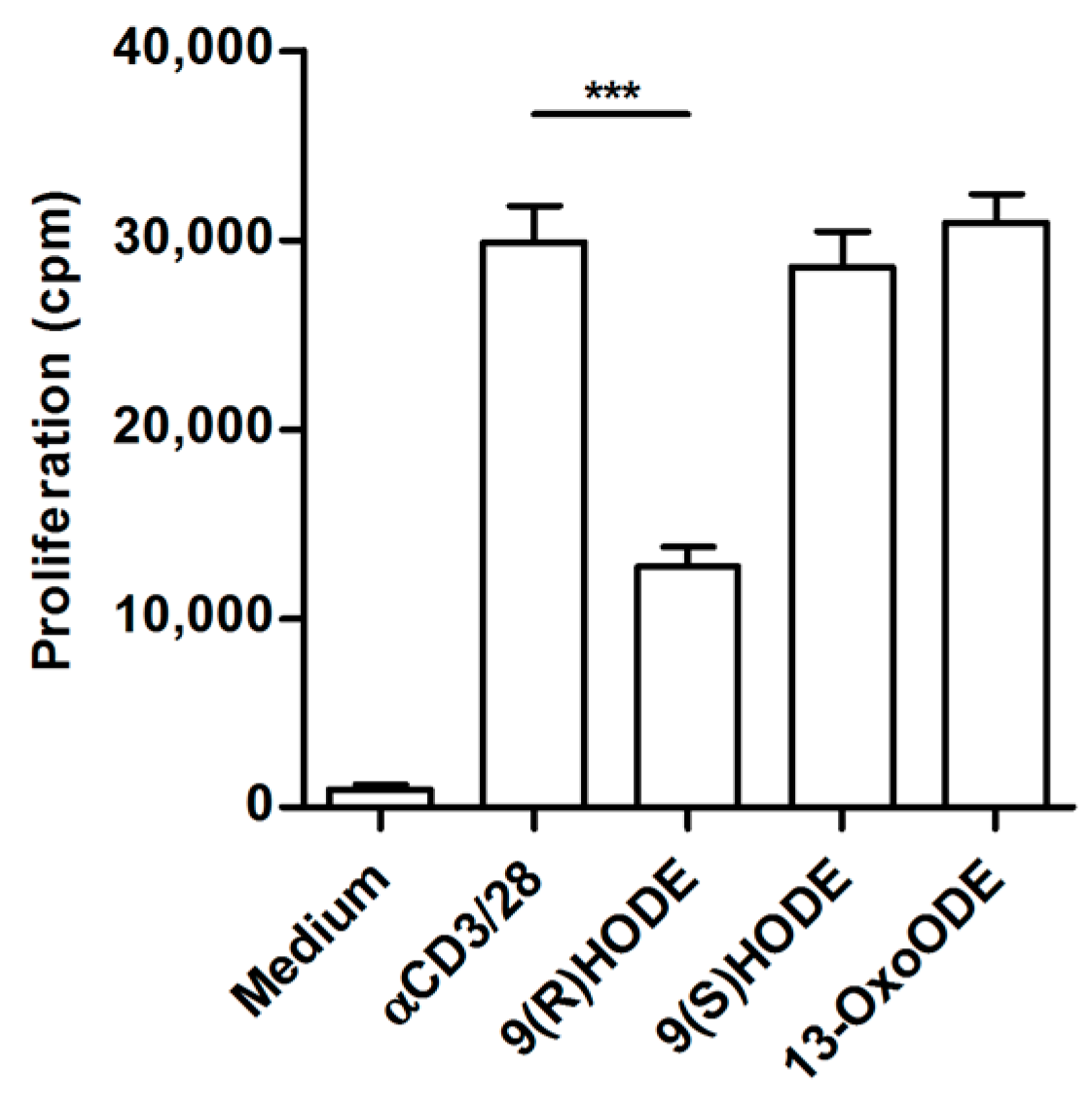
2.6. Altered Lipid Metabolites in Suppressive Ascites
3. Discussion
4. Materials and Methods
4.1. Human Subjects
4.2. Antibodies
4.3. Cell Isolation and Activation
4.4. Cytokine Measurement
4.5. Flow Cytometric Analysis
4.6. Lipid Mediator Analysis of Abdominal Fluid
4.7. Statistics
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| TRegs | Regulatory T cells |
| TIL | Tumor infiltrating lymphocytes |
| PBLs | Peripheral blood lymphocytes |
| PBMCs | Peripheral blood mononuclear cells |
| MNCs | Mononuclear cells |
| LOX | Lipooxygenase |
| Cpm | Counts per minute |
| IFN | Interferon |
| TGF | Tumor growth factor |
| TNF | Tumor necrosis factor |
| IL | Interleukin |
| IL-2R | Interleukin 2 receptor |
| JAK3 | Janus kinase 3 |
| STAT5 | Signal transducer and activator of transcription 5 |
| MAPK | Mitogen-activated kinase |
| FACS | Fluorescent activated cell sorting |
| PPAR | Peroxisome proliferator-activated receptor |
| NFAT | Nuclear factor of activated T cells |
References
- Ferlay, J.; Steliarova-Foucherm, E.; Lortet-Tieulent, J.; Rosso, J.; Coebergh, J.W.W.; Comber, H.; Forman, D.; Bray, F. Cancer incidence and mortality patterns in Europe: Estimates for 40 countries in 2012. Eur. J. Cancer 2013, 49, 1374–1403. [Google Scholar] [CrossRef] [PubMed]
- Reid, B.M.; Permuth, J.B.; Sellers, T.A. Epidemiology of ovarian cancer: A review. Cancer Biol. Med. 2017, 14, 9–32. [Google Scholar] [PubMed]
- Kipps, E.; Tan, D.S.; Kaye, S.B. Meeting the challenge of ascites in ovarian cancer: New avenues for therapy and research. Nat. Rev. Cancer 2013, 13, 273–282. [Google Scholar] [CrossRef] [PubMed]
- Hasby, E.A. Weapons Ovarian Epithelial Tumors May Use in Immune Escape: An Immunohistochemical Correlational Study. Pathol. Oncol. Res. 2012, 18, 509–518. [Google Scholar] [CrossRef] [PubMed]
- Preston, C.C.; Goode, E.L.; Hartmann, L.C.; Kalli, K.R.; Knutson, K.L. Immunity and immune suppression in human ovarian cancer. Immunotherapy 2011, 3, 539–556. [Google Scholar] [CrossRef] [PubMed]
