The Complete Mitogenome of the Wood-Feeding Cockroach Cryptocercus meridianus (Blattodea: Cryptocercidae) and Its Phylogenetic Relationship among Cockroach Families

Abstract
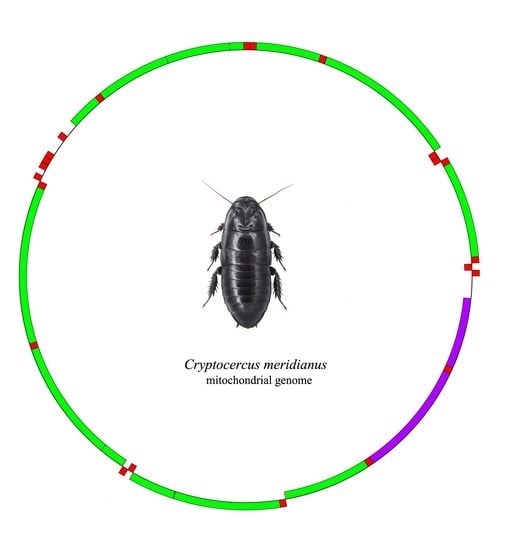
:
1. Introduction
2. Results and Discussion
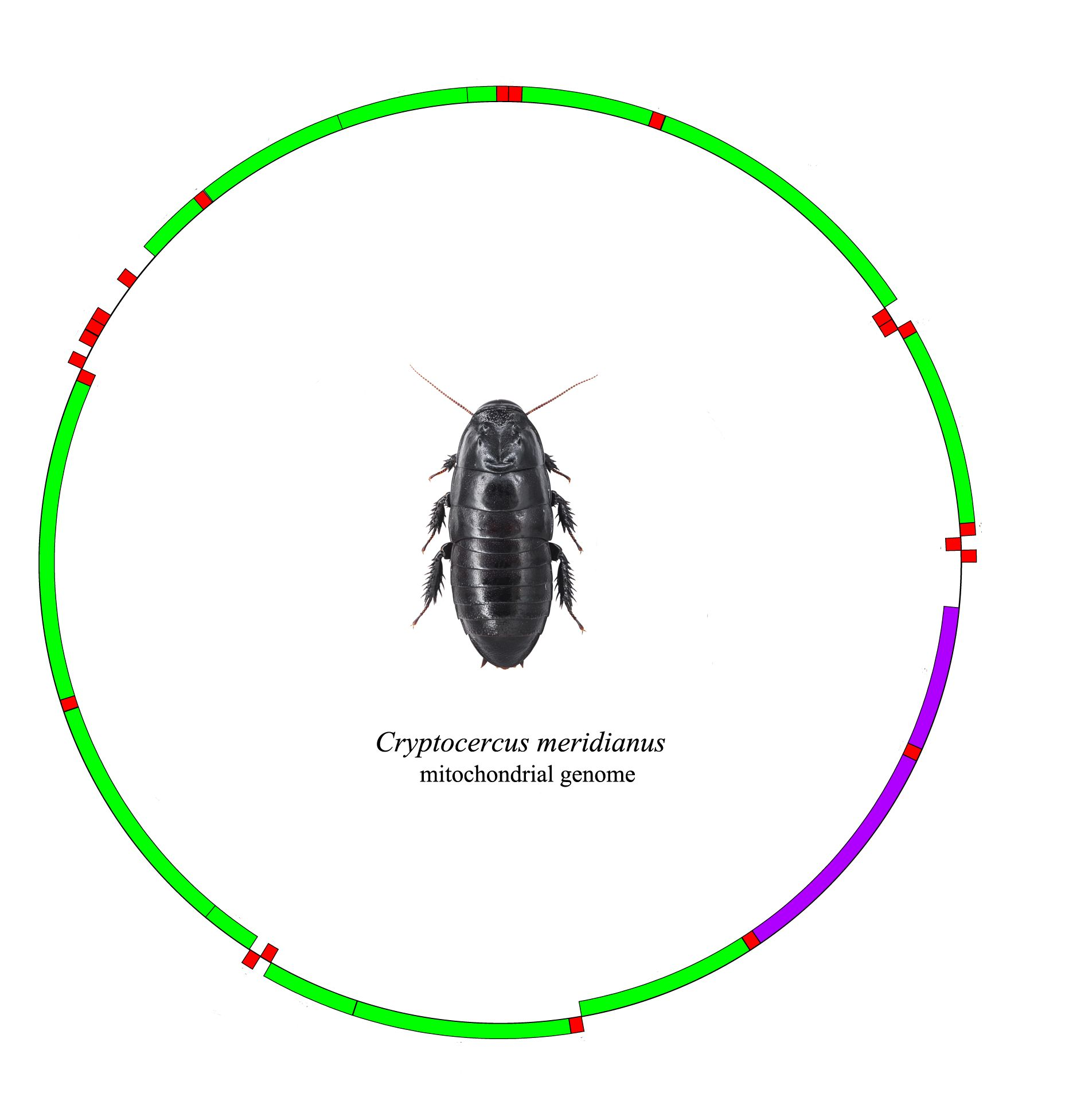
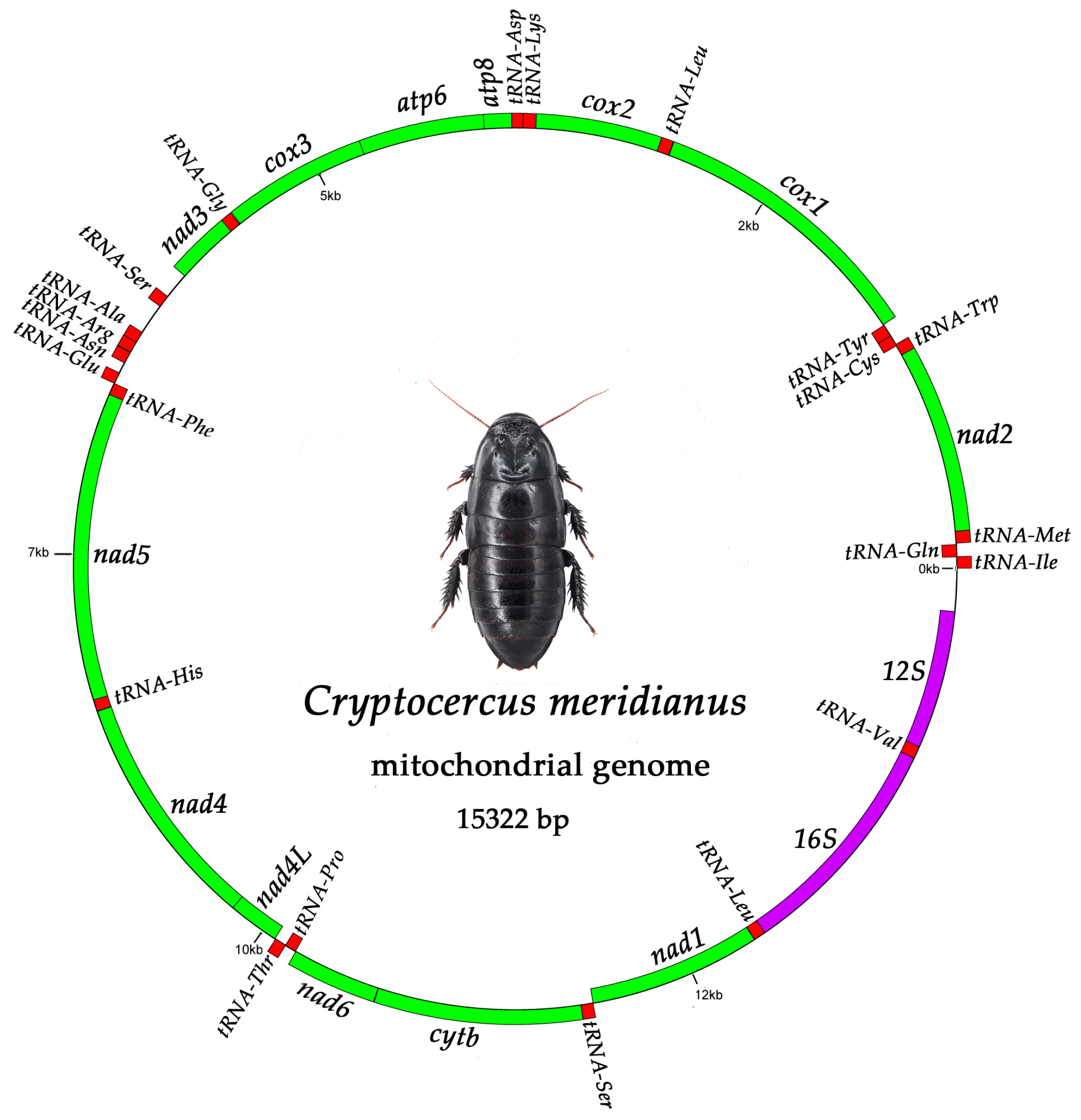
2.1. Mitogenome Organization and Nucleotide Composition
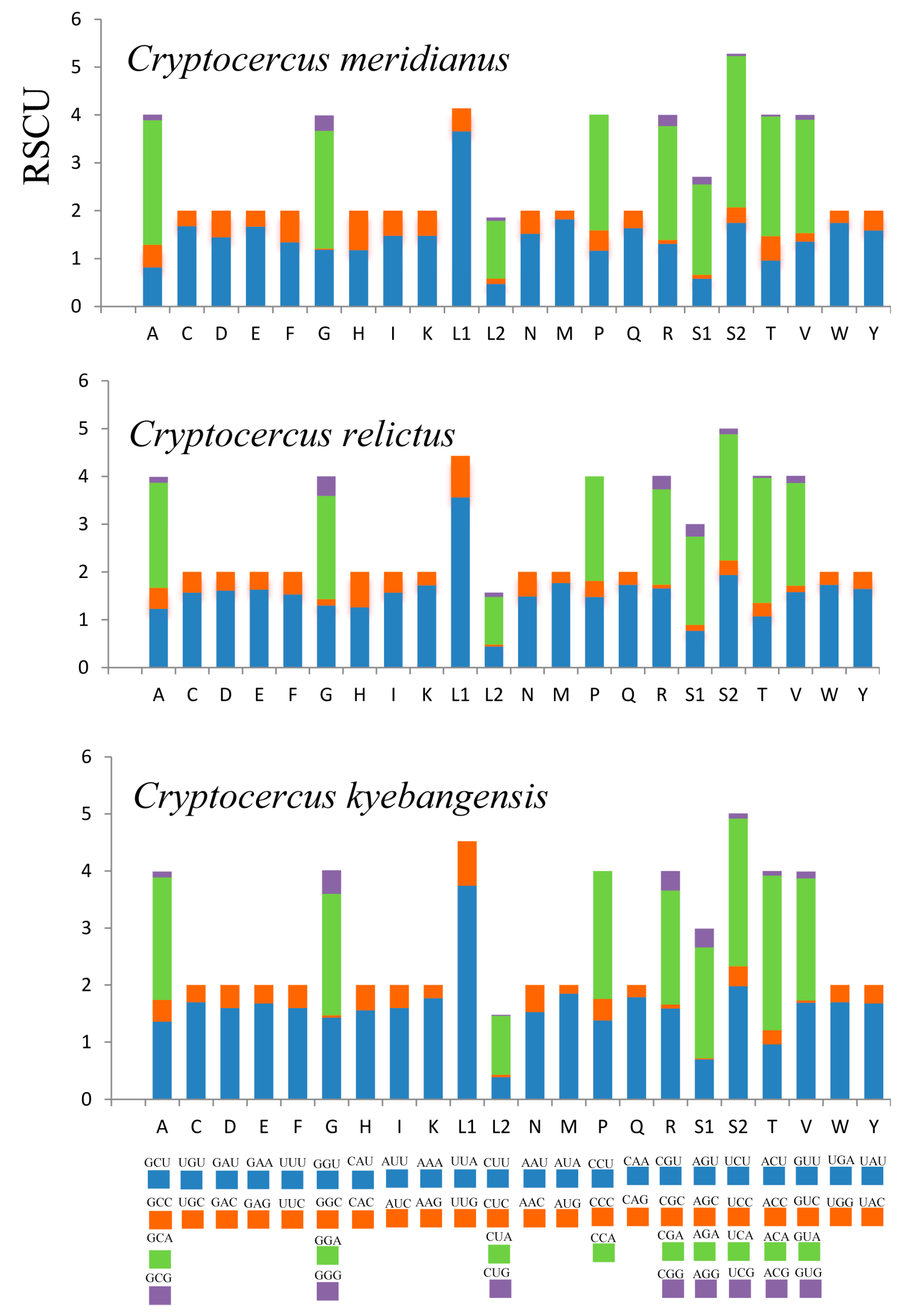
2.2. PCGs (Protein-Coding Genes)
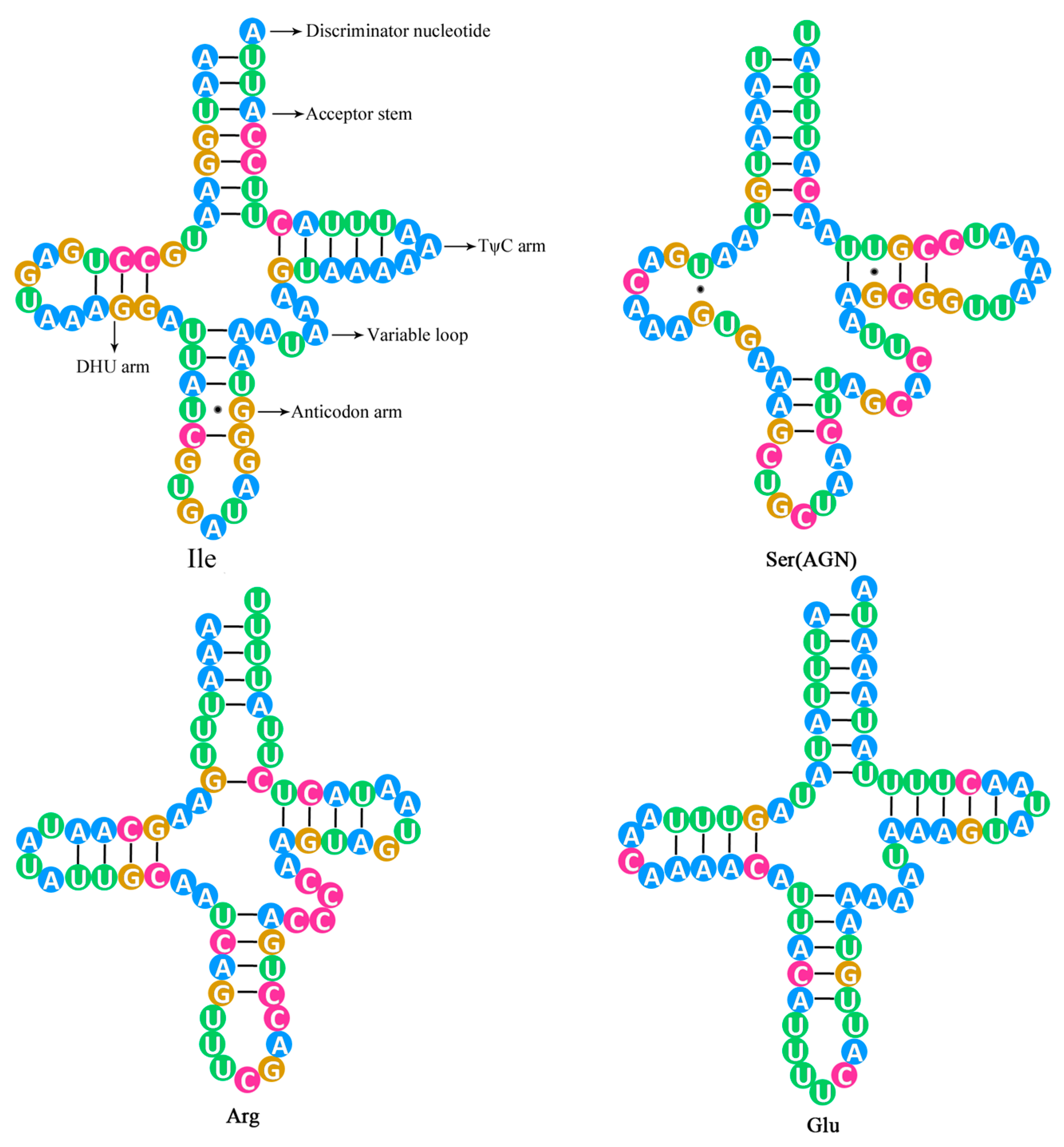
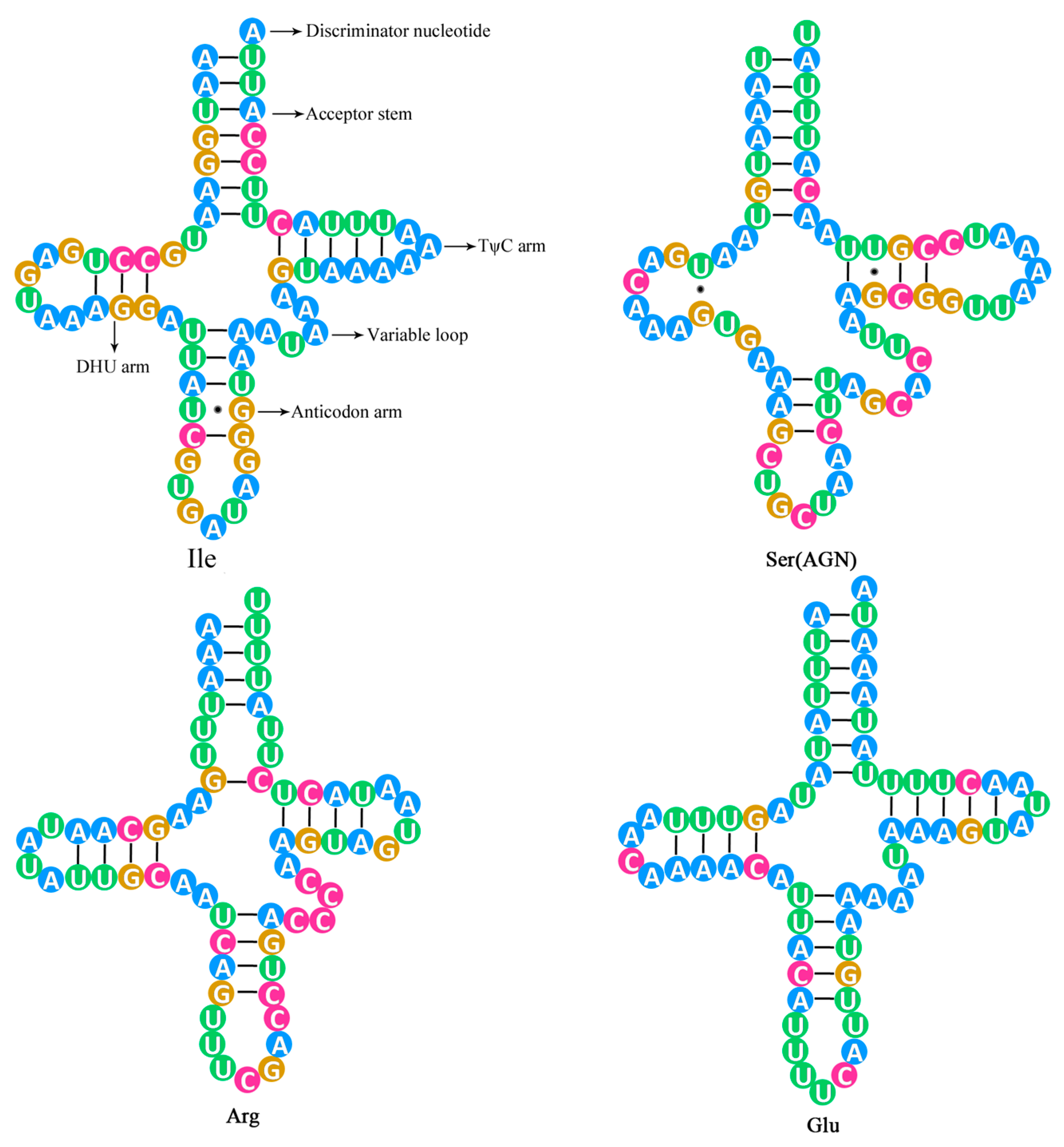
2.3. The tRNA Genes and rRNA Genes
2.4. The D-Loop Region
2.5. Intergenic Spacer and Overlapping Regions
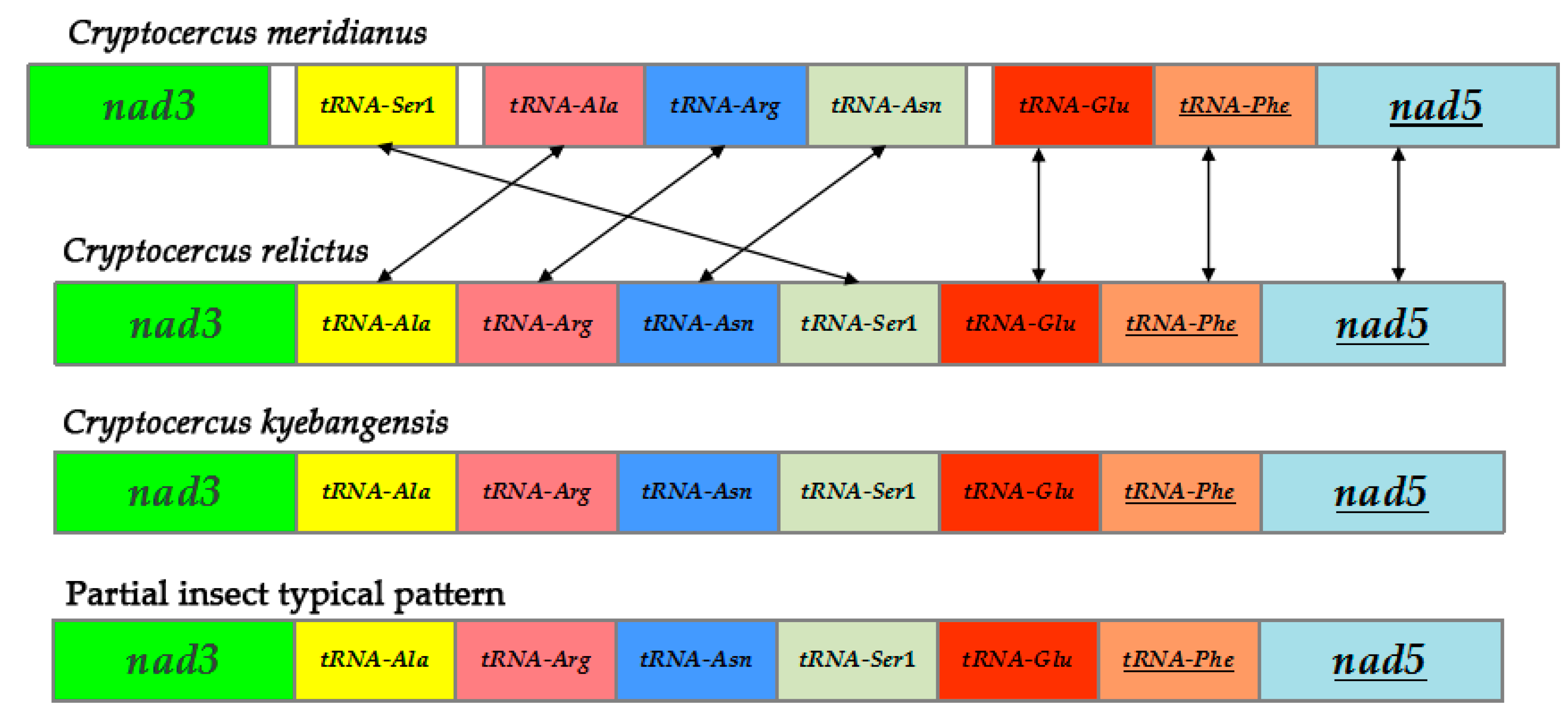
2.6. Rearrangement
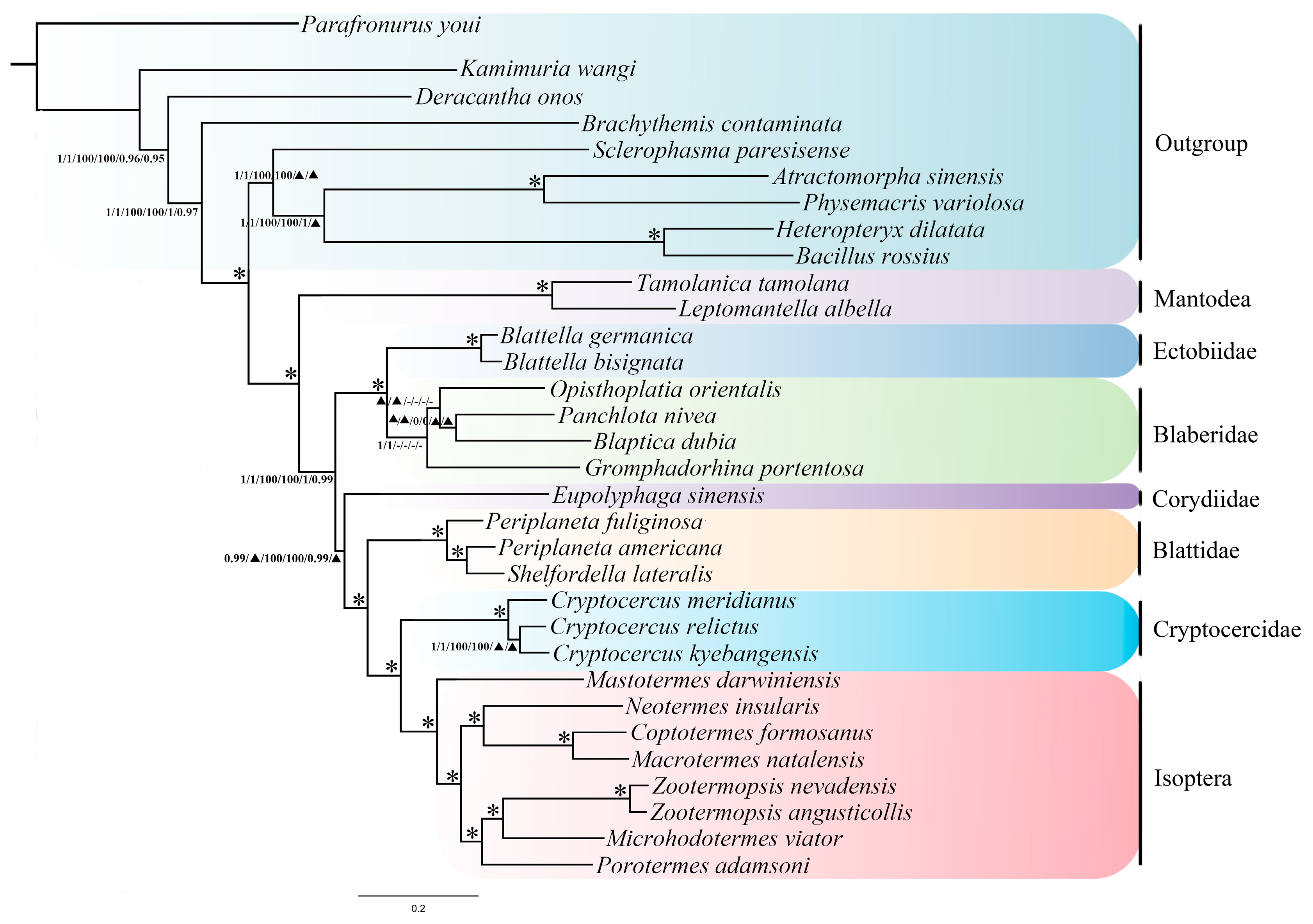
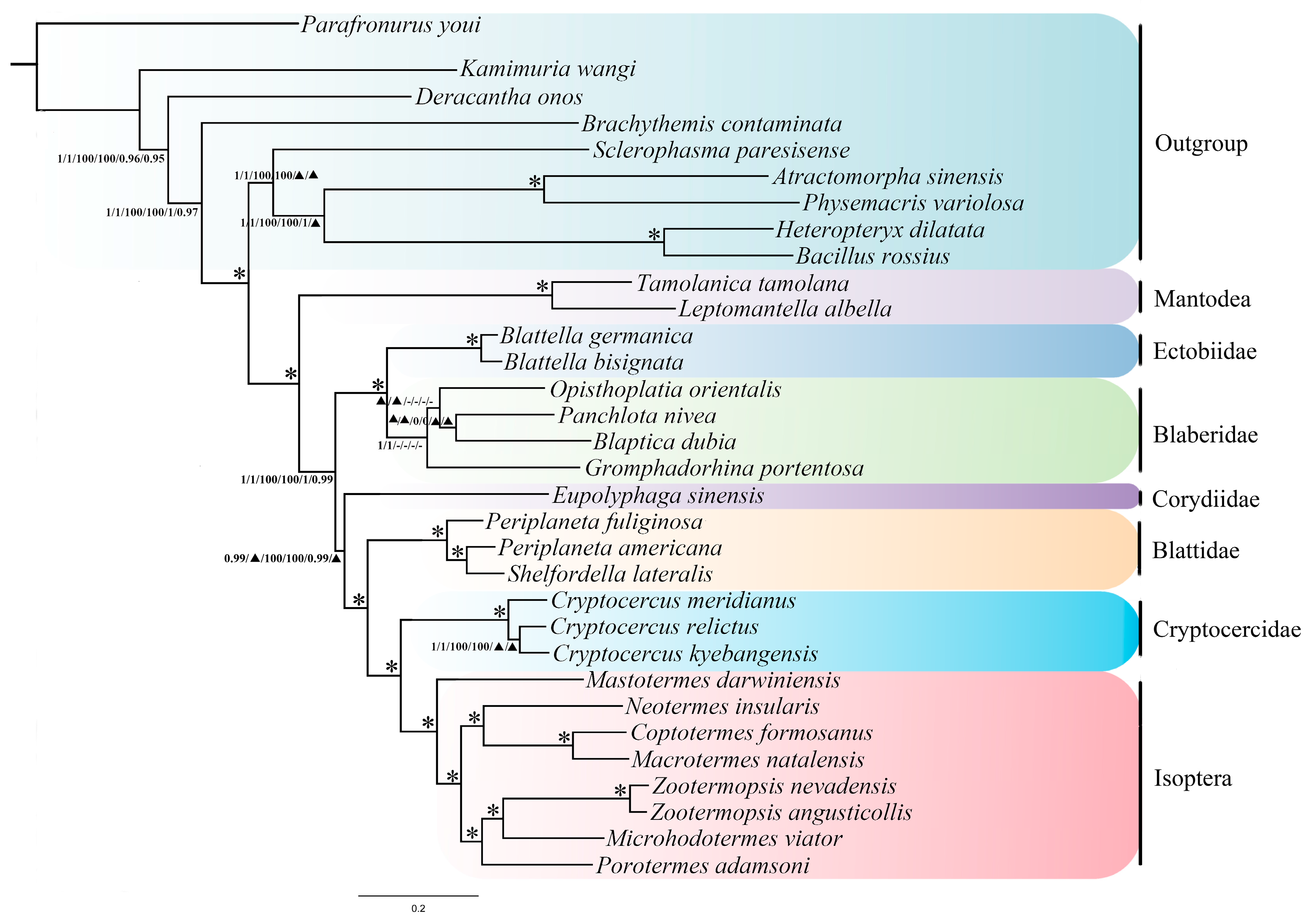
2.7. Phylogenetic Analyses
3. Materials and Methods
3.1. Sampling and DNA Extraction
3.2. PCR Amplification, Sequencing and Sequence Assembly
3.3. Sequence Analysis
3.4. Phylogenetic Analysis
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| atp6 | ATPase subunit 6 |
| atp8 | ATPase subunit 8 |
| cox1-3 | cytochrome c oxidase subunit I-III |
| cytb | cytochrome b |
| nad1-6, 4L | NADH dehydrogenase subunit 1-6, 4L |
| PCGs | protein-coding genes |
| tRNA | transfer RNA genes |
| rRNA | ribosomal RNA genes |
| 12/16S | small and large subunit of ribosomal RNA genes |
| ML | maximum likelihood analyses |
| BI | bayesian inference analyses |
| RSCU | relative synonymous codon usage |
| IGS | intergenic spacer region |
| BPP | bayesian posterior probabilities |
References
- Che, Y.; Wang, D.; Shi, Y.; Du, X.; Zhao, Y.; Lo, N.; Wang, Z. A global molecular phylogeny and timescale of evolution for Cryptocercus, woodroaches. Mol. Phylogenet. Evol. 2016, 98, 201–209. [Google Scholar] [CrossRef] [PubMed]
- Clark, J.W.; Kambhampati, S. Phylogenetic analysis of Blattabacterium, endosymbiotic bacteria from the wood roach, Cryptocercus (Blattodea: Cryptocercidae), including a description of three new species. Mol. Phylogenet. Evol. 2003, 26, 82–88. [Google Scholar] [CrossRef]
- Park, Y.C.; Maekawa, K.; Matsumoto, T.; Santoni, R.; Choe, J.C. Molecular phylogeny and biogeography of the Korean woodroaches Cryptocercus spp. Mol. Phylogenet. Evol. 2004, 30, 450–464. [Google Scholar] [CrossRef]
- Maekawa, K.; Nalepa, C.A. Biogeography and phylogeny of wood-feeding cockroaches in the genus Cryptocercus. Insects 2011, 2, 354–368. [Google Scholar] [CrossRef] [PubMed]
- Lo, N.; Beninati, T.; Stone, F.; Walker, J.; Sacchi, L. Cockroaches that lack Blattabacterium endosymbionts: The phylogenetically divergent genus Nocticola. Biol. Lett. 2007, 3, 327–330. [Google Scholar] [CrossRef] [PubMed]
