PARP1 in Carcinomas and PARP1 Inhibitors as Antineoplastic Drugs
by
 ,
,
Luyao Wang
1,2,3,4,†, 
Chao Liang
1,2,3,4,†,
Fangfei Li
1,2,3,4,†,
Daogang Guan
1,2,3,4,
Xiaoqiu Wu
1,2,3,4,
Xuekun Fu
1,2,3,4,
Aiping Lu
1,2,3,4,* and
Ge Zhang
1,2,3,4,* 1
Law Sau Fai Institute for Advancing Translational Medicine in Bone and Joint Diseases, School of Chinese Medicine, Hong Kong Baptist University, Hong Kong, China
2
Institute of Integrated Bioinfomedicine and Translational Science, School of Chinese Medicine, Hong Kong Baptist University, Hong Kong, China
3
Institute of Precision Medicine and Innovative Drug Discovery, School of Chinese Medicine, Hong Kong Baptist University, Hong Kong, China
4
Shenzhen Lab of Combinatorial Compounds and Targeted Drug Delivery, HKBU Institute of Research and Continuing Education, Shenzhen 518000, China
*
Authors to whom correspondence should be addressed.
†
These authors contributed equally to this work.
Int. J. Mol. Sci. 2017, 18(10), 2111; https://doi.org/10.3390/ijms18102111
Submission received: 14 September 2017
/
Revised: 30 September 2017
/
Accepted: 2 October 2017
/
Published: 8 October 2017
(This article belongs to the Section Biochemistry)
Abstract
:Poly (ADP-ribose) polymerase 1 (PARP1), the best-studied isoform of the nuclear enzyme PARP family, plays a pivotal role in cellular biological processes, such as DNA repair, gene transcription, and so on. PARP1 has been found to be overexpressed in various carcinomas. These all indicate the clinical potential of PARP1 as a therapeutic target of human malignancies. Additionally, multiple preclinical research studies and clinical trials demonstrate that inhibition of PARP1 can repress tumor growth and metastasis. Up until now, PARP1 inhibitors are clinically used not only for monotherapy to suppress various tumors, but also for adjuvant therapy, to maintain or enhance therapeutic effects of mature antineoplastic drugs, as well as protect patients from chemotherapy and surgery-induced injury. To supply a framework for understanding recent research progress of PARP1 in carcinomas, we review the structure, expression, functions, and mechanisms of PARP1, and summarize the clinically mature PARP1-related anticancer agents, to provide some ideas for the development of other promising PARP1 inhibitors in antineoplastic therapy.
1. Introduction
Poly (ADP-ribose) polymerase 1 (PARP1) is increasingly attractive as an anticancer therapeutic target in both preclinical studies and clinical trials. Many PARP1 inhibitors have been approved for treatment of human malignancies or under clinical investigation, such as olaparib (AZD2281), veliparib (ABT-888), and rucaparib (AG-014699, PF-01367338), for treatment of ovarian cancer, breast cancer, prostate cancer, pancreatic cancer, and unspecified solid tumors [1,2,3]. PARP1 has an essential role in cell proliferation, survival, and death, due to its effects on regulation of multiple biological processes [4,5]. A variety of tumor tissues have shown elevated expression of PARP1 protein, which may associate with deterioration, metastasis, and angiogenesis in tumors [6,7]. However, the role of PARP1 in various tumors remains obscure. In this paper, the structure, expression, and functions of PARP1 in cancers will be reviewed, followed by the presentation and analysis of some mature PARP1 inhibitors against human malignancies.
PARP1, the base excision repair (BER) protein, is widely known for its role in sensing damaged DNA and catalyzing DNA repair [8,9]. Initial studies report that PARP1 is a promising target for treatment of BRCA-deficient carcinomas. BRCA-mutant carcinomas, which are homologous recombination (HR) deficient and rely on PARP1-BER for survival, are highly sensitive to PARP1 inhibitors [10,11]. Array data also demonstrates good tumoricidal efficiency of PARP1 inhibitors on these DNA repair defective carcinomas [5,12,13].