- Clarke, B.; Tinker, A.V.; Lee, C.H.; Subramanian, S.; van de Rijn, M.; Turbin, D.; Kalloger, S.; Han, G.; Ceballos, K.; Cadungog, M.G.; et al. Intraepithelial T cells and prognosis in ovarian carcinoma: Novel associations with stage, tumor type, and BRCA1 loss. Mod. Pathol. 2008, 22, 393–402. [Google Scholar] [CrossRef] [PubMed]
- Sato, E.; Olson, S.H.; Ahn, J.; Bundy, B.; Nishikawa, H.; Qian, F.; Jungbluth, A.A.; Frosina, D.; Gnjatic, S.; Ambrosone, C.; et al. Intraepithelial CD8+ tumor-infiltrating lymphocytes and a high CD8+/regulatory T cell ratio are associated with favorable prognosis in ovarian cancer. Proc. Natl. Acad. Sci. USA 2005, 102, 18538–18543. [Google Scholar] [CrossRef] [PubMed]
- Webb, J.R.; Wick, D.A.; Nielsen, J.S.; Tran, E.; Milne, K.; McMurtrie, E.; Nelson, B.H. Profound elevation of CD8+ T cells expressing the intraepithelial lymphocyte marker CD103 (αE/β7 Integrin) in high-grade serous ovarian cancer. Gynecol. Oncol. 2010, 118, 228–236. [Google Scholar] [CrossRef] [PubMed]
- Curiel, T.J.; Coukos, G.; Zou, L.; Alvarez, X.; Cheng, P.; Mottram, P.; Evdemon-Hogan, M.; Conejo-Garcia, J.R.; Zhang, L.; Burow, M.; et al. Specific recruitment of regulatory T cells in ovarian carcinoma fosters immune privilege and predicts reduced survival. Nat. Med. 2004, 10, 942–949. [Google Scholar] [CrossRef] [PubMed]
- Yigit, R.; Massuger, L.F.; Figdor, C.G.; Torensma, R. Ovarian cancer creates a suppressive microenvironment to escape immune elimination. Gynecol. Oncol. 2010, 117, 366–372. [Google Scholar] [CrossRef] [PubMed]
- Tran, E.; Nielsen, J.S.; Wick, D.A.; Ng, A.V.; Johnson, L.D.; Nesslinger, N.J.; McMurtrie, E.; Webb, J.R.; Nelson, B.H. Polyfunctional T-cell responses are disrupted by the ovarian cancer ascites environment and only partially restored by clinically relevant cytokines. PLoS ONE 2010, 5, e15625. [Google Scholar] [CrossRef] [PubMed]
- Jayson, G.C.; Kohn, E.C.; Kitchener, H.C.; Ledermann, J.A. Ovarian cancer. Lancet 2014, 384, 1376–1388. [Google Scholar] [CrossRef]
- Bast, R.C., Jr.; Hennessy, B.; Mills, G.B. The biology of ovarian cancer: New opportunities for translation. Nat. Rev. Cancer 2009, 9, 415–428. [Google Scholar] [CrossRef] [PubMed]