- Inward, D.; Beccaloni, G.; Eggleton, P. Death of an order: A comprehensive molecular phylogenetic study confirms that termites are eusocial cockroaches. Biol. Lett. 2007, 3, 331–335. [Google Scholar] [CrossRef] [PubMed]
- Djernæs, M.; Klass, K.; Picker, M.D.; Damgaard, J. Phylogeny of cockroaches (Insecta, Dictyoptera, Blattodea), with placement of aberrant taxa and exploration of out-group sampling. Syst. Entomol. 2012, 37, 65–83. [Google Scholar] [CrossRef]
- Djernæs, M.; Klass, K.D.; Eggleton, P. Identifying possible sister groups of Cryptocercidae + Isoptera: A combined molecular and morphological phylogeny of Dictyoptera. Mol. Phylogenet. Evol. 2015, 84, 284–303. [Google Scholar] [CrossRef] [PubMed]
- Klass, K.D.; Nalepa, C.; Lo, N. Wood-feeding cockroaches as models for termite evolution (Insecta: Dictyoptera): Cryptocercus vs. Parasphaeria boleiriana. Mol. Phylogenet. Evol. 2008, 46, 809–817. [Google Scholar] [CrossRef] [PubMed]
- Avise, J.C. (Ed.) Molecular Markers, Natural History and Evolution; Chapman and Hall: New York, NY, USA, 1994. [Google Scholar]
- Saccone, C.; Gissi, C.; Reyes, A.; Larizza, A.; Sbisà, E.; Pesole, G. Mitochondrial DNA in metazoa: Degree of freedom in a frozen event. Gene 2002, 286, 3–12. [Google Scholar] [CrossRef]
- Mi, G.J.; Yung, C.P. The complete mitogenome of the wood-feeding cockroach Cryptocercus kyebangensis (Blattodea: Cryptocercidae) and phylogenetic relations among cockroach families. Anim. Cells Syst. 2015, 19, 432–438. [Google Scholar]
- Grandcolas, P.; Legendre, F.; Park, Y.C.; Bellés, X.; Murienne, J.; Pellens, R. The genus Cryptocercus in East Asia: Distribution and new species (Insecta, Dictyoptera, Blattaria, Polyphagidae). Zoosystema 2005, 27, 725–732. [Google Scholar]
- Conant, G.C.; Wolfe, K.H. GenomeVx: Simple web-based creation of editable circular chromosome maps. Bioinformatics 2008, 24, 861–862. [Google Scholar] [CrossRef] [PubMed]
- Wolstenholme, D.R. Animal mitochondrial DNA: Structure and evolution. Int. Rev. Cytol. 1992, 141, 173–216. [Google Scholar] [PubMed]
- Boore, J.L.; Lavrov, D.V.; Brown, W.M. Gene translocation links insects and crustaceans. Nature 1998, 392, 667–668. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Qiang, Y.; Peng, X.Y.; Qian, Z.Q.; Wang, Z.Z. The complete mitochondrial genome of the Sara Longwing Heliconius sara (Insecta: Lepidoptera: Nymphalidae). Mitochondrial DNA 2016, 27, 3167–3168. [Google Scholar] [PubMed]
- Ojala, D.; Montoya, J.; Attardi, G. tRNA punctuation model of RNA processing in human mitochondria. Nature 1981, 290, 470–474. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.T.; Du, Y.Z. Complete mitochondrial genome of Capnia zijinshana (Plecoptera: Capniidae) and phylogenetic analysis among stoneflies. J. Asia Pac. Entomol. 2017, 20, 305–312. [Google Scholar] [CrossRef]