As PARP1 research goes more in-depth, functions and mechanisms of PARP1 have been found to be more complicated, and even double-faced. Furthermore, many PARP1 inhibitors were demonstrated to be not only effective against familial DNA repair defective carcinomas, but also against HR-proficient cancers, like HR-proficient HER2+ breast carcinoma and Ewing’s sarcoma, in pre-clinical or clinical studies [14,15,16]. Currently, design of PARP1 inhibitors and PARP1 inhibitors under clinical trial receive considerable attention [17,18]. In this paper, we will not only review PARP1 inhibitors, but also introduce the role of PARP1 in DNA repair, gene transcription, inflammation, cell cycling, and angiogenesis during tumor development.
2. PARP1 Structure and Expression in Carcinomas
PARP1 consists of four functional domains: the amino (N)-terminal DNA-binding domain (DBD), auto-modification domain (AD), WGR domain, and the carboxy (C)-terminal catalytic domain (CAT) [4,19]. DBD comprises two zinc fingers (ZFs: ZF1 and ZF2) for binding of PARP1 to single- and double-strand DNA breaks (SSBs, DSBs), and a nuclear localization signal (NLS), which is a DNA-nick sensor [20]. There is a third ZF (ZF3) which has been found exclusively in PARP1, rather than other PARP isoforms. It may be dispensable for DNA-binding, but plays an important role in PARP1 catalytic activity via relaying of “PARP1-DNA binding” signal to the catalytic domain [18,21]. AD is a central regulating domain, mainly containing a breast-cancer-susceptibility protein-carboxy terminus (BRCT) motif for auto-ADP ribosylation and mediating PARP1–protein interaction [4,22]. WGR, defined in accordance with its conserved Trp, Gly, and Arg residues, is an essential domain whose function remains unclear [19]. CAT consists of a helical subdomain (HD) and a conserved ADP-ribosyl transferase subdomain (ART). ART is composed of vital histidine (H) and tyrosine (Y) residues for NAD+ binding, and a glutamic acid (E) residue for polymerase activity [19,21,23,24] (Figure 1a).
PARP1, a nuclear enzyme, has now been identified in both the nucleus and cytoplasm of cancer cells. Investigation of PARP1 expression level in various tumors is essential to evaluate potential therapeutic effects, and side effects of PAPR1 inhibitors on each subtype of carcinomas.
A multitude of tumors have been found to be related with upregulated PARP1, when compared to adjacent normal tissues. Ossovskaya’s group investigated more than 8000 surgical tissue samples from malignant patients and healthy individuals. Results showed that PARP1 is overexpressed significantly in malignant tissues of BRCA-mutant, triple negative (TN) and receptor-positive breast carcinoma (BRCA-mutant/triple negative (TN) > receptor-positive), as well as uterine carcinoma, ovarian carcinoma, lung carcinoma, skin carcinoma, and non-Hodgkin’s lymphoma [25]. Of these, breast cancer has attracted the most attention. Clinical data of Domagala’s group showed that nuclear PARP1 expression was upregulated in most breast tumors, while the overexpression of nuclear–cytoplasmic PARP1 was present only in a small percentage of breast tumors [26]. Additionally, nuclear and nuclear–cytoplasmic PARP1 expressions were clinically found to be related to undesirable poorer prognosis and shorter overall survival in lymph node-negative early breast carcinoma [27,28]. Compared to non-small cell lung carcinoma (NSCLC), PARP1 protein level was higher in small cell lung cancer (SCLC), leading to higher sensitivity of SCLC to PARP1 inhibitors [29]. Brenner and members found that EWS-FLI1 genes in Ewing’s sarcoma could maintain PARP1 expression via a positive feedback loop [15]. Thus, Ewing’s sarcoma was significantly sensitive to PARP1 inhibitors. Galia’s group identified the upregulation of PARP1 expression in cellular nuclear compartments of glioblastoma multiforme, but not in the cytoplasm, via immunohistochemistry [30]. There were also other studies that demonstrated the overexpression of PARP1 in prostate carcinoma, colorectal carcinoma, pediatric central nervous system carcinomas, and testicular germ cell tumors, respectively [31,32,33,34] (Table 1). The above studies suggest that the expression level of PARP1 in cancer cells should be related to the sensitivity of various carcinomas to PARP1 inhibitors.