- Berek, J.; Taylor, P.; McGuire, W.; Smith, L.M.; Schultes, B.; Nicodemus, C.F. Oregovomab maintenance monoimmunotherapy does not improve outcomes in advanced ovarian cancer. J. Clin. Oncol. 2009, 27, 418–425. [Google Scholar] [CrossRef] [PubMed]
- Salani, R.; Backes, F.J.; Fung Kee Fung, M.; Holschneider, C.H.; Parker, L.P.; Bristow, R.E.; Goff, B.A. Posttreatment surveillance and diagnosis of recurrence in women with gynecologic malignancies: Society of Gynecologic Oncologists recommendations. Am. J. Obstet. Gynecol. 2011, 204, 466–478. [Google Scholar] [CrossRef] [PubMed]
- Buckley, C.D.; Gilroy, D.W.; Serhan, C.N. Proresolving lipid mediators and mechanisms in the resolution of acute inflammation. Immunity 2014, 40, 315–327. [Google Scholar] [CrossRef] [PubMed]
- Fullerton, J.N.; O’Brien, A.J.; Gilroy, D.W. Lipid mediators in immune dysfunction after severe inflammation. Trends Immunol. 2014, 35, 12–21. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, T. Lipid Mediators in Health and Disease: Enzymes and Receptors as Therapeutic Targets for the Regulation of Immunity and Inflammation. Annu. Rev. Pharmacol. Toxicol. 2009, 49, 123–150. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Kurupati, R.; Liu, L.; Zhou, X.Y.; Zhang, G.; Hudaihed, A.; Filisio, F.; Giles-Davis, W.; Xu, X.; Karakousis, G.C.; et al. Enhancing CD8+ T Cell Fatty Acid Catabolism within a Metabolically Challenging Tumor Microenvironment Increases the Efficacy of Melanoma Immunotherapy. Cancer Cell 2017, 32, 377–391. [Google Scholar] [CrossRef] [PubMed]
- Suraneni, M.V.; Moore, J.R.; Zhang, D.; Badeaux, M.; Macaluso, M.D.; DiGiovanni, J.; Kusewitt, D.; Tang, D.G. Tumor-suppressive functions of 15-Lipoxygenase-2 and RB1CC1 in prostate cancer. Cell Cycle 2014, 13, 1798–1810. [Google Scholar] [CrossRef] [PubMed]
- Gollob, J.A.; Schnipper, C.P.; Murphy, E.A.; Ritz, J.; Frank, D.A. The functional synergy between IL-12 and IL-2 involves p38 mitogen-activated protein kinase and is associated with the augmentation of STAT serine phosphorylation. J. Immunol. 1999, 162, 4472–4481. [Google Scholar] [PubMed]
- De Jong, R.; Janson, A.A.; Faber, W.R.; Naafs, B.; Ottenhoff, T.H. IL-2 and IL-12 act in synergy to overcome antigen-specific T cell unresponsiveness in mycobacterial disease. J. Immunol. 1997, 159, 786–793. [Google Scholar] [PubMed]