- Cheng, X.F.; Zhang, L.P.; Yu, D.N.; Storey, K.B.; Zhang, J.Y. The complete mitochondrial genomes of four cockroaches (Insecta: Blattodea) and phylogenetic analyses within cockroaches. Gene 2016, 586, 115–122. [Google Scholar] [CrossRef] [PubMed]
- Taanman, J.W. The mitochondrial genome: Structure, transcription, translation and replication. Biochim. Biophys. Acta 1999, 1410, 103–123. [Google Scholar] [CrossRef]
- Inohira, K.; Hara, T.; Matsuura, E.T. Nucleotide sequence divergence in the A + T-rich region of mitochondrial DNA in Drosophila simulans and Drosophila mauritiana. Mol. Biol. Evol. 1997, 14, 814–822. [Google Scholar] [CrossRef] [PubMed]
- Yang, F.; Du, Y.Z.; Wang, L.P.; Cao, J.M.; Yu, W.W. The complete mitochondrial genome of the leafminer Liriomyza sativae (Diptera: Agromyzidae): Great difference in the A + T-rich region compared to Liriomyza trifolii. Gene 2011, 485, 7–15. [Google Scholar] [CrossRef] [PubMed]
- Salvato, P.; Simonato, M.; Battisti, A.; Negrisolo, E. The complete mitochondrial genome of the bag-shelter moth Ochrogaster lunifer (Lepidoptera, Notodontidae). BMC Genom. 2008, 9, 331–345. [Google Scholar] [CrossRef] [PubMed]
- Boore, J.L. Animal mitochondrial genomes. Nucleic Acids Res. 1999, 27, 1767–1780. [Google Scholar] [CrossRef] [PubMed]
- Pepe, G.; Holtrop, M.; Gadaleta, G.; Kroon, A.M.; Cantatore, P.; Gallerani, R.; De Benedetto, C.; Quagliariello, C.; Sbisa, E.; Saccone, C. Non-random patterns ofnucleotide substitutions and codon strategy in the mammalian mitochondrial genes coding for identified and unidentified reading frames. Biochem. Int. 1983, 6, 553–563. [Google Scholar] [PubMed]
- Wei, S.J. Progress in research on the comparative mitogenomics of insects. Chin. J. Appl. Entomol. 2011, 48, 1573–1585. [Google Scholar]
- Moritz, C.; Brown, W.M. Tandem duplications in animal mitochondrial DNAs: Variation in incidence and gene content among lizards. Proc. Natl. Acad. Sci. USA 1987, 84, 7183–7187. [Google Scholar] [CrossRef] [PubMed]
- Cantatore, P.; Gadaleta, M.N.; Roberti, M.; Saccone, C.; Wilson, A.C. Duplication and remoulding of tRNA genes during the evolutionary rearrangement of mitochondrial genomes. Nature 1987, 329, 853–855. [Google Scholar] [CrossRef] [PubMed]
- Poulton, J.; Deadman, M.E.; Bindoff, L.; Morten, K.; Land, J.; Brown, G. Families of mtDNA rearrangements can be detected in patients with mtDNA deletions: Duplications may be a transient intermediate form. Hum. Mol. Genet. 1993, 2, 23–30. [Google Scholar] [CrossRef] [PubMed]
- Lunt, D.H.; Hyman, B.C. Animal mitochondrial DNA recombination. Nature 1997, 387, 247. [Google Scholar] [CrossRef] [PubMed]
