
Significance of Wild-Type p53 Signaling in Suppressing Apoptosis in Response to Chemical Genotoxic Agents: Impact on Chemotherapy Outcome


{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Biological Outputs Orchestrated by Wild-Type p53
2.1. p53 Functions
2.2. p53 Regulation in the Absence of Genotoxic Stress
2.3. p53 Regulation Following Genotoxic Stress
2.4. p53 Dynamics Following Genotoxic Stress
2.5. A Threshold Mechanism Determines the Choice Between p53-Mediated Growth Arrest versus Apoptosis
2.6. Anti-Apoptotic Property of p53 Signaling under Physiological Conditions
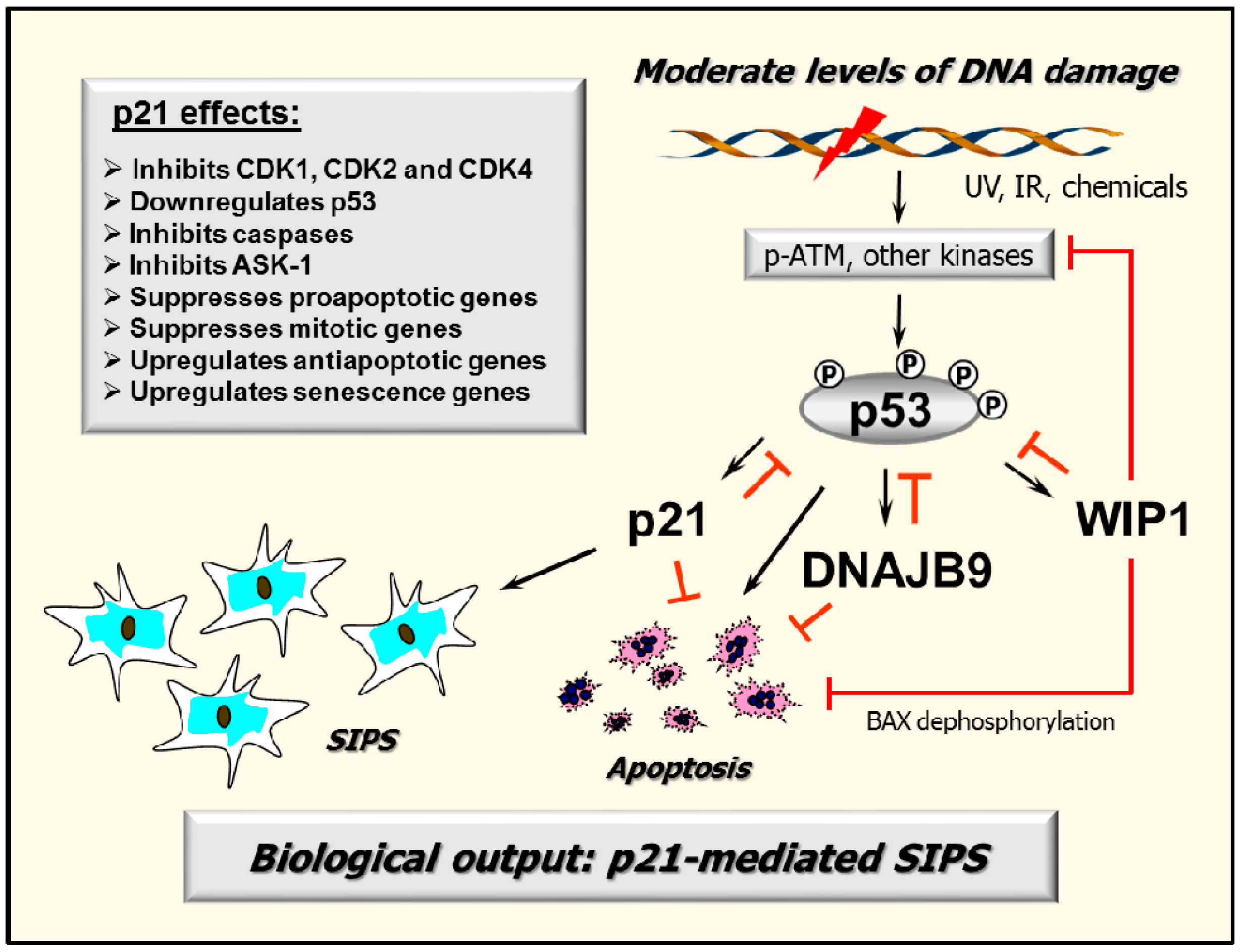
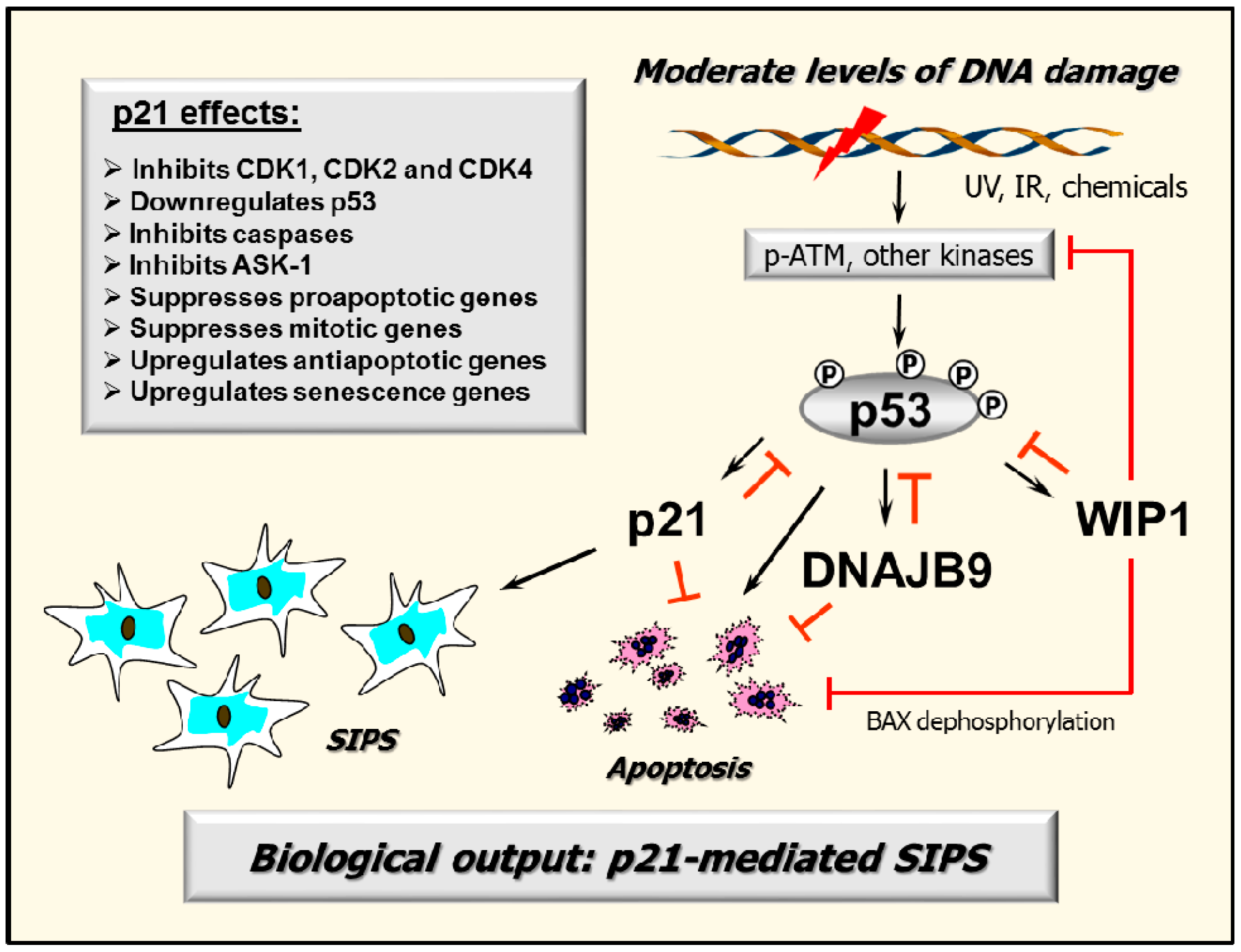
2.7. p53–DNAJB9 Regulatory Loop: Impact on Apoptosis
2.8. Multiple Functions of p21: Downregulating p53 and More
3. Activation of Apoptotic Signaling Does Not Always Lead to Cell Death: Impact on Chemosensitivity Assessment
4. Extrapolating Results Obtained in Overexpression Studies to Clinically Relevant Conditions
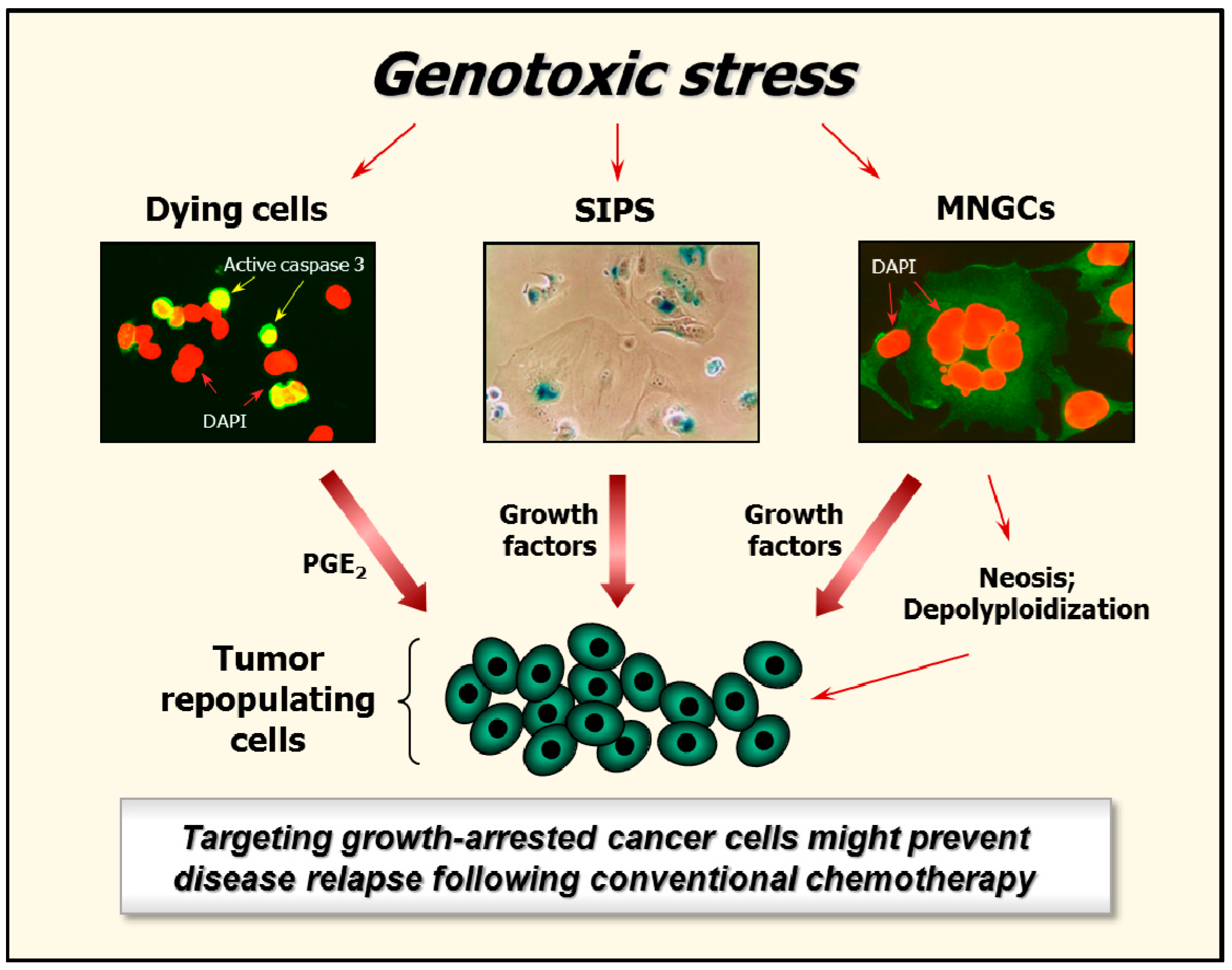
5. Fate of Growth-Arrested Cancer Cells
5.1. Clinically Relevant Doses of Chemotherapeutic Agents Predominantly Trigger Cancer Cell Dormancy Rather Than Cell Death
5.2. Hypoxia and the Creation of MNGCs
5.3. Genome Reduction and Neosis of MNGCs
5.4. Is SIPS Reversible?
6. Targeting Growth-Arrested Cancer Cells as a Potential Therapeutic Strategy
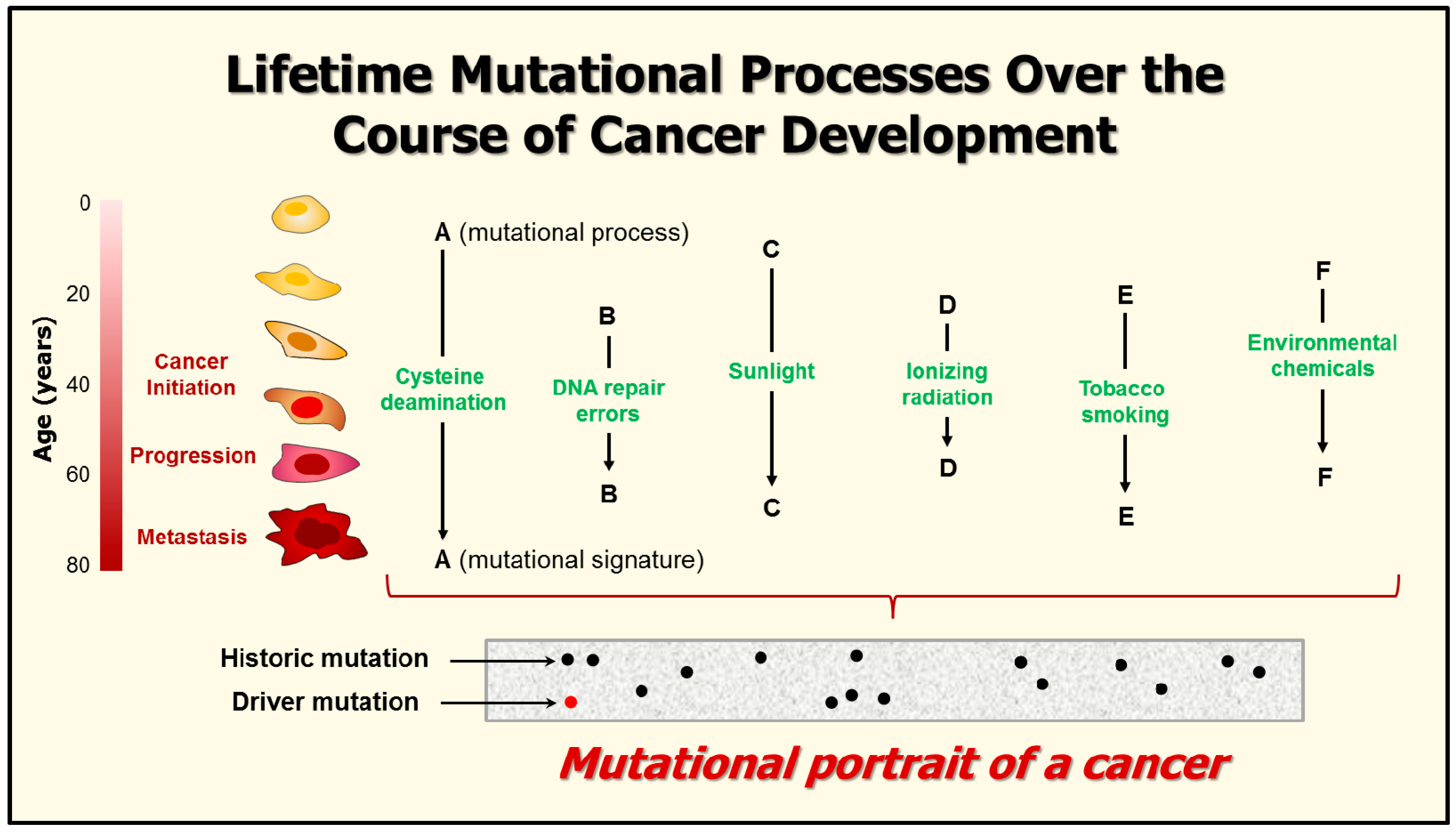
7. Mutational Signatures in Human Cancers
8. Conclusions
Acknowledgments
Conflicts of Interest
Abbreviations
| UV | Ultraviolet light |
| DSBs | DNA double-strand breaks |
| SA-β-gal | Senescence-associated β-galactosidase |
| SIPS | Stress-induced premature senescence |
| DNAJB9 | DNAJ homolog subfamily B member 9 |
| MDM2 | Murine double minute-2 homologue |
| ATM | Ataxia telangiectasia mutated |
| ATR | ATM and RAD3-related |
| CHK | Checkpoint kinase |
| WIP1 | Wild-type p53-induced phosphatase 1 |
| BAX | BCL-2-associated X protein |
| PUMA | p53 upregulated modulator of apoptosis |
| WAF1 | Wild-type p53-activated fragment 1 |
| CIP1 | CDK-interacting protein 1 |
| SDI1 | Senescent cell-derived inhibitor 1 |
| CDK | Cyclin dependent kinase |
| SAPK | Stress-activated protein kinase |
| ASK1 | Apoptosis signal-regulating kinase 1 |
| HPV | Human papillomavirus |
| PGE2 | Prostaglandin E2 |
| SASP | Senescence-associated secretory phenotype |
| SKP2 | S-phase kinase-associated protein 2 |
| EMT | Epithelial-mesenchymal transition |
| MTT | 3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyl-tetrazolium bromide |
| IC50 | Inhibiting concentration, 50% |
| NCCD | Nomenclature Committee on Cell Death |
References
- De Bont, R.; van Larebeke, N. Endogenous DNA damage in humans: A review of quantitative data. Mutagenesis 2004, 19, 169–185. [Google Scholar] [CrossRef] [PubMed]
- Hou, L.; Zhang, X.; Wang, D.; Baccarelli, A. Environmental chemical exposures and human epigenetics. Int. J. Epidemiol. 2012, 41, 79–105. [Google Scholar] [CrossRef] [PubMed]
- Coelho, M.M.V.; Matos, T.R.; Apetato, M. The dark side of the light: Mechanisms of photocarcinogenesis. Clin. Dermatol. 2016, 34, 563–570. [Google Scholar] [CrossRef] [PubMed]
- Lai, Y.; Yu, R.; Hartwell, H.J.; Moeller, B.C.; Bodnar, W.M.; Swenberg, J.A. Measurement of endogenous versus exogenous formaldehyde-induced DNA-protein crosslinks in animal tissues by stable isotope labeling and ultrasensitive mass spectrometry. Cancer Res. 2016, 76, 2652–2661. [Google Scholar] [CrossRef] [PubMed]
- Saintigny, Y.; Chevalier, F.; Bravard, A.; Dardillac, E.; Laurent, D.; Hem, S.; Dépagne, J.; Radicella, J.P.; Lopez, B.S. A threshold of endogenous stress is required to engage cellular response to protect against mutagenesis. Sci. Rep. 2016, 6, 29412. [Google Scholar] [CrossRef] [PubMed]
- Mirzayans, R.; Andrais, B.; Scott, A.; Wang, Y.W.; Murray, D. Ionizing radiation-induced responses in human cells with differing TP53 status. Int. J. Mol. Sci. 2013, 14, 22409–22435. [Google Scholar] [CrossRef] [PubMed]
- Uversky, V.N. p53 proteoforms and intrinsic disorder: An illustration of the protein structure-function continuum concept. Int. J. Mol. Sci. 2016, 17, 1874. [Google Scholar] [CrossRef] [PubMed]
- Vousden, K.H.; Prives, C. Blinded by the light: The growing complexity of p53. Cell 2009, 137, 413–431. [Google Scholar] [CrossRef] [PubMed]
- Stegh, A.H. Targeting the p53 signaling pathway in cancer therapy—The promises, challenges, and perils. Expert Opin. Ther. Targets 2012, 16, 67–83. [Google Scholar] [CrossRef] [PubMed]
