
Impaired Insulin Signaling is Associated with Hepatic Mitochondrial Dysfunction in IR+/−-IRS-1+/− Double Heterozygous (IR-IRS1dh) Mice

 ,
,
Abstract
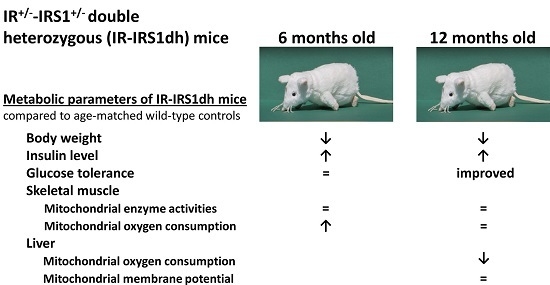
:
1. Introduction
2. Results
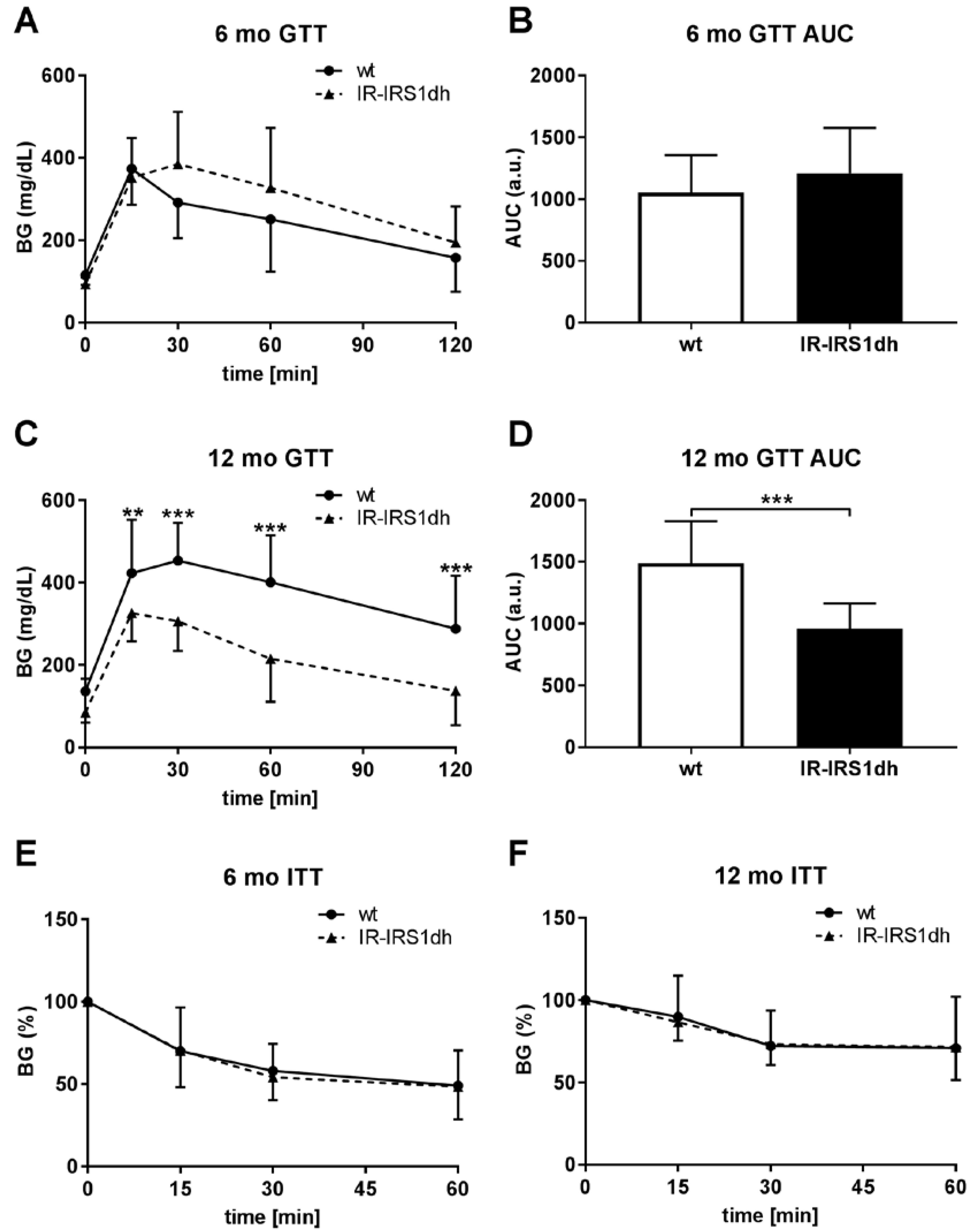
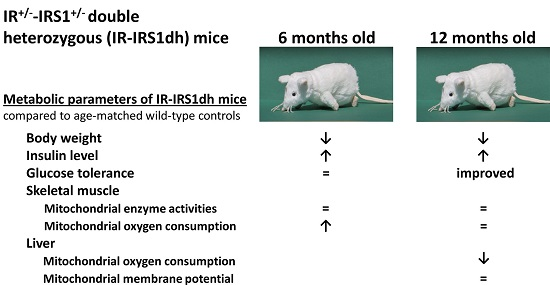
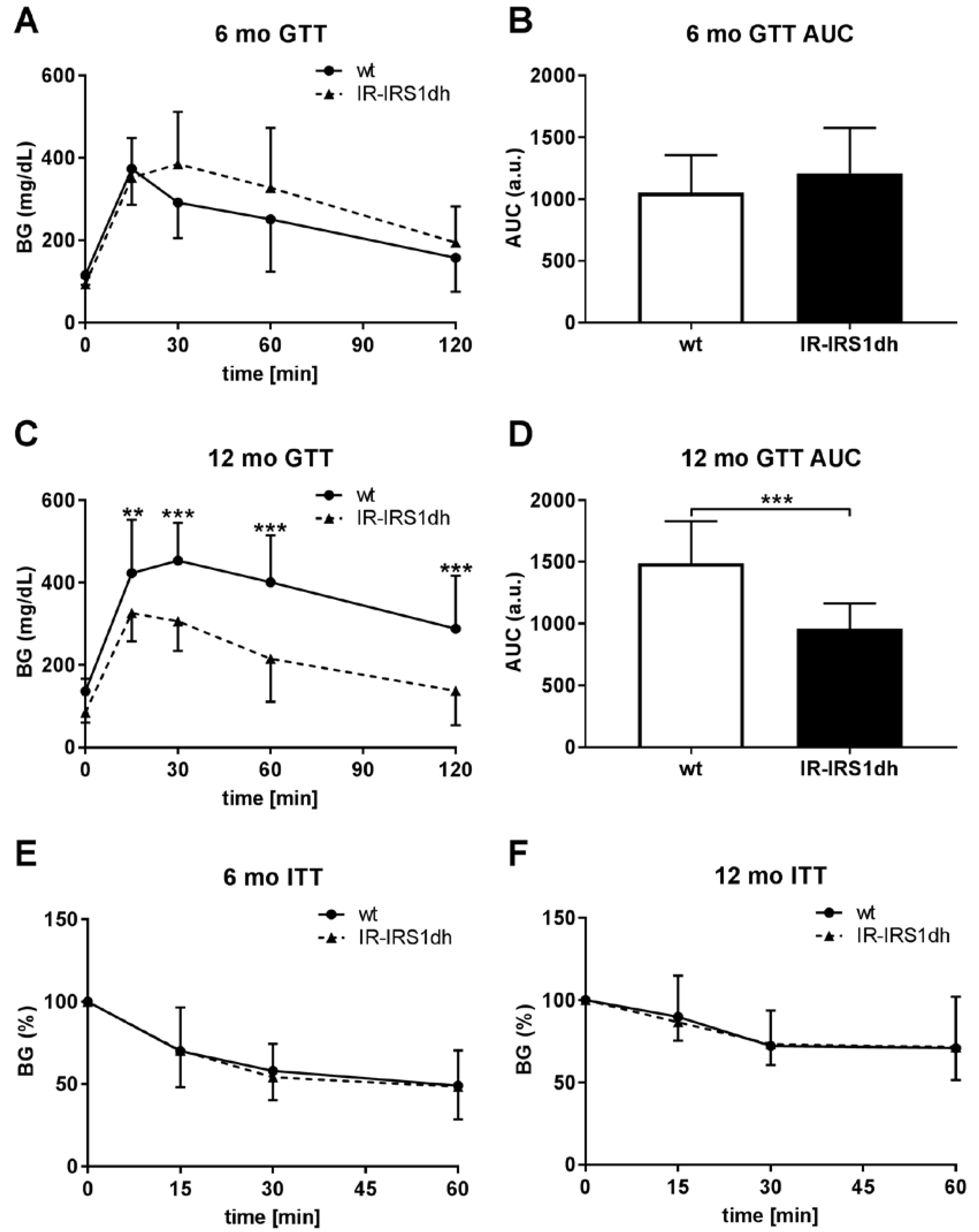
2.1. IR+/−-IRS-1+/− Double Heterozygous (IR-IRS1dh) Mice
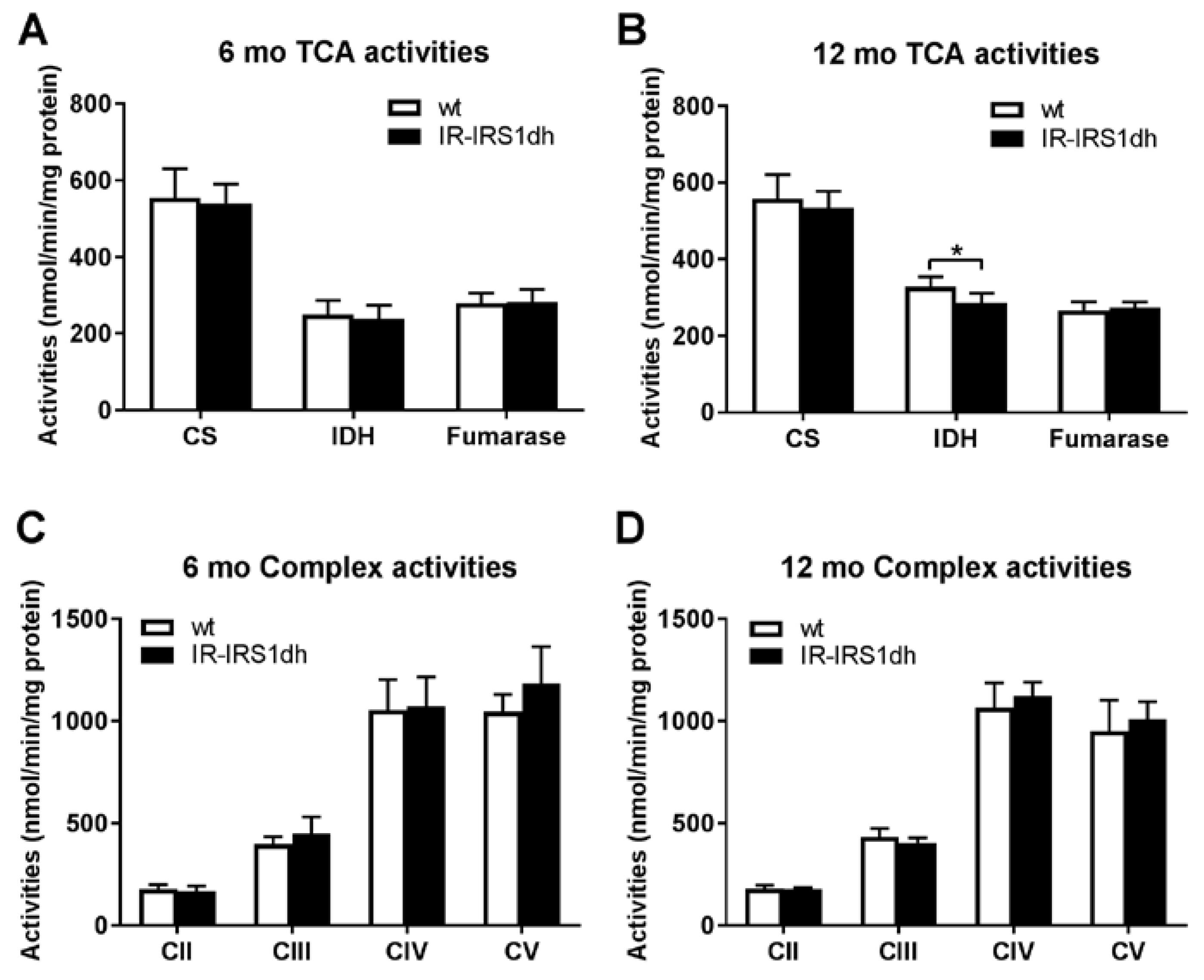
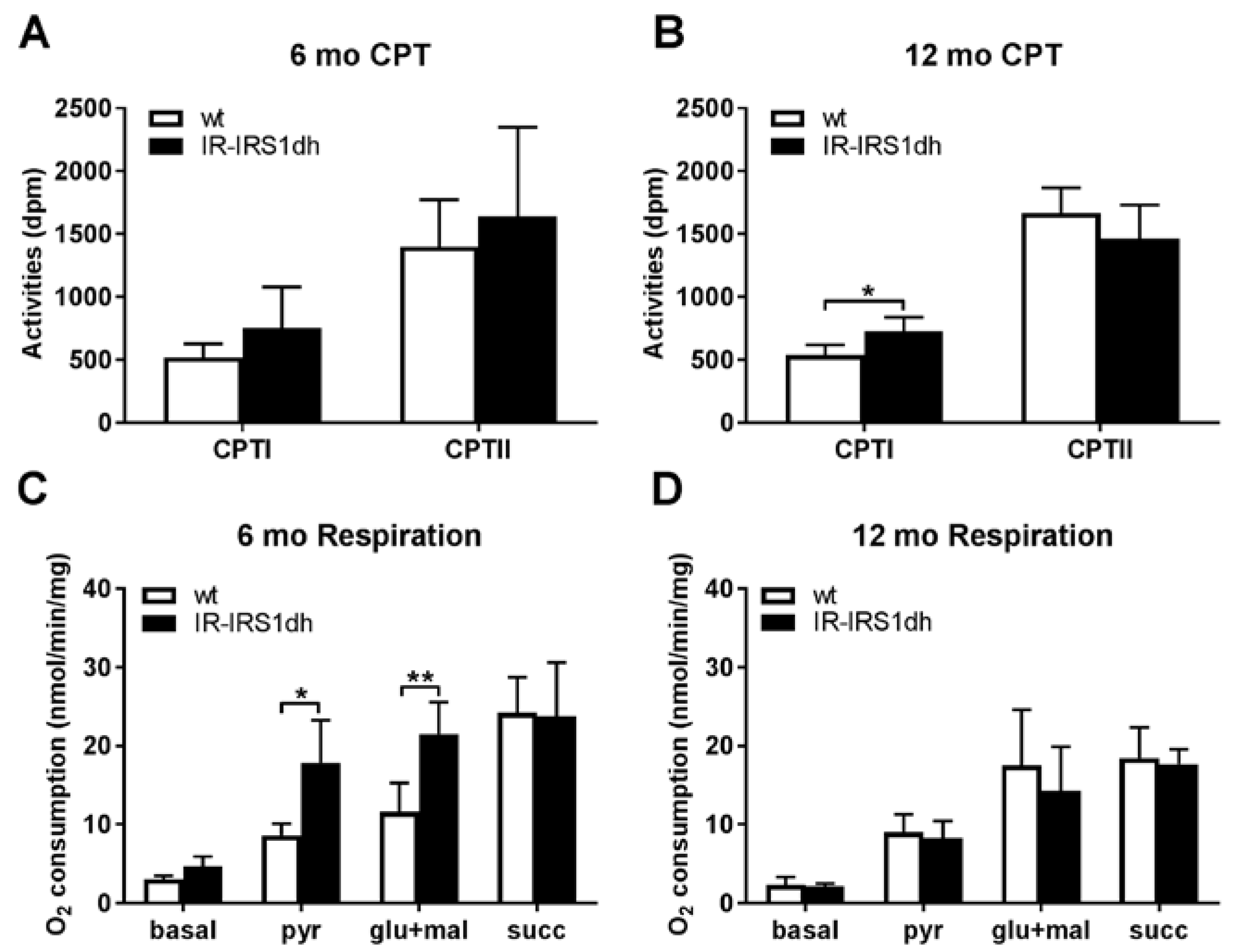
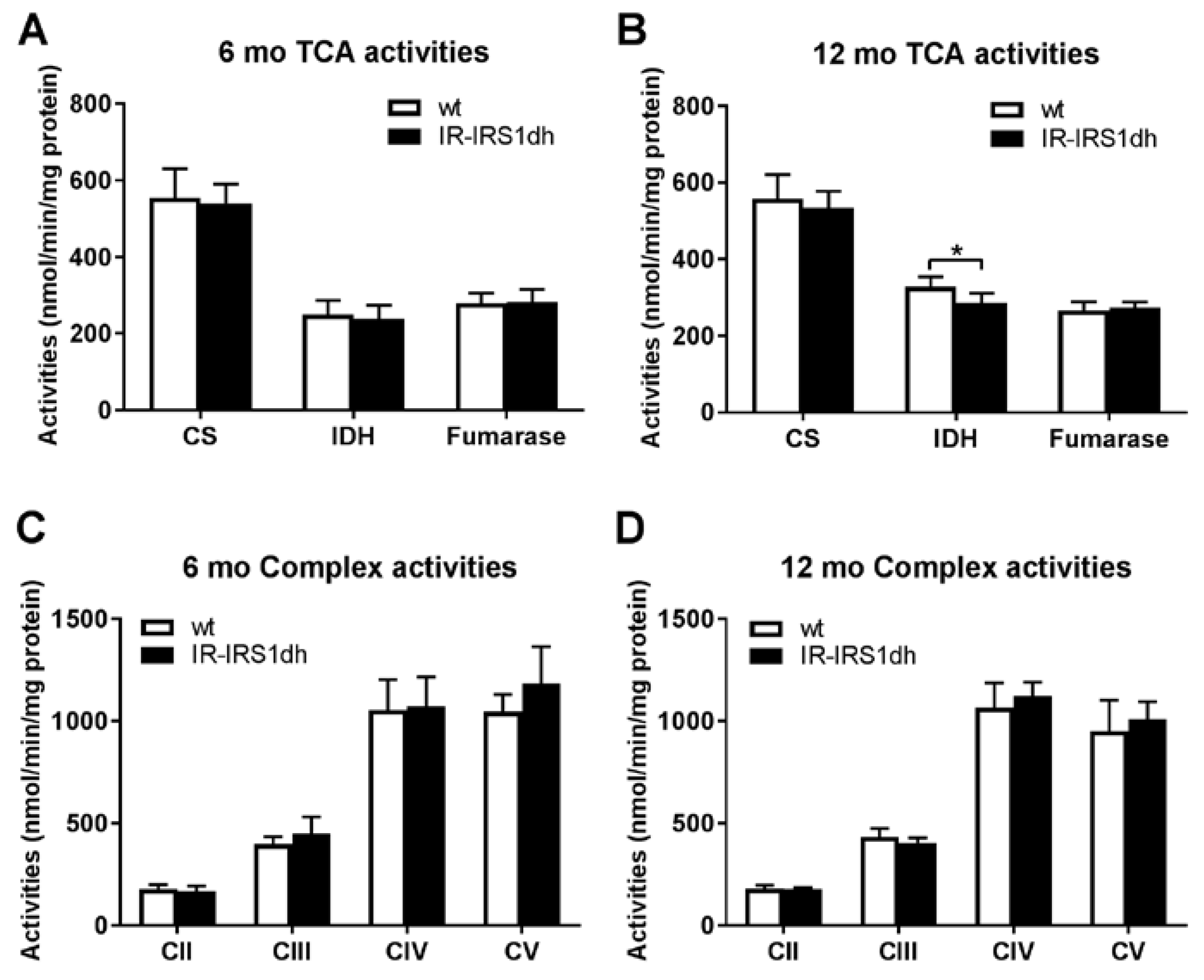
2.2. Mitochondrial Performance in Skeletal Muscle of IR-IRS1dh Mice
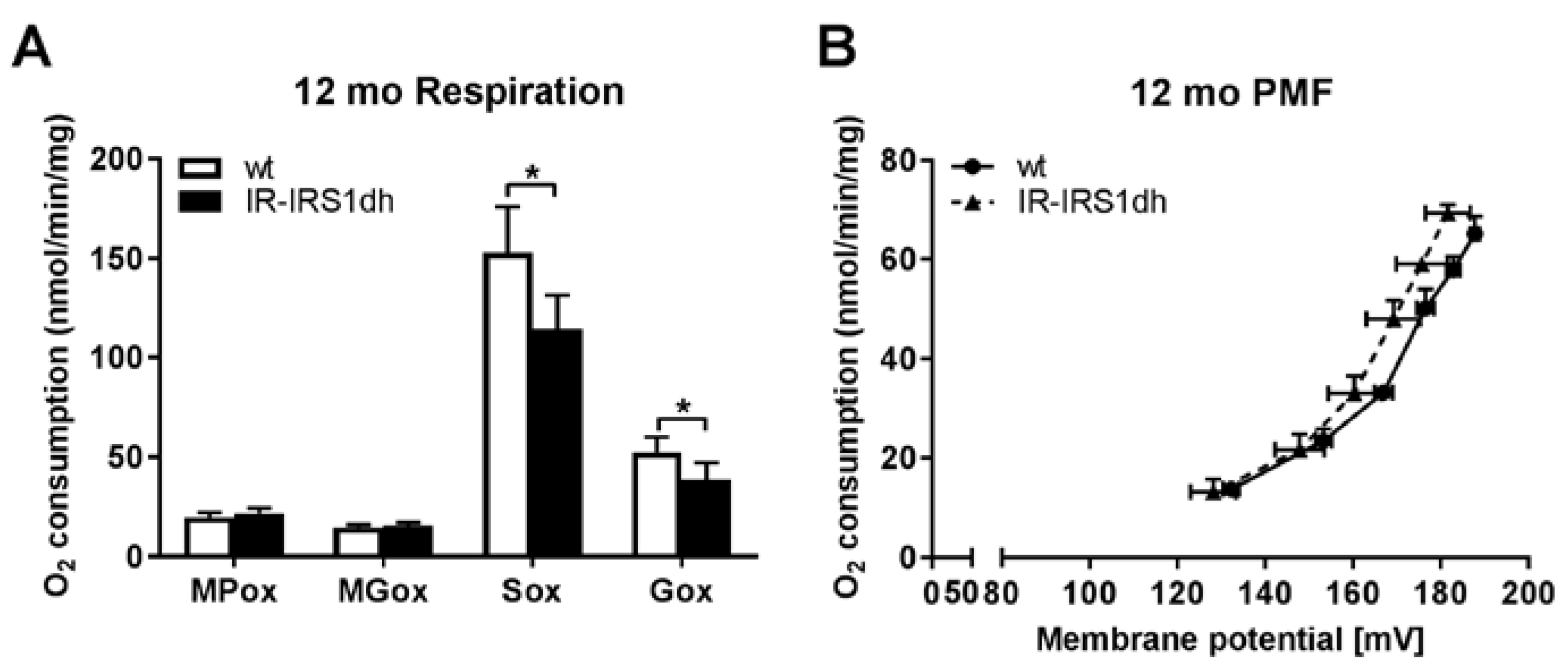
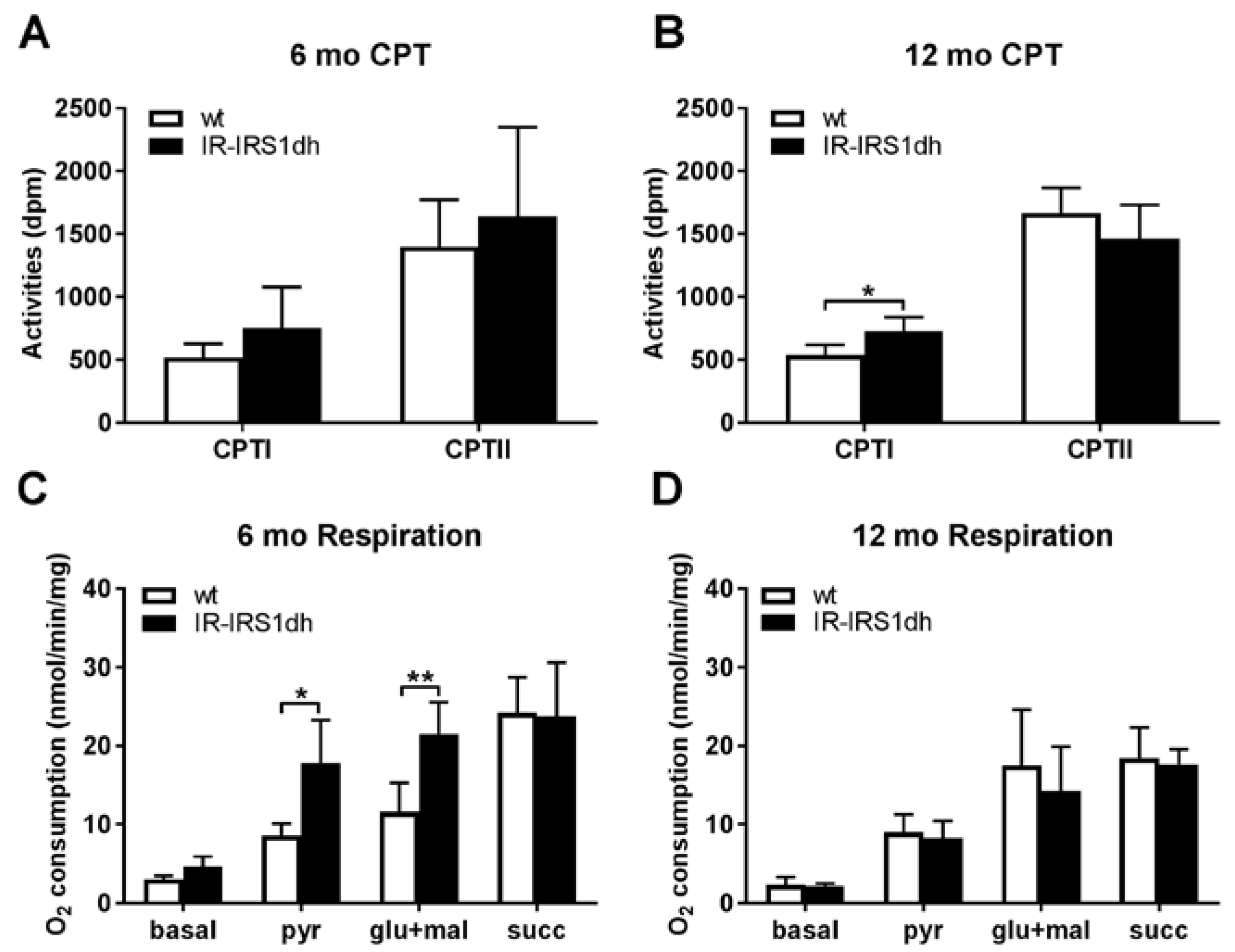
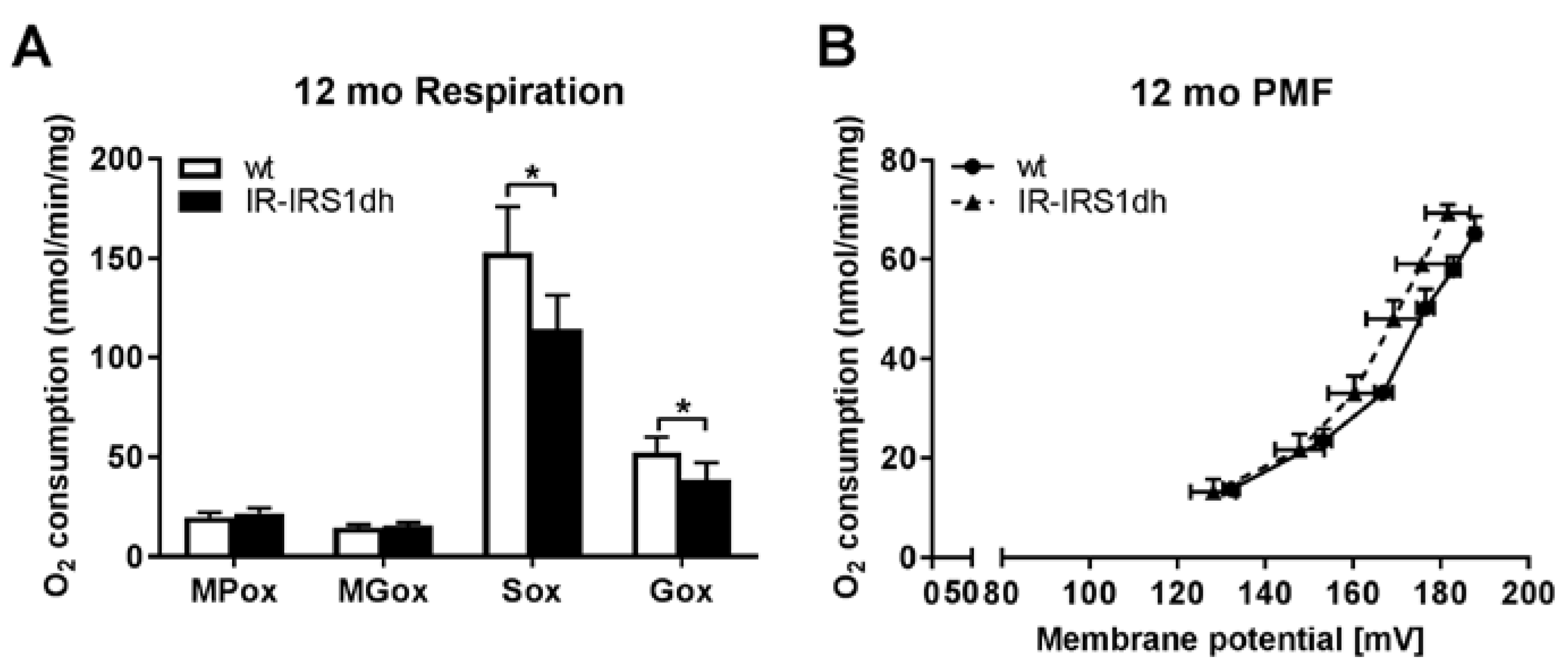
2.3. Mitochondrial Performance in Liver of IR-IRS1dh Mice
3. Discussion
4. Materials and Methods
4.1. Animal Studies
4.2. Mitochondrial Enzyme Activities
4.3. High Resolution Respirometry
4.4. Polarography
4.5. Proton Motive Force Measurement
4.6. Statistics
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| wt | wild-type |
| IR | insulin receptor |
| IRS-1 | insulin receptor substrate-1 |
| IR-IRS1dh mice | insulin receptor (IR)+/–-insulin receptor substrate-1 (IRS-1)+/– double heterozygous mice |
| mo | months |
| TCA | tricarboxylic cycle |
| KO | knockout |
| NAFLD | non-alcoholic fatty liver disease |
| NASH | non-alcoholic steatohepatitis |
| PPAR | peroxisome proliferator-activated receptor |
| PGC1α | peroxisome proliferator-activated receptor gamma coactivator 1-alpha |
References