- Dowton, M.; Campbell, N.J.H. Intramitochondrial recombination—Is it why some mitochondrial genes sleep around? Trends Ecol. Evol. 2001, 16, 269–271. [Google Scholar] [CrossRef]
- Lavrov, D.V.; Boore, J.L.; Brown, W.M. Complete mtDNA sequences of two millipedes suggest a new model for mitochondrial gene rearrangements: Duplication and nonrandom loss. Mol. Biol. Evol. 2002, 19, 163–169. [Google Scholar] [CrossRef] [PubMed]
- Yamauchi, M.M.; Miya, M.U.; Nishida, M. Complete mitochondrial DNA sequence of the swimming crab, Portunus trituberculatus (Crustacea: Decapoda: Brachyura). Gene 2003, 311, 129–135. [Google Scholar] [CrossRef]
- Cameron, S.L.; Lo, N.; Bourguignon, T.; Svenson, G.J.; Evans, T.A. A mitochondrial genome phylogeny of termites (Blattodea: Termitoidae): Robust support for interfamilial relationships and molecular synapomorphies define major clades. Mol. Phylogenet. Evol. 2012, 65, 163–173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lowe, T.M.; Eddy, S.R. tRNAscan-SE: A program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 1997, 25, 955–964. [Google Scholar] [CrossRef] [PubMed]
- Laslett, D.; Canback, B. ARWEN, a program to detect tRNA genes in metazoan mitochondrial nucleotide sequences. Bioinformatics 2008, 24, 172–175. [Google Scholar] [CrossRef] [PubMed]
- Tamura, K.; Peterson, D.; Peterson, N.; Stecher, G.; Nei, M.; Kumar, S. MEGA 5: Molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance and maximum parsimony methods. Mol. Biol. Evol. 2011, 28, 2731–2739. [Google Scholar] [CrossRef] [PubMed]
- Librado, P.; Rozas, J. DnaSP v5: A software for comprehensive analysis of DNA polymorphism data. Bioinformatics 2009, 25, 1451–1452. [Google Scholar] [CrossRef] [PubMed]
- Zou, Z.; Min, Q.; Cheng, S.; Xin, T.; Xia, B. The complete mitochondrial genome of Thitarodes sejilaensis, (Lepidoptera: Hepialidae), a host insect of Ophiocordyceps sinensis, and its implication in taxonomic revision of Hepialus, adopted in China. Gene 2017, 601, 44–55. [Google Scholar] [CrossRef] [PubMed]
- Perna, N.T.; Kocher, T.D. Patterns of nucleotide composition at four-fold degenerate sites of animal mitochondrial genomes. J. Mol. Evol. 1995, 41, 353–358. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Zhou, C.; Gai, Y.; Song, D.; Zhou, K. The complete mitochondrial genome of Parafronurus youi, (Insecta: Ephemeroptera) and phylogenetic position of the Ephemeroptera. Gene 2008, 424, 18–24. [Google Scholar] [CrossRef] [PubMed]
- Abascal, F.; Zardoya, R.; Telford, M.J. TranslatorX: Multiple alignment of nucleotide sequences guided by amino acid translations. Nucleic Acids Res. 2010, 38, 7–13. [Google Scholar] [CrossRef] [PubMed]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [PubMed]