- Desilet, N.; Campbell, T.N.; Choy, F.Y.M. p53-based anti-cancer therapies: An empty promise? Curr. Issues Mol. Biol. 2010, 12, 143–146. [Google Scholar] [PubMed]
- Mirzayans, R.; Andrais, B.; Kumar, P.; Murray, D. The growing complexity of cancer cell response to DNA-damaging agents: Caspase 3 mediates cell death or survival? Int. J. Mol. Sci. 2016, 17, 708. [Google Scholar] [CrossRef] [PubMed]
- Zuco, V.; Zunino, F. Cyclic pifithrin-α sensitizes wild type p53 tumor cells to antimicrotubule agent-induced apoptosis. Neoplasia 2008, 10, 587–596. [Google Scholar] [CrossRef] [PubMed]
- Amin, A.R.; Thakur, V.S.; Gupta, K.; Jackson, M.W.; Harada, H.; Agarwal, M.K.; Shin, D.M.; Wald, D.N.; Agarwal, M.L. Restoration of p53 functions protects cells from concanavalin a-induced apoptosis. Mol. Cancer Ther. 2010, 9, 471–479. [Google Scholar] [CrossRef] [PubMed]
- Waye, S.; Naeem, A.; Choudhry, M.U.; Parasido, E.; Tricoli, L.; Sivakumar, A.; Mikhaiel, J.P.; Yenugonda, V.; Rodriguez, O.C.; Karam, S.D.; et al. The p53 tumor suppressor protein protects against chemotherapeutic stress and apoptosis in human medulloblastoma cells. Aging 2015, 7, 854–868. [Google Scholar] [CrossRef] [PubMed]
- Sengupta, S.; Harris, C.C. p53: Traffic cop at the crossroads of DNA repair and recombination. Nat. Rev. Mol. Cell Biol. 2005, 6, 44–55. [Google Scholar] [CrossRef] [PubMed]
- Levine, A.J.; Hu, W.; Feng, Z. The p53 pathway: What questions remain to be explored? Cell Death Differ. 2006, 13, 1027–1036. [Google Scholar] [CrossRef] [PubMed]
- Roninson, I.B. Tumor cell senescence in cancer treatment. Cancer Res. 2003, 63, 2705–2715. [Google Scholar] [PubMed]
- Mirzayans, R.; Andrais, B.; Scott, A.; Paterson, M.C.; Murray, D. Single-cell analysis of p16INK4a and p21WAF1 expression suggests distinct mechanisms of senescence in normal human and Li-Fraumeni Syndrome fibroblasts. J. Cell. Physiol. 2010, 223, 57–67. [Google Scholar] [PubMed]
- Wang, M.; Morsbach, F.; Sander, D.; Gheorghiu, L.; Nanda, A.; Benes, C.; Kriegs, M.; Krause, M.; Dikomey, E.; Baumann, M.; et al. EGF receptor inhibition radiosensitizes NSCLC cells by inducing senescence in cells sustaining DNA double-strand breaks. Cancer Res. 2011, 71, 6261–6269. [Google Scholar] [CrossRef] [PubMed]
- Hernández-Vargas, H.; Ballestar, E.; Carmona-Saez, P.; von Kobbe, C.; Bañón-Rodrίguez, I.; Esteller, M.; Moreno-Bueno, G.; Palacios, J. Transcriptional profiling of MCF7 breast cancer cells in response to 5-Fluorouracil: Relationship with cell cycle changes and apoptosis, and identification of novel targets of p53. Int. J. Cancer 2006, 119, 1164–1175. [Google Scholar] [CrossRef] [PubMed]
- Alani, R.M.; Young, A.Z.; Shifflett, C.B. Id1 regulation of cellular senescence through transcriptional repression of p16/Ink4a. Proc. Natl. Acad. Sci. USA 2001, 98, 7812–7816. [Google Scholar] [CrossRef] [PubMed]
- Polsky, D.; Young, A.Z.; Busam, K.J.; Alani, R.M. The transcriptional repressor of p16/Ink4a, Id1, is up-regulated in early melanomas. Cancer Res. 2001, 61, 6008–6011. [Google Scholar] [PubMed]
- Leong, W.F.; Chau, J.F.L.; Li, B. p53 deficiency leads to compensatory up-regulation of p16INK4a. Mol. Cancer Res. 2009, 7, 354–360. [Google Scholar] [CrossRef] [PubMed]
- Ljungman, M. Dial 9-1-1 for p53: Mechanisms of p53 activation by cellular stress. Neoplasia 2000, 2, 208–225. [Google Scholar] [CrossRef] [PubMed]
- Efeyan, A.; Serrano, M. p53: Guardian of the genome and policeman of the oncogenes. Cell Cycle 2007, 6, 1006–1010. [Google Scholar] [CrossRef] [PubMed]
- Bulavin, D.V.; Saito, S.; Hollander, M.C.; Sakaguchi, K.; Anderson, C.W.; Appella, E.; Fornace, A.J., Jr. Phosphorylation of human p53 by p38 kinase coordinates N-terminal phosphorylation and apoptosis in response to UV radiation. EMBO J. 1999, 18, 6845–6854. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.; Ma, W.Y.; Maxiner, A.; Sun, Y.; Dong, Z. p38 kinase mediates UV-induced phosphorylation of p53 protein at serine 389. J. Biol. Chem. 1999, 274, 12229–12235. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Prieto, R.; Rojas, J.M.; Taya, Y.; Gutkind, J.S. A role for the p38 mitogen-activated protein kinase pathway in the transcriptional activation of p53 on genotoxic stress by chemotherapeutic agents. Cancer Res. 2000, 60, 2464–2472. [Google Scholar] [PubMed]