- Wanet, A.; Arnould, T.; Najimi, M.; Renard, P. Connecting Mitochondria, Metabolism, and Stem Cell Fate. Stem Cells Dev. 2015, 24, 1957–1971. [Google Scholar] [CrossRef] [PubMed]
- Jelenik, T.; Roden, M. Mitochondrial plasticity in obesity and diabetes mellitus. Antioxid. Redox Signal. 2013, 19, 258–268. [Google Scholar] [CrossRef] [PubMed]
- Hesselink, M.K.; Schrauwen-Hinderling, V.; Schrauwen, P. Skeletal muscle mitochondria as a target to prevent or treat type 2 diabetes mellitus. Nat. Rev. Endocrinol. 2016, 12, 633–645. [Google Scholar] [CrossRef] [PubMed]
- Franko, A.; von Kleist-Retzow, J.C.; Bose, M.; Sanchez-Lasheras, C.; Brodesser, S.; Krut, O.; Kunz, W.S.; Wiedermann, D.; Hoehn, M.; Stohr, O.; et al. Complete failure of insulin-transmitted signaling, but not obesity-induced insulin resistance, impairs respiratory chain function in muscle. J. Mol. Med. 2012, 90, 1145–1160. [Google Scholar] [CrossRef] [PubMed]
- Franko, A.; von Kleist-Retzow, J.C.; Neschen, S.; Wu, M.; Schommers, P.; Bose, M.; Kunze, A.; Hartmann, U.; Sanchez-Lasheras, C.; Stoehr, O.; et al. Liver adapts mitochondrial function to insulin resistant and diabetic states in mice. J. Hepatol. 2014, 60, 816–823. [Google Scholar] [CrossRef] [PubMed]
- Franko, A.; Hrabe de Angelis, M.; Wiesner, R.J. Mitochondrial Function, Dysfunction and Adaptation in the Liver during the Development of Diabetes. In Mitochondria in Liver Disease; Han, D., Kaplowitz, N., Eds.; CRC Press: Abingdon, UK, 2015; pp. 383–411. [Google Scholar]
- Boucher, J.; Kleinridders, A.; Kahn, C.R. Insulin receptor signaling in normal and insulin-resistant states. Cold Spring Harb. Perspect. Biol. 2014, 6, a009191. [Google Scholar] [CrossRef] [PubMed]