3. PARP1 Function and Mechanism in Carcinomas
3.1. Multifaceted Function on DNA Repair
PARP1-assisted DNA repair in cancers is complicated [4,14]. On one hand, tumor cells always harbor DNA repair defects to maintain the DNA lesions that can foster carcinogenesis. Additionally, damaging DNA of cancer cells is always used as the treatment mechanism of some anticancer agents. PARP1 inhibitors can further suppress the DNA repair process and drive cancer cell death, subsequently inhibiting cancers independently, or as anticancer assistant agents. On the other hand, DNA lesion in cells induced by excessive reactive oxygen species (ROS) is one of the major factors of carcinogenesis. In microenvironment homeostasis of cellular, this can be averted by PARP1-BER pathway. However, oxidative clustered DNA lesions (OCDLs), which mean excessive DNA lesions occurred frequently in tumors, may disturb the microenvironment homeostasis and lead to more severe DNA insult. Sustained activation of PARP1 and insufficient DNA repair will lead to further mutagenesis, metastasis, and energy-depleted necrosis of tumors. In this condition, PARP1 inhibitors could suppress mutagenesis and metastasis, as well as turn necrosis to apoptosis aiming to avoid inflammation-mediated extra cytotoxicity (Figure 2).
PARP1-assisted DNA repair is a nicotinamide adenine dinucleotide (NAD+)-related energy consuming process. After DNA-binding domain (DBD) of PARP1 binding to the damaged DNA, the NAD+ will be cleaved into ADP-ribose and nicotinamide, which is catalyzed by the catalytic domain (CAT) of PARP1. Then poly (ADP-ribose) (pADPr) is synthesized on acceptor proteins (e.g., PARP1, histones, or transcription factors) through the combination of ADP-ribose, also assisted by the catalysis of CAT. Subsequently, PARP1 leaves damaged DNA, owing to the dense negative charge of pADPr, allowing the recruitment of related repair proteins and replication [35] (Figure 1b). This process downregulates NAD+ and ATP levels in the cell, which can be recycled in a normal microenvironment [18]. However, over-activation of PARP1-related DNA repair will disturb the energetic balance in the cell, leading to rapid energy depletion, and then, cell necrosis [14,18]. Inhibition of PARP1 prevents the exhaustion of ATP in cells, subsequently minimizing related side effects resulting from chemotherapy and surgery-induced injury.
Nowadays, a progressively increasing number of studies focus on investigating the structural aspect of the mechanism of PARP1-related DNA repair. The X-ray co-crystal structure between PARP1 and NAD+ revealed the extensive hydrogen bonding interactions between the amide moiety of NAD+ with the hydroxyl of Ser904, and carbonyl and NH group of Gly863. Additionally, other interactions in the network of PARP1 and NAD+, such as π–π stacking interactions between the amide moiety of NAD+ and Tyr907 of PARP1, were also discovered [18] (Figure 1b). Langelier and members provided the X-ray crystal structure to illustrate the binding mode of human PARP1 (ZF1, ZF3, and WGR-CAT) with DNA DSBs, and advised that the upregulation of CAT domain dynamics is essential to DNA-dependent activation of PARP1 [19] (Figure 1c). Recent study of David Neuhaus’ research group [24] demonstrated the complexity of ZF1–ZF2 domain, with dumbbell DNA as the minimal structural unit for first-stage interaction between PARP1 and SSBs. The binding ability of ZF1–ZF2 domain to SSBs is slightly less strong than PARP1. In this study, NMR/X-ray hybrid structure of ZF1–ZF2 bound to an SSB was presented, which showed how two flexibly linked zinc fingers of PARP1 bound to the break of damaged DNA. Nuclear Overhauser effect (NOE) contacts within ZF1–ZF2, ZF1-5′ stem/T23 of DNA, and ZF2-3′ stem of DNA indicated the interaction interfaces of domain–domain and protein–DNA. Gibson’s group identified two gatekeeper residues of PARP1, and depicted the structural interaction between gatekeeper residues and NAD+. They also demonstrated that inhibition of PARP1 could promote the pausing of RNA polymerase II, which is another key factor of carcinomas [36].