- Lu, X.; Nguyen, T.A.; Moon, S.H.; Darlington, Y.; Sommer, M.; Donehower, L.A. The type 2C phosphatase Wip1: An oncogenic regulator of tumor suppressor and DNA damage response pathways. Cancer Metastasis Rev. 2008, 27, 123–135. [Google Scholar] [CrossRef] [PubMed]
- Lahav, G.; Rosenfeld, N.; Sigal, A.; Geva-Zatorsky, N.; Levine, A.J.; Elowitz, M.B.; Alon, U. Dynamics of the p53-MDM2 feedback loop in individual cells. Nat. Genet. 2004, 36, 147–150. [Google Scholar] [CrossRef] [PubMed]
- Batchelor, E.; Mock, C.S.; Bhan, I.; Loewer, A.; Lahav, G. Recurrent initiation: A mechanism for triggering p53 pulses in response to DNA damage. Mol. Cell 2008, 9, 277–289. [Google Scholar] [CrossRef] [PubMed]
- Mirzayans, R.; Andrais, B.; Scott, A.; Murray, D. New insights into p53 signaling and cancer-cell response to DNA damage: Implications for cancer therapy. J. Biomed. Biotechnol. 2012, 2012, 170325. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.H.; Sladek, T.L.; Liu, X.; Butler, B.R.; Froelich, C.J.; Thor, A.D. Reconstitution of caspase 3 sensitizes MCF-7 breast cancer cells to doxorubicin- and etoposide-induced apoptosis. Cancer Res. 2001, 61, 348–354. [Google Scholar] [PubMed]
- Jänicke, R.U. MCF-7 breast carcinoma cells do not express caspase-3. Breast. Cancer Res. Treat. 2009, 117, 219–221. [Google Scholar] [CrossRef] [PubMed]
- Essmann, F.; Engels, I.H.; Totzke, G.; Schulze-Osthoff, K.; Jänicke, R.U. Apoptosis resistance of MCF-7 breast carcinoma cells to ionizing radiation is independent of p53 and cell cycle control but caused by the lack of caspase-3 and a caffeine-inhibitable event. Cancer Res. 2004, 64, 7065–7072. [Google Scholar] [CrossRef] [PubMed]
- Purvis, J.E.; Karhohs, K.W.; Mock, C.; Batchelor, E.; Loewer, A.; Lahav, G. p53 dynamics control cell fate. Science 2012, 336, 1440–1444. [Google Scholar] [CrossRef] [PubMed]
- Sun, T.; Cui, J. Dynamics of p53 in response to DNA damage: Mathematical modeling and perspective. Prog. Biophys. Mol. Biol. 2015, 119, 175–182. [Google Scholar] [CrossRef] [PubMed]
- Jin, S.; Levine, A.J. The p53 functional circuit. J. Cell Sci. 2001, 114, 4139–4140. [Google Scholar] [PubMed]
- Haupt, S.; Berger, M.; Goldberg, Z.; Haupt, Y. Apoptosis—The p53 network. J. Cell Sci. 2003, 116, 4077–4085. [Google Scholar] [CrossRef] [PubMed]
- Oren, M. Decision making by p53: Life, death and cancer. Cell Death Differ. 2003, 10, 431–442. [Google Scholar] [CrossRef] [PubMed]
- Kracikova, M.; Akiril, G.; George, A.; Sachidanandam, R.; Aaronson, S.A. A threshold mechanism mediates p53 cell fate decision between growth arrest and apoptosis. Cell Death Differ. 2013, 20, 576–588. [Google Scholar] [CrossRef] [PubMed]
- Schug, T.T. Awakening p53 in senescent cells using nutlin-3. Aging 2009, 1, 842–844. [Google Scholar] [CrossRef] [PubMed]
- Shen, H.; Maki, C.G. Persistent p21 expression after nutlin-3a removal is associated with senescence-like arrest in 4N cells. J. Biol. Chem. 2010, 285, 23105–23114. [Google Scholar] [CrossRef] [PubMed]
- Arya, A.K.; El-Fert, A.; Devling, T.; Eccles, R.M.; Aslam, M.A.; Rubbi, C.P.; Vlatković, N.; Fenwick, J.; Lloyd, B.H.; Sibson, D.R.; et al. Nutlin-3, the small-molecule inhibitor of MDM2, promotes senescence and radiosensitises laryngeal carcinoma cells harbouring wild-type p53. Br. J. Cancer 2010, 103, 186–195. [Google Scholar] [CrossRef] [PubMed]
- Polański, R.; Noon, A.P.; Blaydes, J.; Phillips, A.; Rubbi, C.P.; Parsons, K.; Vlatković, N.; Boyd, M.T. Senescence induction in renal carcinoma cells by Nutlin-3: A potential therapeutic strategy based on MDM2 antagonism. Cancer Lett. 2014, 353, 211–219. [Google Scholar] [CrossRef] [PubMed]
- Jänicke, R.U.; Sohn, D.; Schulze-Osthoff, K. The dark side of a tumor suppressor: Anti-apoptotic p53. Cell Death Differ. 2008, 15, 959–976. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.J.; Kim, J.M.; Kim, K.H.; Heo, J.I.; Kwak, S.J.; Han, J.A. Genotoxic stress/p53-induced DNAJB9 inhibits the pro-apoptotic function of p53. Cell Death Differ. 2015, 22, 86–95. [Google Scholar] [CrossRef] [PubMed]
- Mirzayans, R.; Murray, D. Cellular Senescence: Implications for Cancer Therap, 1st ed.; Nova Science Publishers: New York, NY, USA, 2009; pp. 1–130. [Google Scholar]
- Warfel, N.A.; El-Deiry, W.S. p21WAF1 and tumourigenesis: 20 years after. Curr. Opin. Oncol. 2013, 25, 52–58. [Google Scholar] [CrossRef] [PubMed]