- Biddinger, S.B.; Kahn, C.R. From mice to men: Insights into the insulin resistance syndromes. Annu. Rev. Physiol. 2006, 68, 123–158. [Google Scholar] [CrossRef] [PubMed]
- Bruning, J.C.; Winnay, J.; Bonner-Weir, S.; Taylor, S.I.; Accili, D.; Kahn, C.R. Development of a novel polygenic model of NIDDM in mice heterozygous for IR and IRS-1 null alleles. Cell 1997, 88, 561–572. [Google Scholar] [CrossRef]
- Partridge, L.; Alic, N.; Bjedov, I.; Piper, M.D. Ageing in Drosophila: The role of the insulin/Igf and TOR signalling network. Exp. Gerontol. 2011, 46, 376–381. [Google Scholar] [CrossRef] [PubMed]
- Kulkarni, R.N.; Jhala, U.S.; Winnay, J.N.; Krajewski, S.; Montminy, M.; Kahn, C.R. PDX-1 haploinsufficiency limits the compensatory islet hyperplasia that occurs in response to insulin resistance. J. Clin. Investig. 2004, 114, 828–836. [Google Scholar] [CrossRef] [PubMed]
- Xue, B.; Kim, Y.B.; Lee, A.; Toschi, E.; Bonner-Weir, S.; Kahn, C.R.; Neel, B.G.; Kahn, B.B. Protein-tyrosine phosphatase 1B deficiency reduces insulin resistance and the diabetic phenotype in mice with polygenic insulin resistance. J. Biol. Chem. 2007, 282, 23829–23840. [Google Scholar] [CrossRef] [PubMed]
- Michael, M.D.; Kulkarni, R.N.; Postic, C.; Previs, S.F.; Shulman, G.I.; Magnuson, M.A.; Kahn, C.R. Loss of insulin signaling in hepatocytes leads to severe insulin resistance and progressive hepatic dysfunction. Mol. Cell 2000, 6, 87–97. [Google Scholar] [CrossRef]