- Stamatakis, A.; Hoover, P.; Rougemont, J. A rapid bootstrap algorithm for the RAxML web servers. Syst. Biol. 2008, 57, 758–771. [Google Scholar] [CrossRef] [PubMed]
- Ronquist, F.; Teslenko, M.; Vander Mark, P.; Ayres, D.L.; Darling, A.; Hohna, S.; Larget, B.; Liu, L.; Suchard, M.A.; Huelsenbeck, J.P. MrBayes 3.2: Efficient Bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 2012, 61, 539–542. [Google Scholar] [CrossRef] [PubMed]
- Lanfear, R.; Calcott, B.; Ho, S.Y.; Guindon, S. Partitionfinder: Combined selection of partitioning schemes and substitution models for phylogenetic analyses. Mol. Biol. Evol. 2012, 29, 1695–1701. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Chu, Q.; Wei, D.D.; Qiu, Y.J.; Shang, F.; Dou, W.; Wang, J.J. The mitochondrial genome of booklouse, Liposcelis sculptilis (Psocoptera: Liposcelididae) and the evolutionary timescale of Liposcelis. Sci. Rep. 2016, 6, 30660. [Google Scholar] [CrossRef] [PubMed]
- Yu, P.; Cheng, X.; Ma, Y.; Yu, D.; Zhang, J. The complete mitochondrial genome of Brachythemis contaminata (Odonata: Libellulidae). Mitochondrial DNA 2014, 27, 2272–2273. [Google Scholar] [PubMed]
- Cameron, S.L.; Barker, S.C.; Whiting, M.F. Mitochondrial genomics and the new insect order Mantophasmatodea. Mol. Phylogenet. Evol. 2006, 38, 274–279. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; Huang, Y.; Shi, F.; Ye, H. The complete mitochondrial genome of Deracantha onos (Orthoptera: Bradyporidae). Mol. Biol. Rep. 2007, 36, 7–12. [Google Scholar] [CrossRef] [PubMed]
- Sheffield, N.C.; Hiatt, K.D.; Valentine, M.C.; Song, H.; Whiting, M.F. Mitochondrial genomics in Orthoptera using MOSAS. Mitochondrial DNA 2010, 21, 87–104. [Google Scholar] [CrossRef] [PubMed]
- Ding, F.M.; Shi, H.W.; Huang, Y. Complete mitochondrial genome and secondary structures of lrRNA and srRNA of Atractomorpha sinensis (Orthoptera, Pyrgomorphidae). Zool. Res. 2007, 28, 580–588. [Google Scholar]
- Qian, Y.H.; Wu, H.Y.; Ji, X.Y.; Yu, W.W.; Du, Y.Z. Mitochondrial genome of the Stonefly Kamimuria wangi (Plecoptera: Perlidae) and phylogenetic position of plecoptera based on mitogenomes. PLoS ONE 2014, 9, e86328. [Google Scholar]
- Plazzi, F.; Ricci, A.; Passamonti, M. The mitochondrial genome of Bacillus stick insects (Phasmatodea) and the phylogeny of orthopteroid insects. Mol. Phylogenet. Evol. 2011, 58, 304–316. [Google Scholar] [CrossRef] [PubMed]
- Kômoto, N.; Yukuhiro, K.; Ueda, K.; Tomita, S. Exploring the molecular phylogeny of phasmids with whole mitochondrial genome sequences. Mol. Phylogenet. Evol. 2011, 58, 43–52. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Yu, P.; Ma, Y.; Cheng, H.; Zhang, J. The complete mitochondrial genome of L. albella (Mantodea: Iridopterygidae). Mitochondrial DNA 2016, 27, 465–466. [Google Scholar] [CrossRef] [PubMed]