- Boyman, O.; Sprent, J. The role of interleukin-2 during homeostasis and activation of the immune system. Nat. Rev. Immunol. 2012, 12, 180–190. [Google Scholar] [CrossRef] [PubMed]
- Kolenko, V.; Wang, Q.; Riedy, M.C.; O’Shea, J.; Ritz, J.; Cathcart, M.K.; Rayman, P.; Tubbs, R.; Edinger, M.; Novick, A.; et al. Tumor-induced suppression of T lymphocyte proliferation coincides with inhibition of Jak3 expression and IL-2 receptor signaling: Role of soluble products from human renal cell carcinomas. J. Immunol. 1997, 159, 3057–3067. [Google Scholar] [PubMed]
- Edwards, I.J.; O’Flaherty, J.T. Omega-3 Fatty Acids and PPARgamma in Cancer. PPAR Res. 2008, 2008, 358052. [Google Scholar] [CrossRef] [PubMed]
- Lengyel, E. Ovarian cancer development and metastasis. Am. J. Pathol. 2010, 177, 1053–1064. [Google Scholar] [CrossRef] [PubMed]
- Tan, D.S.P.; Agarwal, R.; Kaye, S.B. Mechanisms of transcoelomic metastasis in ovarian cancer. Lancet Oncol. 2006, 7, 925–934. [Google Scholar] [CrossRef]
- Bailis, W.; Shyer, J.A.; Chiorazzi, M.; Flavell, R.A. No Oxygen? No Glucose? No Problem: Fatty Acid Catabolism Enhances Effector CD8+ TILs. Cancer Cell 2017, 32, 280–281. [Google Scholar] [CrossRef] [PubMed]
- Sharfe, N.; Dadi, H.K.; Shahar, M.; Roifman, C.M. Human immune disorder arising from mutation of the α chain of the interleukin-2 receptor. Proc. Natl. Acad. Sci. USA 1997, 94, 3168–3171. [Google Scholar] [CrossRef] [PubMed]
- Mortarini, R.; Vegetti, C.; Molla, A.; Arienti, F.; Ravagnani, F.; Maurichi, A.; Patuzzo, R.; Santinami, M.; Anichini, A. Impaired STAT phosphorylation in T cells from melanoma patients in response to IL-2: Association with clinical stage. Clin. Cancer Res. 2009, 15, 4085–4094. [Google Scholar] [CrossRef] [PubMed]
- Nam, E.J.; Kim, Y.T. Alteration of cell-cycle regulation in epithelial ovarian cancer. Int. J. Gynecol. Cancer 2008, 18, 1169–1182. [Google Scholar] [CrossRef] [PubMed]
- Beloribi-Djefaflia, S.; Vasseur, S.; Guillaumond, F. Lipid metabolic reprogramming in cancer cells. Oncogenesis 2016, 5, e189. [Google Scholar] [CrossRef] [PubMed]