- Javelaud, D.; Besançon, F. Inactivation of p21WAF1 sensitizes cells to apoptosis via an increase of both p14ARF and p53 levels and an alteration of the Bax/BCL-2 ratio. J. Biol. Chem. 2002, 277, 37849–37954. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Xiong, Y.; Yarbrough, W.G. ARF promotes MDM2 degradation and stabilizes p53: ARF-INK4a locus deletion impairs both the Rb and p53 tumor suppression pathways. Cell 1998, 92, 725–734. [Google Scholar] [CrossRef]
- Honda, R.; Yasuda, H. Association of p19 (ARF) with MDM2 inhibits ubiquitin ligase activity of MDM2 for tumor suppressor p53. EMBO J. 1999, 18, 22–27. [Google Scholar] [CrossRef] [PubMed]
- Mirzayans, R.; Andrais, B.; Scott, A.; Wang, Y.W.; Weiss, R.H.; Murray, D. Spontaneous γH2AX foci in human solid tumor-derived cell lines in relation to p21WAF1 and WIP1 expression. Int. J. Mol. Sci. 2015, 16, 11609–11628. [Google Scholar] [CrossRef] [PubMed]
- Sohn, D.; Essmann, F.; Schulze-Osthoff, K.; Jänicke, R.U. p21 blocks irradiation-induced apoptosis downstream of mitochondria by inhibition of cyclin-dependent kinase-mediated caspase-9 activation. Cancer Res. 2006, 66, 11254–11262. [Google Scholar] [CrossRef] [PubMed]
- Roninson, I.B. Oncogenic functions of tumour suppressor p21 (Waf1/Cip1/Sdi1): Association with cell senescence and tumour-promoting activities of stromal fibroblasts. Cancer Lett. 2002, 179, 1–14. [Google Scholar] [CrossRef]
- Dotto, G.P. p21WAF1/Cip1: More than a break to the cell cycle? Biochim. Biophys. Acta 2000, 1471, M43–M56. [Google Scholar] [CrossRef]
- Pang, L.Y.; Scott, M.; Hayward, R.L.; Mohammed, H.; Whitelaw, C.B.; Smith, G.C.; Hupp, T.R. p21WAF1 is component of a positive feedback loop that maintains the p53 transcriptional program. Cell Cycle 2011, 10, 932–950. [Google Scholar] [CrossRef] [PubMed]
- Hill, R.; Leidal, A.M.; Madureira, P.A.; Gillis, L.D.; Waisman, D.M.; Chiu, A.; Lee, P.W. Chromium-mediated apoptosis: Involvement of DNA-dependent protein kinase (DNA-PK) and differential induction of p53 target genes. DNA Repair 2008, 7, 1484–1499. [Google Scholar] [CrossRef] [PubMed]
- Broude, E.V.; Demidenko, Z.N.; Vivo, C.; Swift, M.E.; Davis, B.M.; Blagosklonny, M.V.; Roninson, I.B. p21 (CDKN1A) is a negative regulator of p53 stability. Cell Cycle 2007, 6, 1468–1471. [Google Scholar] [CrossRef] [PubMed]
- Murray, D.; Mirzayans, R. Role of therapy-induced cellular senescence in tumor cells and its modification in radiotherapy; the good, the bad and the ugly. J. Nucl. Med. Radiat. Ther. 2013, 6, 018. [Google Scholar]
- Crescenzi, E.; Palumbo, G.; de Boer, J.; Brady, H.J. Ataxia telangiectasia mutated and p21CIP1 modulate cell survival of drug-induced senescent tumor cells: Implications for chemotherapy. Clin. Cancer Res. 2008, 14, 1877–1887. [Google Scholar] [CrossRef] [PubMed]
- Samuel, T.; Weber, H.O.; Rauch, P.; Verdoodt, B.; Eppe, J.T.; McShea, A.; Hermeking, H.; Funk, J.O. The G2/M regulator 14-3-3δ prevents apoptosis through sequestration of Bax. J. Biol. Chem. 2001, 276, 45201–45206. [Google Scholar] [CrossRef] [PubMed]
- Nomura, M.; Shimizu, S.; Sugiyama, T.; Narita, M.; Ito, T.; Matsuda, H.; Tsujimoto, Y. 14-3-3δ interacts directly with and negatively regulates pro-apoptotic Bax. J. Biol. Chem. 2003, 278, 2058–2065. [Google Scholar] [CrossRef] [PubMed]
- Datta, S.R.; Katsov, A.; Hu, L.; Petros, A.; Fesik, S.W.; Yaffe, M.B.; Greenberg, M.E. 14-3-3 proteins and survival kinases cooperate to inactivate BAD by BH3 domain phosphorylation. Mol. Cell 2000, 6, 41–51. [Google Scholar] [CrossRef]
- Chiang, C.W.; Harris, G.; Ellig, C.; Masters, S.C.; Subramanian, R.; Shenolikar, S.; Wadzinski, B.E.; Yang, E. Protein phosphatase 2A activates the proapoptotic function of BAD in interleukin-3-dependent lymphoid cells by a mechanism requiring 14-3-3 dissociation. Blood 2001, 97, 1289–1297. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, R.R.; Masters, S.C.; Zhang, H.; Fu, H. Functional conservation of 14-3-3 isoforms in inhibiting bad-induced apoptosis. Exp. Cell Res. 2001, 271, 142–151. [Google Scholar] [CrossRef] [PubMed]
- Ichim, G.; Tait, S.W.G. A fate worse than death: Apoptosis as an oncogenic process. Nat. Rev. Cancer 2016, 16, 539–548. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; He, Z.; Shen, J.; Huang, Q.; Li, W.; Liu, X.; He, Y.; Wolf, F.; Li, C.Y. Apoptotic caspases regulate induction of iPSCs from human fibroblasts. Cell Stem Cell 2010, 7, 508–520. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Huang, Q.; Chen, J.; Peng, Y.; Roop, D.R.; Bedford, J.S.; Li, C.Y. Apoptotic cells activate the “phoenix rising” pathway to promote wound healing and tissue regeneration. Sci. Signal. 2010, 3, ra13. [Google Scholar] [CrossRef] [PubMed]