- Qian, Z.Q. The complete mitogenome of the dampwood termite Zootermopsis nevadensis (Insecta: Isoptera: Termopsidae). Mitochondrial DNA 2016, 27, 1163–1164. [Google Scholar] [CrossRef] [PubMed]
- Meng, Z.; Jiang, S.; Chen, X.; Lei, C. The complete mitochondrial genome of fungus-growing termite, Macrotermes natalensis (Isoptera: Macrotermitinae). Mitochondrial DNA 2014, 27, 1728–1729. [Google Scholar] [CrossRef] [PubMed]
- Tokuda, G.; Isagawa, H.; Sugio, K. The complete mitogenome of the Formosan termite, Coptotermes formosanus, Shiraki. Insectes Soc. 2012, 59, 17–24. [Google Scholar] [CrossRef]
- Xiao, B.; Chen, A.H.; Zhang, Y.Y.; Jiang, G.F.; Hu, C.C.; Zhu, C.D. Complete mitochondrial genomes of two cockroaches, Blattella germanica and Periplaneta americana, and the phylogenetic position of termites. Curr. Genet. 2012, 58, 65–77. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.Y.; Xuan, W.J.; Zhao, J.L.; Zhu, C.D.; Jiang, G.F. The complete mitochondrial genome of the cockroach Eupolyphaga sinensis (Blattaria: Polyphagidae) and the phylogenetic relationships within the Dictyoptera. Mol. Biol. Rep. 2010, 37, 3509–3516. [Google Scholar] [CrossRef] [PubMed]
- Chen, A.H. Complete mitochondrial genome of the double-striped cockroach Blattella bisignata (Insecta: Blattaria: Blaberoidea). Mitochondrial DNA 2013, 24, 14–16. [Google Scholar] [CrossRef] [PubMed]
- Yamauchi, M.M.; Miya, M.U.; Nishida, M. Use of a PCR-based approach for sequencing whole mitochondrial genomes of insects: Two examples (cockroach and dragonfly) based on the method developed for decapod crustaceans. Insect Mol. Biol. 2004, 13, 435–442. [Google Scholar] [CrossRef] [PubMed]
- Tian, X.; Ma, G.; Cui, Y.; Dong, P.; Zhu, Y.; Gao, X. The complete mitochondrial genomes of Opisthoplatia orientalis and Blaptica dubia (Blattodea: Blaberidae). Mitochondrial DNA 2015, 28, 139–140. [Google Scholar] [CrossRef] [PubMed]
- Kück, P.; Meid, S.A.; Groß, C.; Wägele, J.W.; Misof, B. AliGROOVE—Visualization of heterogeneous sequence divergence within multiple sequence alignments and detection of inflated branch support. BMC Bioinform. 2014, 15, 294. [Google Scholar] [CrossRef] [PubMed]
- Song, F.; Li, H.; Jiang, P.; Zhou, X.Z.; Liu, J.P.; Sun, C.H.; Vogler, A.P.; Cai, W.Z. Capturing the phylogeny of Holometabola with mitochondrial genome data and Bayesian site-heterogeneous mixture models. Genome Biol. Evol. 2016, 8, 1411–1426. [Google Scholar] [CrossRef] [PubMed]
- Lartillot, N.; Rodrigue, N.; Stubbs, D.; Richer, J. PhyloBayes MPI: Phylogenetic reconstruction with infinite mixtures of profiles in a parallel environment. Syst. Biol. 2013, 62, 611–615. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | C. meridianus | C. relictus | C. kyebangensis | |||