- Feige, J.N.; Gelman, L.; Michalik, L.; Desvergne, B.; Wahli, W. From molecular action to physiological outputs: Peroxisome proliferator-activated receptors are nuclear receptors at the crossroads of key cellular functions. Prog. Lipid Res. 2006, 45, 120–159. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.Y.; Wang, L.H.; Chen, T.; Hodge, D.R.; Resau, J.H.; DaSilva, L.; Farrar, W.L. Activation of human T lymphocytes is inhibited by peroxisome proliferator-activated receptor gamma (PPARγ) agonists. PPARgamma co-association with transcription factor NFAT. J. Biol. Chem. 2000, 275, 4541–4544. [Google Scholar] [CrossRef] [PubMed]
- Siu, K.T.; Rosner, M.R.; Minella, A.C. An integrated view of cyclin E function and regulation. Cell Cycle 2012, 11, 57–64. [Google Scholar] [CrossRef] [PubMed]
- Rowell, E.A.; Walsh, M.C.; Wells, A.D. Opposing Roles for the Cyclin-Dependent Kinase Inhibitor p27kip1 in the Control of CD4+ T Cell Proliferation and Effector Function. J. Immunol. 2005, 174, 3359–3368. [Google Scholar] [CrossRef] [PubMed]
- O’Flaherty, J.T.; Rogers, L.C.; Paumi, C.M.; Hantgan, R.R.; Thomas, L.R.; Clay, C.E.; High, K.; Chen, Y.Q.; Willingham, M.C.; Smitherman, P.K.; et al. 5-Oxo-ETE analogs and the proliferation of cancer cells. Biochim. Biophys. Acta 2005, 1736, 228–236. [Google Scholar] [CrossRef] [PubMed]
- Hampel, J.K.; Brownrigg, L.M.; Vignarajah, D.; Croft, K.D.; Dharmarajan, A.M.; Bentel, J.M.; Puddey, I.B.; Yeap, B.B. Differential modulation of cell cycle, apoptosis and PPARgamma2 gene expression by PPARgamma agonists ciglitazone and 9-hydroxyoctadecadienoic acid in monocytic cells. Prostaglandins Leukot. Essent. Fatty Acids 2006, 74, 283–293. [Google Scholar] [CrossRef] [PubMed]
- Obinata, H.; Hattori, T.; Nakane, S.; Tatei, K.; Izumi, T. Identification of 9-hydroxyoctadecadienoic acid and other oxidized free fatty acids as ligands of the G protein-coupled receptor G2A. J. Biol. Chem. 2005, 280, 40676–40683. [Google Scholar] [CrossRef] [PubMed]
- Weng, Z.; Fluckiger, A.C.; Nisitani, S.; Wahl, M.I.; Le, L.Q.; Hunter, C.A.; Fernal, A.A.; Le Beau, M.M.; Witte, O.N. A DNA damage and stress inducible G protein-coupled receptor blocks cells in G2/M. Proc. Natl. Acad. Sci. USA 1998, 95, 12334–12339. [Google Scholar] [CrossRef] [PubMed]