- Huang, Q.; Li, F.; Liu, X.; Li, W.; Shi, W.; Liu, F.F.; O’Sullivan, B.; He, Z.; Peng, Y.; Tan, A.C.; et al. Caspase 3-mediated stimulation of tumor cell repopulation during cancer radiotherapy. Nat. Med. 2011, 17, 860–866. [Google Scholar] [CrossRef] [PubMed]
- Boland, K.; Flanagan, L.; Prehn, J.H. Paracrine control of tissue regeneration and cell proliferation by caspase-3. Cell Death Dis. 2013, 4, e725. [Google Scholar] [CrossRef] [PubMed]
- Mao, P.; Smith, L.; Xie, W.; Wang, M. Dying endothelial cells stimulate proliferation of malignant glioma cells via a caspase 3-mediated pathway. Oncol. Lett. 2013, 5, 1615–1620. [Google Scholar] [PubMed]
- Liu, Y.R.; Sun, B.; Zhao, X.L.; Gu, Q.; Liu, Z.Y.; Dong, X.Y.; Che, N.; Mo, J. Basal caspase-3 activity promotes migration, invasion, and vasculogenic mimicry formation of melanoma cells. Melanoma Res. 2013, 23, 243–253. [Google Scholar] [CrossRef] [PubMed]
- Donato, A.L.; Huang, Q.; Liu, X.; Li, F.; Zimmerman, M.A.; Li, C.Y. Caspase 3 promotes surviving melanoma tumor cell growth after cytotoxic therapy. J. Invest. Dermatol. 2014, 134, 1686–1692. [Google Scholar] [CrossRef] [PubMed]
- Cheng, J.; Tian, L.; Ma, J.; Gong, Y.; Zhang, Z.; Chen, Z.; Xu, B.; Xiong, H.; Li, H.; Huang, Q. Dying tumor cells stimulate proliferation of living tumor cells via caspase-dependent protein kinase C-δ activation in pancreatic ductal adenocarcinoma. Mol. Oncol. 2015, 9, 105–114. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; He, Y.; Li, F.; Huang, Q.; Kato, T.A.; Hall, R.P.; Li, C.Y. Caspase-3 promotes genetic instability and carcinogenesis. Mol. Cell 2015, 58, 284–296. [Google Scholar] [CrossRef] [PubMed]
- Feng, X.; Yu, Y.; He, S.; Cheng, J.; Gong, Y.; Zhang, Z.; Yang, X.; Xu, B.; Liu, X.; Li, C.Y.; et al. Dying glioma cells establish a proangiogenic microenvironment through a caspase 3 dependent mechanism. Cancer Lett. 2017, 385, 12–20. [Google Scholar] [CrossRef] [PubMed]
- Kroemer, G.; Galluzzi, L.; Vandenabeele, P.; Abrams, J.; Alnemri, E.S.; Baehrecke, E.H.; Blagosklonny, M.V.; El-Deiry, W.S.; Golstein, P.; Green, D.R.; et al. Classification of cell death: Recommendations of the Nomenclature Committee on Cell Death 2009. Cell Death Differ. 2009, 16, 3–11. [Google Scholar] [CrossRef] [PubMed]
- Berndtsson, M.; Hägg, M.; Panaretakis, T.; Havelka, A.M.; Shoshan, M.C.; Linder, S. Acute apoptosis by cisplatin requires induction of reactive oxygen species but is not associated with damage to nuclear DNA. Int. J. Cancer 2007, 120, 175–180. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.L.; Hong, S.W.; Shin, J.S.; Kim, J.S.; Ko, S.G.; Hong, N.J.; Kim, D.J.; Lee, W.J.; Jin, D.H.; Lee, M.S. p34SEI-1 inhibits doxorubicin-induced senescence through a pathway mediated by protein kinase C-δ and c-Jun-NH2-kinase 1 activation in human breast cancer MCF7 cells. Mol. Cancer Res. 2009, 7, 1845–1853. [Google Scholar] [CrossRef] [PubMed]
- Sliwinska, M.A.; Mosieniak, G.; Wolanin, K.; Babik, A.; Piwocka, K.; Magalska, A.; Szczepanowska, J.; Fronk, J.; Sikora, E. Induction of senescence with doxorubicin leads to increased genomic instability of HCT116 cells. Mech. Ageing Dev. 2009, 130, 24–32. [Google Scholar] [CrossRef] [PubMed]
- Puig, P.E.; Guilly, M.N.; Bouchot, A.; Droin, N.; Cathelin, D.; Bouyer, F.; Favier, L.; Ghiringhelli, F.; Kroemer, G.; Solary, E.; et al. Tumor cells can escape DNA-damaging cisplatin through DNA endoreduplication and reversible polyploidy. Cell Biol. Int. 2008, 32, 1031–1043. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Zhang, D.; Yang, Z.; Zhang, X. Tumor budding, micropapillary pattern, and polyploidy giant cancer cells in colorectal cancer: Current status and future prospects. Stem Cells Int. 2016, 2016, 4810734. [Google Scholar] [CrossRef] [PubMed]
- Niu, N.; Zhang, J.; Zhang, N.; Mercado-Uribe, I.; Tao, F.; Han, Z.; Pathak, S.; Multani, A.S.; Kuang, J.; Yao, J.; et al. Linking genomic reorganization to tumor initiation via the giant cell cycle. Oncogenesis 2016, 5, e281. [Google Scholar] [CrossRef] [PubMed]
- Mo, J.; Sun, B.; Zhao, X.; Gu, Q.; Dong, X.; Liu, Z.; Ma, Y.; Zhao, N.; Tang, R.; Liu, Y.; et al. Hypoxia-induced senescence contributes to the regulation of microenvironment in melanomas. Pathol. Res. Pract. 2013, 209, 640–647. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Wang, D.; Li, H. Initiation of premature senescence by BCL-2 in hypoxic condition. Int. J. Clin. Exp. Pathol. 2014, 7, 2446–2453. [Google Scholar] [PubMed]