- Kulkarni, R.N.; Almind, K.; Goren, H.J.; Winnay, J.N.; Ueki, K.; Okada, T.; Kahn, C.R. Impact of genetic background on development of hyperinsulinemia and diabetes in insulin receptor/insulin receptor substrate-1 double heterozygous mice. Diabetes 2003, 52, 1528–1534. [Google Scholar] [CrossRef] [PubMed]
- Tschop, M.H.; Speakman, J.R.; Arch, J.R.; Auwerx, J.; Bruning, J.C.; Chan, L.; Eckel, R.H.; Farese, R.V., Jr.; Galgani, J.E.; Hambly, C.; et al. A guide to analysis of mouse energy metabolism. Nat. Methods 2012, 9, 57–63. [Google Scholar] [CrossRef] [PubMed]
- Kenyon, C. The plasticity of aging: Insights from long-lived mutants. Cell 2005, 120, 449–460. [Google Scholar] [CrossRef] [PubMed]
- Bluher, M.; Kahn, B.B.; Kahn, C.R. Extended longevity in mice lacking the insulin receptor in adipose tissue. Science 2003, 299, 572–574. [Google Scholar] [CrossRef] [PubMed]
- White, M.F. IRS2 integrates insulin/IGF1 signalling with metabolism, neurodegeneration and longevity. Diabetes Obes. Metab. 2014, 16, 4–15. [Google Scholar] [CrossRef] [PubMed]
- Bluher, M.; Michael, M.D.; Peroni, O.D.; Ueki, K.; Carter, N.; Kahn, B.B.; Kahn, C.R. Adipose tissue selective insulin receptor knockout protects against obesity and obesity-related glucose intolerance. Dev. Cell 2002, 3, 25–38. [Google Scholar] [CrossRef]
- Sleigh, A.; Raymond-Barker, P.; Thackray, K.; Porter, D.; Hatunic, M.; Vottero, A.; Burren, C.; Mitchell, C.; McIntyre, M.; Brage, S.; et al. Mitochondrial dysfunction in patients with primary congenital insulin resistance. J. Clin. Investig. 2011, 121, 2457–2461. [Google Scholar] [CrossRef] [PubMed]
- Lowell, B.B.; Shulman, G.I. Mitochondrial dysfunction and type 2 diabetes. Science 2005, 307, 384–387. [Google Scholar] [CrossRef] [PubMed]