- Martin, H. Role of PPAR-gamma in inflammation. Prospects for therapeutic intervention by food components. Mutat. Res. 2010, 690, 57–63. [Google Scholar] [CrossRef] [PubMed]
- Gelman, L.; Feige, J.N.; Desvergne, B. Molecular basis of selective PPARgamma modulation for the treatment of Type 2 diabetes. Biochim. Biophys. Acta 2007, 1771, 1094–1107. [Google Scholar] [CrossRef] [PubMed]
- Balgoma, D.; Yang, M.; Sjodin, M.; Snowden, S.; Karimi, R.; Levanen, B.; Merikallio, H.; Kaarteenaho, R.; Palmberg, L.; Larsson, K.; et al. Linoleic acid-derived lipid mediators increase in a female-dominated subphenotype of COPD. Eur. Respir. J. 2016, 47, 1645–1656. [Google Scholar] [CrossRef] [PubMed]
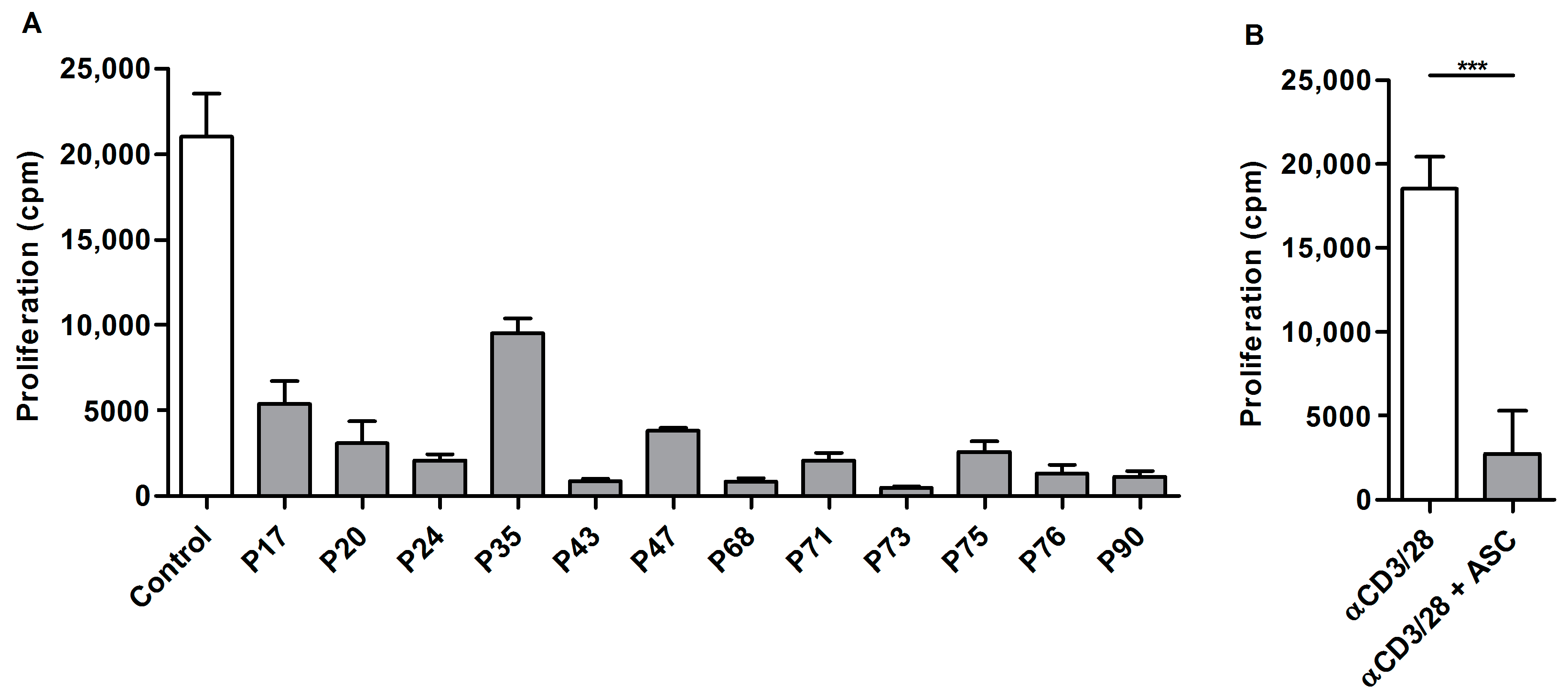

{kind=link}
{kind=link}
{kind=link}
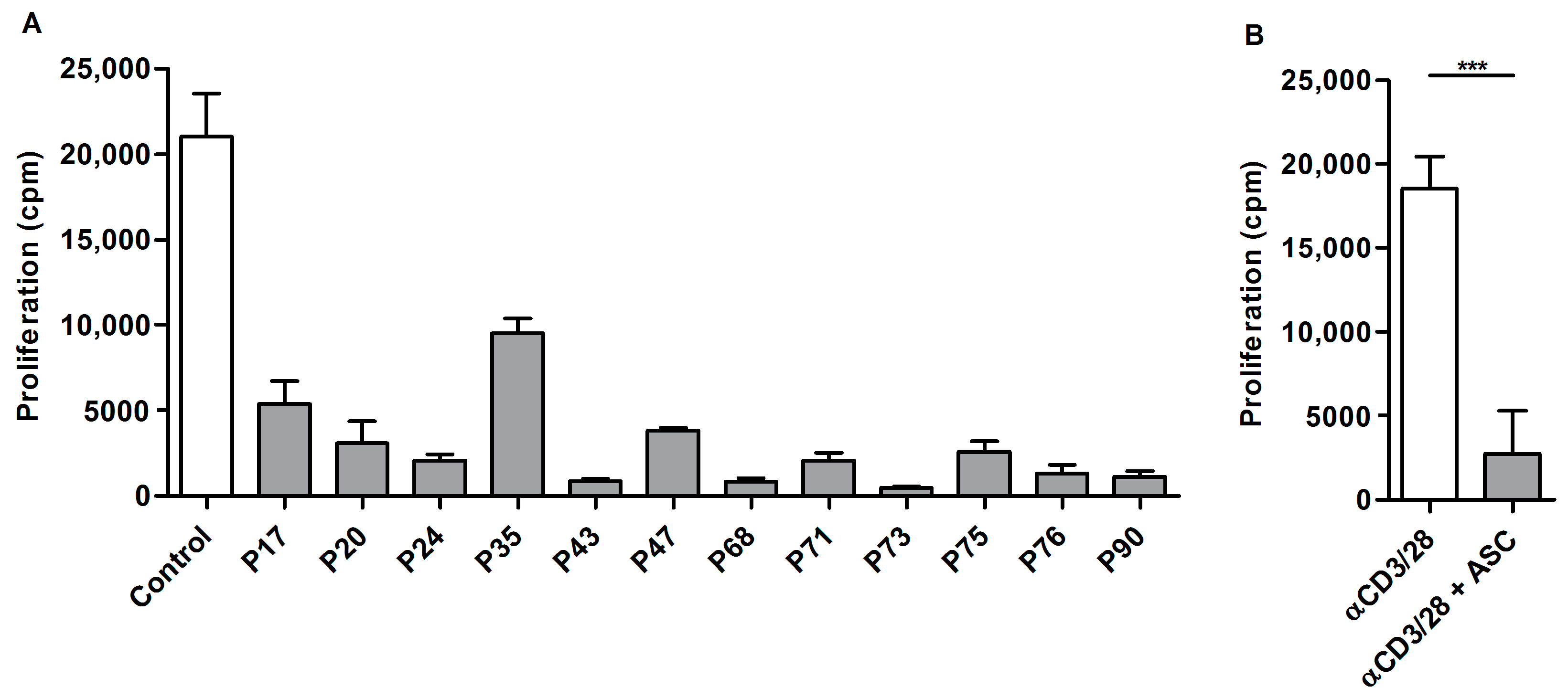{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample (ng/mL) | Mean | SD | Mean | SD | Ratio |
|---|---|---|---|---|---|
| Controls (n = 5) | Patients (n = 5) | Patients/Controls | |||
| 5-HETE | 85.26 | 81.8 | 289.69 | 58.2 | 3.4 |
| EPA | 101 | 16.4 | 169.51 | 81 | 1.7 |
| 4-HDoHE | 38.66 | 40 | 120.22 | 20.8 | 3.1 |
| 13-HODE | 24.34 | 6.1 | 109.87 | 64.4 | 4.5 |
| 9-HODE | 6.07 | 3.6 | 74.4 | 46.3 | 12.3 |
| 9-HETE | 2.75 | 2.4 | 65.47 | 35.3 | 23.8 |
| 13-KODE | 1.34 | 0.8 | 45.9 | 29 | 34.3 |
| 5-HEPE | 10.34 | 12.4 | 45.61 | 15 | 4.4 |
| 11-HETE | 2.3 | 1.5 | 38.75 | 18.1 | 16.9 |
| 12-HETE | 6.4 | 5.8 | 35.95 | 16.1 | 5.6 |
| 8-HETE | 1.99 | 1.5 | 32.24 | 17.9 | 16.2 |
| 9-KODE | 0.76 | 0.5 | 30.94 | 26.6 | 40.5 |
| 6-trans-LTB4 | 1.56 | 1.8 | 29.5 | 13.2 | 18.9 |
| 8-HDoHE | 1.41 | 1.2 | 25.36 | 12 | 17.9 |
| 11-HDoHE | 1.46 | 1.1 | 25.08 | 12.9 | 17.2 |
| 5,15-DiHETE | 2.47 | 2.6 | 23.63 | 10 | 9.6 |
| 17-HDoHE | 10.3 | 5 | 22.67 | 8.1 | 2.2 |
| PGD2 | 0.85 | 0.7 | 20.57 | 8.1 | 24.3 |
| PGD1 | 0.45 | 0.4 | 17.27 | 9.5 | 38.5 |
| 5-HETrE | 3.05 | 3.4 | 16.5 | 7.2 | 5.4 |
| 8,15-DiHETE | 0.26 | 0.2 | 16.31 | 8.5 | 63.8 |
| 15-HETE | 10.05 | 4.8 | 15.14 | 4.5 | 1.5 |