- Fei, F.; Zhang, D.; Yang, Z.; Wang, S.; Wang, X.; Wu, Z.; Wu, Q.; Zhang, S. The number of polyploid giant cancer cells and epithelial-mesenchymal transition-related proteins are associated with invasion and metastasis in human breast cancer. J. Exp. Clin. Cancer Res. 2015, 34, 158. [Google Scholar] [CrossRef] [PubMed]
- Lv, H.; Shi, Y.; Zhang, L.; Zhang, D.; Liu, G.; Yang, Z.; Li, Y.; Fei, F.; Zhang, S. Polyploid giant cancer cells with budding and the expression of cyclin E, S-phase kinase-associated protein 2, stathmin associated with the grading and metastasis in serous ovarian tumor. BMC Cancer 2014, 14, 576. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Mercado-Uribe, I.; Hanash, S.; Liu, J. iTRAQ-based proteomic analysis of polyploid giant cancer cells and budding progeny cells reveals several distinct pathways for ovarian cancer development. PLoS ONE 2013, 8, e80120. [Google Scholar] [CrossRef] [PubMed]
- Zhivotovsky, B.; Kroemer, G. Apoptosis and genomic instability. Nat. Rev. Mol. Cell Biol. 2004, 5, 752–762. [Google Scholar] [CrossRef] [PubMed]
- Coppé, J.P.; Desprez, P.Y.; Krtolica, A.; Campisi, J. The senescence-associated secretory phenotype: The dark side of tumor suppression. Annu. Rev. Pathol. 2010, 5, 99–118. [Google Scholar] [CrossRef] [PubMed]
- Davalos, A.R.; Coppé, J.P.; Campisi, J.; Desprez, P.Y. Senescent cells as a source of inflammatory factors for tumor progression. Cancer Metastasis Rev. 2010, 29, 273–283. [Google Scholar] [CrossRef] [PubMed]
- Sikora, E.; Mosieniak, G.; Sliwinska, M.A. Morphological and functional characteristic of senescent cancer cells. Curr. Drug Targets 2016, 17, 377–387. [Google Scholar] [CrossRef] [PubMed]
- Castro-Vega, L.J.; Jouravleva, K.; Ortiz-Montero, P.; Liu, W.Y.; Galeano, J.L.; Romero, M.; Popova, T.; Bacchetti, S.; Vernot, J.P.; Londoño-Vallejo, A. The senescent microenvironment promotes the emergence of heterogeneous cancer stem-like cells. Carcinogenesis 2015, 36, 1180–1192. [Google Scholar] [CrossRef] [PubMed]
- Cantor, D.J.; David, G. SIN3B, the SASP, and pancreatic cancer. Mol. Cell. Oncol. 2014, 1, e969167. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, M.; Boothman, D.A. Stress-induced premature senescence (SIPS)—Influence of SIPS on radiotherapy. J. Radiat. Res. 2008, 49, 105–112. [Google Scholar] [CrossRef] [PubMed]
- Muscat, A.; Popovski, D.; Jayasekara, W.S.; Rossello, F.J.; Ferguson, M.; Marini, K.D.; Alamgeer, M.; Algar, E.M.; Downie, P.; Watkins, D.N.; et al. Low-dose histone deacetylase inhibitor treatment leads to tumor growth arrest and multi-lineage differentiation of malignant rhabdoid tumors. Clin. Cancer Res. 2016, 22, 3560–3570. [Google Scholar] [CrossRef] [PubMed]
- Foerster, F.; Chen, T.; Altmann, K.H.; Vollmar, A.M. Actin-binding doliculide causes premature senescence in p53 wild type cells. Bioorg. Med. Chem. 2016, 24, 123–129. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.S.; Yu, Y.C.; Lee, Y.J.; Chen, J.H.; Hsu, H.Y.; Chiu, S.J. Depletion of securin induces senescence after irradiation and enhances radiosensitivity in human cancer cells regardless of functional p53 expression. Int. J. Radiat. Oncol. Biol. Phys. 2010, 77, 566–574. [Google Scholar] [CrossRef] [PubMed]
- Fitzgerald, A.L.; Osman, A.A.; Xie, T.X.; Patel, A.; Skinner, H.; Sandulache, V.; Myers, J.N. Reactive oxygen species and p21Waf1/Cip1 are both essential for p53-mediated senescence of head and neck cancer cells. Cell Death Dis. 2015, 6, e1678. [Google Scholar] [CrossRef] [PubMed]
- Maier, P.; Hartmann, L.; Wenz, F.; Herskind, C. Cellular pathways in response to ionizing radiation and their targetability for tumor radiosensitization. Int. J. Mol. Sci. 2016, 17, 102. [Google Scholar] [CrossRef] [PubMed]
- Leikam, C.; Hufnagel, A.L.; Otto, C.; Murphy, D.J.; Mühling, B.; Kneitz, S.; Nanda, I.; Schmid, M.; Wagner, T.U.; Haferkamp, S.; et al. In vitro evidence for senescent multinucleated melanocytes as a source for tumor-initiating cells. Cell Death Dis. 2015, 6, e1711. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Wu, P.C.; Dong, D.Z.; Ivanova, I.; Chu, E.; Zeliadt, S.; Vesselle, H.; Wu, D.Y. Polyploidy road to therapy-induced cellular senescence and escape. Int. J. Cancer 2013, 132, 1505–1515. [Google Scholar] [CrossRef] [PubMed]