- Affourtit, C. Mitochondrial involvement in skeletal muscle insulin resistance: A case of imbalanced bioenergetics. Biochim. Biophys. Acta 2016, 1857, 1678–1693. [Google Scholar] [CrossRef] [PubMed]
- Karakelides, H.; Asmann, Y.W.; Bigelow, M.L.; Short, K.R.; Dhatariya, K.; Coenen-Schimke, J.; Kahl, J.; Mukhopadhyay, D.; Nair, K.S. Effect of insulin deprivation on muscle mitochondrial ATP production and gene transcript levels in type 1 diabetic subjects. Diabetes 2007, 56, 2683–2689. [Google Scholar] [CrossRef] [PubMed]
- Nisr, R.B.; Affourtit, C. Insulin acutely improves mitochondrial function of rat and human skeletal muscle by increasing coupling efficiency of oxidative phosphorylation. Biochim. Biophys. Acta 2014, 1837, 270–276. [Google Scholar] [CrossRef] [PubMed]
- Crescenzo, R.; Bianco, F.; Mazzoli, A.; Giacco, A.; Liverini, G.; Iossa, S. A possible link between hepatic mitochondrial dysfunction and diet-induced insulin resistance. Eur. J. Nutr. 2015. [Google Scholar] [CrossRef] [PubMed]
- Koliaki, C.; Szendroedi, J.; Kaul, K.; Jelenik, T.; Nowotny, P.; Jankowiak, F.; Herder, C.; Carstensen, M.; Krausch, M.; Knoefel, W.T.; et al. Adaptation of hepatic mitochondrial function in humans with non-alcoholic Fatty liver is lost in steatohepatitis. Cell Metab. 2015, 21, 739–746. [Google Scholar] [CrossRef] [PubMed]
- Begriche, K.; Igoudjil, A.; Pessayre, D.; Fromenty, B. Mitochondrial dysfunction in NASH: Causes, consequences and possible means to prevent it. Mitochondrion 2006, 6, 1–28. [Google Scholar] [CrossRef] [PubMed]