|---|---|---|---|---|---|---|
| Size (bp) | A + T (%) | Size (bp) | A + T (%) | Size (bp) | A + T (%) | |
| PCGs | 11,186 | 73.3 | 11,183 | 72.5 | 11,184 | 73.4 |
| tRNAs | 1450 | 74.9 | 1450 | 74.4 | 1451 | 74.6 |
| rRNAs | 2060 | 76.2 | 2063 | 75.8 | 2067 | 76.5 |
| D-loop | 240 | 77.5 | 672 | 80.3 | 1009 | 80.2 |
| Genome | 15,322 | 74.2 | 15,373 | 73.5 | 15,720 | 74.4 |
| Gene Name | Strand | Location | Size (bp) | IGS | Anticodon | Start/Stop Codon | |
|---|---|---|---|---|---|---|---|
| tRNA-Ile | H | 1–65 | 65 | GAT | |||
| tRNA-Gln | L | 64–132 | 69 | −2 | TTG | ||
| tRNA-Met | H | 140–205 | 66 | 7 | CAT | ||
| nad2 | H | 206–1233 | 1028 | 0 | ATG/TA* | ||
| tRNA-Trp | H | 1234–1299 | 66 | 0 | TCA | ||
| tRNA-Cys | L | 1292–1355 | 64 | −8 | GCA | ||
| tRNA-Tyr | L | 1356–1424 | 69 | 0 | GTA | ||
| cox1 | H | 1429–2964 | 1536 | 4 | TTG/TAA | ||
| tRNA-LeuUUR | H | 2967–3032 | 66 | 2 | TAA | ||
| cox2 | H | 3033–3717 | 685 | 0 | ATA/T** | ||
| tRNA-Lys | H | 3718–3787 | 70 | 0 | CTT | ||
| tRNA-Asp | H | 3788–3849 | 62 | 0 | GTC | ||
| atp8 | H | 3850–4008 | 159 | 0 | ATT/TAA | ||
| atp6 | H | 4002–4682 | 681 | −7 | ATG/TAA | ||
| cox3 | H | 4682–5470 | 789 | −1 | ATG/TAA | ||
| tRNA-Gly | H | 5475–5538 | 64 | 4 | TCC | ||
| nad3 | H | 5536–5892 | 357 | −3 | ATA/TAA | ||
| tRNA-SerAGN | H | 6040–6108 | 69 | 147 | GCT | ||
| tRNA-Ala | H | 6283–6347 | 65 | 174 | TGC | ||
| tRNA-Arg | H | 6348–6410 | 63 | 0 | TCG | ||
| tRNA-Asn | H | 6414–6478 | 65 | 3 | GTT | ||
| tRNA-Glu | H | 6541–6604 | 64 | 62 | TTC | ||
| tRNA-Phe | L | 6605–6671 | 67 | 0 | GAA | ||
| nad5 | L | 6672–8400 | 1729 | 0 | ATG/T** | ||
| tRNA-His | L | 8401–8464 | 64 | 0 | GTG | ||
| nad4 | L | 8467–9807 | 1341 | 2 | ATG/TAA | ||
| nad4L | L | 9801–10,088 | 288 | −7 | ATG/TAA | ||
| tRNA-Thr | H | 10,093–10,157 | 65 | 4 | TGT | ||
| tRNA-Pro | L | 10,158–10,221 | 64 | 0 | TGG | ||
| nad6 | H | 10,224–10,721 | 498 | 2 | ATT/TAA | ||
| cytb | H | 10,724–11,855 | 1132 | 2 | ATG/T** | ||
| tRNA-SerUCN | H | 11,856–11,923 | 68 | 0 | TGA | ||
| nad1 | L | 11,920–12,886 | 967 | −4 | ATG/T** | ||
| tRNA-LeuCUN | L | 12,888–12,954 | 67 | 1 | TAG | ||
| 16S | L | 12,955–14,237 | 1283 | 0 | |||
| tRNA-Val | L | 14,238–14,305 | 68 | 0 | TAC | ||
| 12S | L | 14,306–15,082 | 777 | 0 | |||
| D-loop | 15,083–15,322 | 240 | 0 | ||||
| Gene | C. meridianus | C. relictus | C. kyebangensis | |||
|---|---|---|---|---|---|---|
| AT Skew | GC Skew | AT Skew | GC Skew | AT Skew | GC Skew | |
| PCGs | 0.22 | −0.23 | 0.24 | −0.23 | 0.23 | −0.23 |
| tRNAs | 0.12 | −0.16 | 0.15 | −0.16 | 0.14 | −0.15 |
| rRNAs | 0.29 | −0.36 | 0.30 | −0.34 | 0.31 | −0.34 |