| 15-KETE | 0.92 | 0.6 | 12.28 | 7.3 | 13.3 |
| 5,6-DiHETE | 0.58 | 0.7 | 10.54 | 4.7 | 18.2 |
| 14-HDoHE | 2.14 | 0.8 | 7.83 | 2.5 | 3.7 |
| 9-HEPE | 0.31 | 0.3 | 7.19 | 3.8 | 23.1 |
| 15-HETrE | 1.2 | 0.4 | 6.65 | 2.3 | 5.6 |
| 5-KETE | 0.57 | 0.8 | 5.8 | 2.7 | 10.2 |
| 12-HEPE | 0.53 | 0.2 | 5.09 | 2.7 | 9.5 |
| 12-KETE | 0.16 | 0.2 | 4.98 | 3 | 30.8 |
| 13-HOTrE | 3.04 | 1.1 | 4.86 | 4.2 | 1.6 |
| 11-HEPE | 0.25 | 0.2 | 4.79 | 2.6 | 19.4 |
| 10,17-DiHDoHE | 0.06 | 0.1 | 4.15 | 2 | 65.1 |
| 5-iPF2a-VI | 0.25 | 0.2 | 3.1 | 1.4 | 12.6 |
| 8-HEPE | 0.23 | 0.2 | 3.09 | 1.8 | 13.7 |
| 8-HETrE | 0.26 | 0.2 | 2.72 | 1 | 10.5 |
| PGE1 | 0.19 | 0.1 | 2.46 | 1.1 | 13.1 |
| 9,10,13-TriHOME | 0.38 | 0.2 | 2.07 | 1.1 | 5.5 |
| 9,12,13-TriHOME | 0.48 | 0.3 | 1.87 | 0.9 | 3.9 |
| 18-HEPE | 0.15 | 0.1 | 1.86 | 0.8 | 12.1 |
| LXB4 | 0.08 | 0.1 | 1.83 | 0.9 | 22.5 |
| 15-HEPE | 0.68 | 0.4 | 1.82 | 0.7 | 2.7 |
| 9-HOTrE | 0.36 | 0.2 | 1.62 | 1.3 | 4.5 |
| 12,13-DiHOME | 4.57 | 1 | 1.53 | 0.9 | 0.3 |
| 13-HOTrE(gamma) | 0.41 | 0.1 | 1.37 | 0.4 | 3.3 |
| EKODE | 0.85 | 0.2 | 1.36 | 0.8 | 1.6 |
| 11-HEDE | 0.29 | 0.1 | 1.36 | 0.8 | 4.7 |
| 9,10-DiHOME | 7.01 | 2.2 | 1.35 | 0.7 | 0.2 |
| 20-COOH-LTB4 | 0 | 0 | 1.16 | 0.8 | - |
| 6-keto-PGF1a | 12.78 | 10.9 | 1.13 | 0.6 | 0.1 |
| 8(9)-EpETrE | 0.05 | 0.1 | 1.1 | 0.4 | 20 |
| 17(18)-EpETE | 0.18 | 0.2 | 1.05 | 0.3 | 5.7 |
| 15-HEDE | 0.1 | 0 | 0.82 | 0.4 | 8.1 |
| 12-HHTrE | 3.29 | 4.7 | 0.78 | 0.5 | 0.2 |
| 8-isoPGE2 | 0.21 | 0.1 | 0.76 | 0.1 | 3.7 |
| LTB4 | 0.06 | 0 | 0.67 | 0.4 | 11.8 |
| 17,18-DiHETE | 1.01 | 0.2 | 0.67 | 0.2 | 0.7 |
| 14,15-DiHETrE | 0.39 | 0 | 0.65 | 0.1 | 1.7 |
| PGE2 | 1.07 | 1.2 | 0.64 | 0.3 | 0.6 |
| 19-HETE | 0.6 | 0.2 | 0.64 | 0 | 1.1 |
| 20-HETE | 0.28 | 0.1 | 0.63 | 0.1 | 2.3 |
| 11,12-DiHETrE | 0.31 | 0.1 | 0.51 | 0 | 1.6 |
| 14,15-DiHETE | 0.23 | 0 | 0.5 | 0.2 | 2.1 |
| 7-Maresin-1 | 0 | 0 | 0.49 | 0.3 | - |
| 12(13)-EpOME | 0.47 | 0.1 | 0.48 | 0.2 | 1 |
| 19,20-DiHDPA | 0.3 | 0 | 0.39 | 0.1 | 1.3 |
| 5,6-DiHETrE | 0.18 | 0 | 0.31 | 0.1 | 1.8 |
| 7,17-hydroxy-DPA | 0.03 | 0 | 0.28 | 0.1 | 9.3 |
| 11-keto-TXB2 | 0.03 | 0 | 0.27 | 0.1 | 10.2 |
| 9(10)-EpOME | 0.22 | 0.1 | 0.26 | 0.1 | 1.2 |