- Koliaki, C.; Roden, M. Alterations of Mitochondrial Function and Insulin Sensitivity in Human Obesity and Diabetes Mellitus. Annu. Rev. Nutr. 2016. [Google Scholar] [CrossRef] [PubMed]
- Theurey, P.; Rieusset, J. Mitochondria-Associated Membranes Response to Nutrient Availability and Role in Metabolic Diseases. Trends Endocrinol. Metab. 2017, 28, 32–45. [Google Scholar] [CrossRef] [PubMed]
- Theurey, P.; Tubbs, E.; Vial, G.; Jacquemetton, J.; Bendridi, N.; Chauvin, M.A.; Alam, M.R.; Le Romancer, M.; Vidal, H.; Rieusset, J. Mitochondria-associated endoplasmic reticulum membranes allow adaptation of mitochondrial metabolism to glucose availability in the liver. J. Mol. Cell. Biol. 2016, 8, 129–143. [Google Scholar] [CrossRef] [PubMed]
- Tubbs, E.; Theurey, P.; Vial, G.; Bendridi, N.; Bravard, A.; Chauvin, M.A.; Ji-Cao, J.; Zoulim, F.; Bartosch, B.; Ovize, M.; et al. Mitochondria-associated endoplasmic reticulum membrane (MAM) integrity is required for insulin signaling and is implicated in hepatic insulin resistance. Diabetes 2014, 63, 3279–3294. [Google Scholar] [CrossRef] [PubMed]
- Hancock, C.R.; Han, D.H.; Chen, M.; Terada, S.; Yasuda, T.; Wright, D.C.; Holloszy, J.O. High-fat diets cause insulin resistance despite an increase in muscle mitochondria. Proc. Natl. Acad. Sci. USA. 2008, 105, 7815–7820. [Google Scholar] [CrossRef] [PubMed]
- Wanagat, J.; Hevener, A.L. Mitochondrial quality control in insulin resistance and diabetes. Curr. Opin. Genet. Dev. 2016, 38, 118–126. [Google Scholar] [CrossRef] [PubMed]