- Puck, T.T.; Marcus, P.I. Action of X-rays on mammalian cells. J. Exp. Med. 1956, 103, 653–666. [Google Scholar] [CrossRef] [PubMed]
- Puck, T.T.; Marcus, P.I. A rapid method for viable cell titration and clonal production with HeLa cells in tissue culture: The use of X-irradiated cells to supply conditioning factors. Proc. Natl. Acad. Sci. USA 1955, 41, 432–437. [Google Scholar] [CrossRef] [PubMed]
- Puck, T.T.; Marcus, P.I.; Cieciura, S.J. Clonal growth of mammalian cells in vitro; growth characteristics of colonies from single HeLa cells with and without a feeder layer. J. Exp. Med. 1956, 103, 273–283. [Google Scholar] [CrossRef] [PubMed]
- Narisawa-Saito, M.; Kiyono, T. Basic mechanisms of high-risk human papillomavirus-induced carcinogenesis: Roles of E6 and E7 proteins. Cancer Sci. 2007, 98, 1505–1511. [Google Scholar] [CrossRef] [PubMed]
- Mirzayans, R.; Andrais, B.; Scott, A.; Wang, Y.W.; Kumar, P.; Murray, D. Multinucleated giant cancer cells produced in response to ionizing radiation retain viability and replicate their genome. Int. J. Mol. Sci. 2017, 18, 360. [Google Scholar] [CrossRef] [PubMed]
- Kizaka-Kondoh, S.; Inoue, M.; Harada, H.; Hiraoka, M. Tumor hypoxia: A target for selective cancer therapy. Cancer Sci. 2003, 94, 1021–1028. [Google Scholar] [CrossRef] [PubMed]
- Muz, B.; de la Puente, P.; Azab, F.; Azab, A.K. The role of hypoxia in cancer progression, angiogenesis, metastasis, and resistance to therapy. Hypoxia 2015, 3, 83–92. [Google Scholar] [CrossRef] [PubMed]
- Aragonés, J.; Fraisl, P.; Baes, M.; Carmeliet, P. Oxygen sensors at the crossroad of metabolism. Cell Metab. 2009, 9, 11–22. [Google Scholar] [CrossRef] [PubMed]
- Ho, V.T.; Bunn, H.F. Effects of transition metals on the expression of the erythropoietin gene: Further evidence that the oxygen sensor is a heme protein. Biochem. Biophys. Res. Commun. 1996, 223, 175–180. [Google Scholar] [CrossRef] [PubMed]
- Piret, J.P.; Mottet, D.; Raes, M.; Michiels, C. CoCl2, a chemical inducer of hypoxia-inducible factor-1, and hypoxia reduce apoptotic cell death in hepatoma cell line HepG2. Ann. N. Y. Acad. Sci. 2002, 973, 443–447. [Google Scholar] [CrossRef] [PubMed]
- Piret, J.P.; Lecocq, C.; Toffoli, S.; Ninane, N.; Raes, M.; Michiels, C. Hypoxia and CoCl2 protect HepG2 cells against serum deprivation- and t-BHP-induced apoptosis: A possible anti-apoptotic role for HIF-1. Exp. Cell Res. 2004, 295, 340–349. [Google Scholar] [CrossRef] [PubMed]
- Weihua, Z.; Lin, Q.; Ramoth, A.J.; Fan, D.; Fidler, I.J. Formation of solid tumors by a single multinucleated cancer cell. Cancer 2011, 117, 4092–4099. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Mercado-Uribe, I.; Xing, Z.; Sun, B.; Kuang, J.; Liu, J. Generation of cancer stem-like cells through the formation of polyploid giant cancer cells. Oncogene 2014, 33, 116–128. [Google Scholar] [CrossRef] [PubMed]
- Erenpreisa, J.A.; Cragg, M.S.; Fringes, B.; Sharakhov, I.; Illidge, T.M. Release of mitotic descendants by giant cells from irradiated Burkitt’s lymphoma cell lines. Cell Biol. Int. 2000, 24, 635–648. [Google Scholar] [CrossRef] [PubMed]
- Illidge, T.M.; Cragg, M.S.; Fringes, B.; Olive, P.; Erenpresia, J.A. Polyploid giant cells provide a survival mechanism of p53 mutant cells after DNA damage. Cell Biol. Int. 2000, 24, 621–633. [Google Scholar] [CrossRef] [PubMed]
- Lagadec, C.; Vlashi, E.; Della Donna, L.; Dekmezian, C.; Pajonk, F. Radiation-induced reprogramming of breast cancer cells. Stem Cells 2012, 30, 833–844. [Google Scholar] [CrossRef] [PubMed]
- Erenpreisa, J.; Cragg, M.S. MOS, aneuploidy and the ploidy cycle of cancer cells. Oncogene 2010, 29, 5447–5451. [Google Scholar] [CrossRef] [PubMed]
- Sundaram, M.; Guernsey, D.L.; Rajaraman, M.M.; Rajaraman, R. Neosis: A novel type of cell division in cancer. Cancer Biol. Ther. 2004, 3, 207–218. [Google Scholar] [CrossRef] [PubMed]
- Walen, K.H. Spontaneous cell transformation: Karyoplasts derived from multinucleated cells produce new cell growth in senescent human epithelial cell cultures. In Vitro Cell Dev. Biol. Anim. 2004, 40, 150–158. [Google Scholar] [CrossRef]
- Navolanic, P.M.; Akula, S.M.; McCubrey, J.A. Neosis and its potential role in cancer development and chemoresistance. Cancer Biol. Ther. 2004, 3, 219–320. [Google Scholar] [CrossRef] [PubMed]
- Rajaraman, R.; Rajaraman, M.M.; Rajaraman, S.R.; Guernsey, R.L. Neosis—A paradigm of self-renewal in cancer. Cell Biol. Int. 2005, 29, 1084–1097. [Google Scholar] [CrossRef] [PubMed]