| D-loop | 0.37 | −0.52 | 0.15 | −0.09 | 0.27 | −0.33 |
| Genome | 0.22 | −0.25 | 0.23 | −0.23 | 0.24 | −0.24 |
| Order | Species | Accession Number | Reference |
|---|---|---|---|
| Ephemeroptera | Parafronurus youi | EU349015 | [42] |
| Odonata | Brachythemis contaminata | NC_026305 | [49] |
| Mantophasmatodea | Sclerophasma paresisense | NC_007701 | [50] |
| Orthoptera | Deracantha onos | EU137664 | [51] |
| Physemacris variolosa | NC_014491 | [52] | |
| Atractomorpha sinensis | NC_011824 | [53] | |
| Plecoptera | Kamimuria wangi | KC894944 | [54] |
| Phasmatodea | Bacillus rossius | GU001956 | [55] |
| Heteropteryx dilatata | AB477468 | [56] | |
| Mantodea | Tamolanica tamolana | NC_007702 | [35] |
| Leptomantella albella | NC_024028 | [57] | |
| Isoptera | Mastotermes darwiniensis | NC_018120 | [35] |
| Microhodotermes viator | NC_018122 | [35] | |
| Neotermes insularis | NC_018124 | [35] | |
| Zootermopsis nevadensis | NC_024658 | [58] | |
| Zootermopsis angusticollis | NC_018123 | [35] | |
| Macrotermes natalensis | NC_025522 | [59] | |
| Coptotermes formosanus | AB626147 | [60] | |
| Porotermes adamsoni | NC_018121 | [35] | |
| Blattodea | Cryptocercus meridianus | MG518617 | this study |
| Cryptocercus relictus | NC_018132 | [35] | |
| Cryptocercus kyebangensis | KP872847 | [12] | |
| Blattella germanica | NC_012901 | [61] | |
| Eupolyphaga sinensis | NC_014274 | [62] | |
| Blattella bisignata | NC_018549 | [63] | |
| Periplaneta americana | NC_016956 | [61] | |
| Periplaneta fuliginosa | AB126004 | [64] | |
| Opisthoplatia orientalis | KT893460 | [65] | |
| Panchlota nivea | KU684412 | [20] | |
| Shelfordella lateralis | KU684413 | [20] | |
| Gromphadorhina | KU684411 | [20] | |
| portentosa | |||
| Blaptica dubia | KU684410 | [20] |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, W.; Wang, Z.; Che, Y. The Complete Mitogenome of the Wood-Feeding Cockroach Cryptocercus meridianus (Blattodea: Cryptocercidae) and Its Phylogenetic Relationship among Cockroach Families. Int. J. Mol. Sci. 2017, 18, 2397. https://doi.org/10.3390/ijms18112397
Li W, Wang Z, Che Y. The Complete Mitogenome of the Wood-Feeding Cockroach Cryptocercus meridianus (Blattodea: Cryptocercidae) and Its Phylogenetic Relationship among Cockroach Families. International Journal of Molecular Sciences. 2017; 18(11):2397. https://doi.org/10.3390/ijms18112397
Chicago/Turabian StyleLi, Weijun, Zongqing Wang, and Yanli Che. 2017. "The Complete Mitogenome of the Wood-Feeding Cockroach Cryptocercus meridianus (Blattodea: Cryptocercidae) and Its Phylogenetic Relationship among Cockroach Families" International Journal of Molecular Sciences 18, no. 11: 2397. https://doi.org/10.3390/ijms18112397