- Franko, A.; Huypens, P.; Neschen, S.; Irmler, M.; Rozman, J.; Rathkolb, B.; Neff, F.; Prehn, C.; Dubois, G.; Baumann, M.; et al. Bezafibrate Improves Insulin Sensitivity and Metabolic Flexibility in STZ-Induced Diabetic Mice. Diabetes 2016, 65, 2540–2552. [Google Scholar] [CrossRef] [PubMed]
- Franko, A.; Neschen, S.; Rozman, J.; Rathkolb, B.; Aichler, M.; Feuchtinger, A.; Brachthauser, L.; Neff, F.; Kovarova, M.; Wolf, E.; et al. Bezafibrate ameliorates diabetes via reduced steatosis and improved hepatic insulin sensitivity in diabetic TallyHo mice. Mol. Metab. 2017, 6, 256–266. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.N.; Zhou, H.Y.; Fu, Y.Y.; Li, Y.Y.; Wu, F.; Gu, M.; Wu, L.Y.; Xia, C.M.; Dong, T.C.; Li, J.Y.; et al. Novel small-molecule PGC-1α transcriptional regulator with beneficial effects on diabetic db/db mice. Diabetes 2013, 62, 1297–1307. [Google Scholar] [CrossRef] [PubMed]
- Rustin, P.; Chretien, D.; Bourgeron, T.; Gerard, B.; Rotig, A.; Saudubray, J.M.; Munnich, A. Biochemical and molecular investigations in respiratory chain deficiencies. Clin. Chim. Acta 1994, 228, 35–51. [Google Scholar] [CrossRef]
- Brand, M.D. Measurement of mitochondrial protonmotive force. Bioenergetics 1995, 154, 39–62. [Google Scholar]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Mouse Parameters | 6 mo wt | 6 mo IR-IRS1dh | 12 mo wt | 12 mo IR-IRS1dh |
|---|---|---|---|---|
| BW (g) | 33.4 ± 5.9 (11) | 28.0 ± 1.7 * (24) | 39.2 ± 6.0 (11) | 29.7 ± 1.9 *** (20) |
| Fasting BG (mg/dL) | 115.8 ± 23.7 (11) | 93.8 ± 16.4 * (24) | 135.8 ± 30.3 (11) | 84.8 ± 24.7 *** (20) |
| Insulin (pg/mL) | 664 ± 565 (11) | 2325 ± 1455 *** (22) | 2891 ± 1656 (8) | 5390 ± 1454 ** (15) |