| LTE4 | 0.12 | 0.1 | 0.2 | 0.1 | 1.7 |
| iPF2a-IV | 0 | 0 | 0.2 | 0.1 | 48.3 |
| PGB2 | 0.34 | 0.4 | 0.17 | 0.1 | 0.5 |
| 8,9-DiHETrE | 0.11 | 0 | 0.17 | 0 | 1.5 |
| 11B-PGF2a | 0.29 | 0.4 | 0.11 | 0 | 0.4 |
| 8-iso-PGF2a | 0.04 | 0 | 0.07 | 0 | 2 |
| 15-KEDE | 0.01 | 0 | 0.07 | 0 | 5.5 |
| 12(13)EpODE | 0.02 | 0 | 0.05 | 0 | 2.5 |
| 15-deoxy-delta-12,14-PGJ2 | 0.01 | 0 | 0 | 0 | 0 |
| LTB3 | 0 | 0 | 0 | 0 | - |
| PGD3 | 0 | 0 | 0 | 0 | - |
| delta-12-PGJ2 | 0 | 0 | 0 | 0 | - |
| 17-RvD1 | 0 | 0 | 0 | 0 | - |
| 11(12)-EpETrE | 0 | 0 | 0 | 0 | - |
| PGF2a | 6.22 | 8.1 | 0 | 0 | - |
| PGE3 | 0 | 0 | 0 | 0 | - |
| PGJ2 | 0 | 0 | 0 | 0 | - |
| RvD1 | 0 | 0 | 0 | 0 | - |
| RvD2 | 0 | 0 | 0 | 0 | - |
| 14(15)-EpETE | 0 | 0 | 0 | 0 | - |
| 14(15)-EpETrE | 0 | 0 | 0 | 0 | - |
| 16(17)-EpDPE | 0 | 0 | 0 | 0 | - |
| 19(20)-EpDPE | 0 | 0 | 0 | 0 | - |
| 5(6)-EpETrE | 0 | 0 | 0 | 0 | - |
| 9-KOTrE | 0 | 0 | 0 | 0 | - |
| 20-OH-LTB4 | 0 | 0 | 0 | 0 | - |
| LXA4 | 0 | 0 | 0 | 0 | - |
| LXA5 | 0 | 0 | 0 | 0 | - |
| tetranor-PGDM | 0 | 0 | 0 | 0 | - |
| 13,14-dihydro-15-keto-PGE2 | 0.01 | 0 | 0 | 0 | 0 |
| 8-iso-PGF3a | 0 | 0 | 0 | 0 | - |
| 11-keto-TXB3 | 0 | 0 | 0 | 0 | - |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wefers, C.; Duiveman-de Boer, T.; Zusterzeel, P.L.M.; Massuger, L.F.A.G.; Fuchs, D.; Torensma, R.; Wheelock, C.E.; De Vries, I.J.M. Different Lipid Regulation in Ovarian Cancer: Inhibition of the Immune System. Int. J. Mol. Sci. 2018, 19, 273. https://doi.org/10.3390/ijms19010273
Wefers C, Duiveman-de Boer T, Zusterzeel PLM, Massuger LFAG, Fuchs D, Torensma R, Wheelock CE, De Vries IJM. Different Lipid Regulation in Ovarian Cancer: Inhibition of the Immune System. International Journal of Molecular Sciences. 2018; 19(1):273. https://doi.org/10.3390/ijms19010273
Chicago/Turabian StyleWefers, Christina, Tjitske Duiveman-de Boer, Petra L. M. Zusterzeel, Leon F. A. G. Massuger, David Fuchs, Ruurd Torensma, Craig E. Wheelock, and I. Jolanda M. De Vries. 2018. "Different Lipid Regulation in Ovarian Cancer: Inhibition of the Immune System" International Journal of Molecular Sciences 19, no. 1: 273. https://doi.org/10.3390/ijms19010273