- Walen, K.H. Budded karyoplasts from multinucleated fibroblast cells contain centrosomes and change their morphology to mitotic cells. Cell Biol. Int. 2005, 29, 1057–1065. [Google Scholar] [CrossRef] [PubMed]
- Rajaraman, R.; Guernsey, D.L.; Rajaraman, M.M.; Rajaraman, S.R. Stem cells, senescence, neosis and self-renewal in cancer. Cell Biol. Int. 2006, 29, 1084–1097. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Q.; Zhang, Q.; Wang, S.; Xie, S.; Fang, W.; Liu, Z.; Liu, J.; Yao, K. A fraction of CD133+ CNE2 cells is made of giant cancer cells with morphological evidence of asymmetric mitosis. J. Cancer 2015, 6, 1236–1244. [Google Scholar] [CrossRef] [PubMed]
- Esmatabadi, M.J.; Bakhshinejad, B.; Motlagh, F.M.; Babashah, S.; Sadeghizadeh, M. Therapeutic resistance and cancer recurrence mechanisms: Unfolding the story of tumour coming back. J. Biosci. 2016, 41, 497–506. [Google Scholar] [CrossRef] [PubMed]
- Mosieniak, G.; Sliwinska, M.A.; Alster, O.; Strzeszewska, A.; Sunderland, P.; Piechota, M.; Was, H.; Sikora, E. Polyploidy formation in doxorubicin-treated cancer cells can favor escape from senescence. Neoplasia 2015, 17, 882–893. [Google Scholar] [CrossRef] [PubMed]
- Shah, O.J.; Lin, X.; Li, L.; Huang, X.; Li, J.; Anderson, M.G.; Tang, H.; Rodriguez, L.E.; Warder, S.E.; McLoughlin, S.; et al. BCL-XL represents a druggable molecular vulnerability during aurora B inhibitor-mediated polyploidization. Proc. Natl. Acad. Sci. USA 2010, 107, 12634–12639. [Google Scholar] [CrossRef] [PubMed]
- Martin, S.K.; Pu, H.; Penticuff, J.C.; Cao, Z.; Horbinski, C.; Kyprianou, N. Multinucleation and mesenchymal-to-epithelial transition alleviate resistance to combined cabazitaxel and antiandrogen therapy in advanced prostate cancer. Cancer Res. 2016, 76, 912–926. [Google Scholar] [CrossRef] [PubMed]
- Nik-Zainal, S.; Alexandrov, L.B.; Wedge, D.C.; van Loo, P.; Greenman, C.D.; Raine, K.; Jones, D.; Hinton, J.; Marshall, J.; Stebbings, L.A.; et al. Mutational processes molding the genomes of 21 breast cancers. Cell 2012, 149, 979–993. [Google Scholar] [CrossRef] [PubMed]
- Alexandrov, L.B.; Nik-Zainal, S.; Wedge, D.C.; Aparicio, S.A.; Behjati, S.; Biankin, A.V.; Bignell, G.R.; Bolli, N.; Borg, A.; Børresen-Dale, A.L.; et al. Signatures of mutational processes in human cancer. Nature 2013, 500, 415–421. [Google Scholar] [CrossRef] [PubMed]
- Alexandrov, L.B.; Nik-Zainal, S.; Wedge, D.C.; Campbell, P.J.; Stratton, M.R. Deciphering signatures of mutational processes operative in human cancer. Cell Rep. 2013, 3, 246–259. [Google Scholar] [CrossRef] [PubMed]
- Helleday, T.; Eshtad, S.; Nik-Zainal, S. Mechanisms underlying mutational signatures in human cancers. Nat. Rev. Genet. 2014, 15, 585–598. [Google Scholar] [CrossRef] [PubMed]
- Alexandrov, L.B.; Jones, P.H.; Wedge, D.C.; Sale, J.E.; Campbell, P.J.; Nik-Zainal, S.; Stratton, M.R. Clock-like mutational processes in human somatic cells. Nat. Genet. 2015, 47, 1402–1407. [Google Scholar] [CrossRef] [PubMed]
- Fox, E.J.; Salk, J.J.; Loeb, L.A. Exploring the implications of distinct mutational signatures and mutation rates in aging and cancer. Genome Med. 2016, 8, 30. [Google Scholar] [CrossRef] [PubMed]
- Behjati, S.; Gundem, G.; Wedge, D.C.; Roberts, N.D.; Tarpey, P.S.; Cooke, S.L.; van Loo, P.; Alexandrov, L.B.; Ramakrishna, M.; Davies, H.; et al. Mutational signatures of ionizing radiation in second malignancies. Nat. Commun. 2016, 7, 12605. [Google Scholar] [CrossRef] [PubMed]
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mirzayans, R.; Andrais, B.; Kumar, P.; Murray, D. Significance of Wild-Type p53 Signaling in Suppressing Apoptosis in Response to Chemical Genotoxic Agents: Impact on Chemotherapy Outcome. Int. J. Mol. Sci. 2017, 18, 928. https://doi.org/10.3390/ijms18050928
Mirzayans R, Andrais B, Kumar P, Murray D. Significance of Wild-Type p53 Signaling in Suppressing Apoptosis in Response to Chemical Genotoxic Agents: Impact on Chemotherapy Outcome. International Journal of Molecular Sciences. 2017; 18(5):928. https://doi.org/10.3390/ijms18050928
Chicago/Turabian StyleMirzayans, Razmik, Bonnie Andrais, Piyush Kumar, and David Murray. 2017. "Significance of Wild-Type p53 Signaling in Suppressing Apoptosis in Response to Chemical Genotoxic Agents: Impact on Chemotherapy Outcome" International Journal of Molecular Sciences 18, no. 5: 928. https://doi.org/10.3390/ijms18050928