| Leptin (pg/mL) | 3138 ± 1589 (14) | 2314 ± 1340 (22) | 5585 ± 853 (7) | 2363 ± 685 *** (15) |
| IGF-1 (pg/mL) | 534 ± 142 (14) | 543 ± 160 (23) | 631 ± 133 (8) | 587 ± 186 (12) |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Franko, A.; Kunze, A.; Böse, M.; Von Kleist-Retzow, J.-C.; Paulsson, M.; Hartmann, U.; Wiesner, R.J. Impaired Insulin Signaling is Associated with Hepatic Mitochondrial Dysfunction in IR+/−-IRS-1+/− Double Heterozygous (IR-IRS1dh) Mice. Int. J. Mol. Sci. 2017, 18, 1156. https://doi.org/10.3390/ijms18061156
Franko A, Kunze A, Böse M, Von Kleist-Retzow J-C, Paulsson M, Hartmann U, Wiesner RJ. Impaired Insulin Signaling is Associated with Hepatic Mitochondrial Dysfunction in IR+/−-IRS-1+/− Double Heterozygous (IR-IRS1dh) Mice. International Journal of Molecular Sciences. 2017; 18(6):1156. https://doi.org/10.3390/ijms18061156
Chicago/Turabian StyleFranko, Andras, Alexander Kunze, Marlen Böse, Jürgen-Christoph Von Kleist-Retzow, Mats Paulsson, Ursula Hartmann, and Rudolf J. Wiesner. 2017. "Impaired Insulin Signaling is Associated with Hepatic Mitochondrial Dysfunction in IR+/−-IRS-1+/− Double Heterozygous (IR-IRS1dh) Mice" International Journal of Molecular Sciences 18, no. 6: 1156. https://doi.org/10.3390/ijms18061156
APA StyleFranko, A., Kunze, A., Böse, M., Von Kleist-Retzow, J.-C., Paulsson, M., Hartmann, U., & Wiesner, R. J. (2017). Impaired Insulin Signaling is Associated with Hepatic Mitochondrial Dysfunction in IR+/−-IRS-1+/− Double Heterozygous (IR-IRS1dh) Mice. International Journal of Molecular Sciences, 18(6), 1156. https://doi.org/10.3390/ijms18061156