3.2. PARP1 in Gene Transcription
PARP1 associates with DNA through binding or interacting with nucleosomes, and other chromatin-related proteins, including gene transcription factors, transcription machinery, and chromatin modulators [23,37]. Dysregulation of gene transcription has been clinically demonstrated as a vital influencing factor of cancer cell proliferation, invasion, metastasis, and response to therapy. PARP1 is carcinogenic in tumors with and without DNA lesions, due to its functions in both DNA repair and transcriptional regulation, positively or negatively. The activation function of PARP1 on the transcription of many oncogenes, such as vascular endothelial growth factor receptor 1 (VEGFR1) gene and hypoxia-inducible factor 1α and 2A (HIP1α and HIF2A) genes, was tumorigenic [5,38,39]. For instance, PARP1, supporting the transcription of androgen receptor in androgen receptor-positive prostate carcinoma, is necessary for tumor cell proliferation [31]. Recent research demonstrated that PARP1 would induce transcriptional activation of the melanocyte-lineage survival oncogene (MITF), indicating its role in melanomagenesis [40]. Similar effect exists were observed when PARP1 binds to transcription factors [5]. For example, the binding of PARP1 to NF-κB could activate the expression of melanoma growth stimulatory activity-regulated protein (CXCL1), subsequently improving the progression of melanoma [41]. Thus, inhibition of PARP1 may be associated with transcriptional silencing of oncogenes. PARP1 does not always modulate transcription of genes positively. In malignancies, downregulating transcription of tumor suppressors by PARP1 can also promote tumor growth and progression [42]. The repressive effects of PARP1 on classical tumor suppressing agents p53 and APC, in cancers, are good examples [43]. Accordingly, PARP1 inhibitors may inhibit tumor progression via recovering the tumor suppressing function of related factors.
PARP1 also plays a significant role in maintaining integrity of genes [37]. Besides the transcription modulation pathway above, PARP1 also regulates gene expression through chromatin remodeling, RNA polymerase II, and DNA methylation pathways [44,45,46]. The functional study of PARP1 on chromatin modulation has lasted for several decades, since 1999 [47]. In general, PARP1 regulates chromatin structure through modulating the activity and location of histone in chromatin [42,48]. The binding of PARP1 and nucleosome can modulate chromatin to a super-nucleosomal structure which suppresses transcription [48]. PARP1 can also disassociate linker histone (H1), subsequently improving RNA polymerase II-mediated gene transcription [49]. DNA methylation is also a common reason for tumorigenesis in various carcinomas, similar to gene mutation. PARP1 inhibition can downregulate the transcription of methylated DNA [50]. PARP1-mediated modulation of gene transcription has been demonstrated to play some vital roles in suppressing tumors. However, the exact effects of PARP1 on transcription and DNA methylation pathways at specific genes are complicated and obscure, indicating the manipulation of gene transcription via PARP-1 in tumors is still too immature and tender for clinical cancer therapy (Figure 3).
3.3. PARP1 in Tumor-Promoting Inflammation
Chronic inflammation, which will result in ROS-induced DNA lesion, may be another major factor of carcinogenesis. Hyperactivated PARP1 upregulates inflammatory signal factors like NF-κB, subsequently leading to inflammation and further tumorigenesis [14]. NF-κB, the classical tumor-promoting inflammatory signal, is a class of transcriptional factor which can activate pro-inflammatory transcription machinery. P65 (RelA), p50, p52, Rel B, and c-Rel all belong to NF-κB family [51]. Additionally, the interaction between NF-κB and PARP1 upregulates the level of pro-inflammatory cytokines like tumor necrosis factor α (TNFα) and interleukin 6 (IL6), which will also initiate tumor-promoting inflammation [52].
On the other hand, NF-κB-mediated chronic inflammation facilitates tumors to some malignant phenotypes which can escape from immune surveillance [53]. NF-κB also plays a significant role in carcinoma progression, metastasis, and angiogenesis [54]. Thus, inhibition of overexpressed PARP1 in human malignances may work on repressing chronic inflammatory-induced cancer progression, through blocking the NF-κB activation effect of PARP1. In addition, studies found that NF-κB was hyperactivated in HER2+ breast carcinoma, and could block cancer cells apoptosis [55]. Interestingly, another study demonstrated that HER2+ breast carcinoma was highly sensitive to PARP1 inhibitors, due to the suppressing effect of PARP1 inhibitors on NF-κB inflammatory signal [16]. This is a good supporting instance for the theory that PARP1 inhibition is also effective in DNA repair proficient carcinomas. PARP1 inhibitors can not only reduce inflammation-related side effects caused by chemotherapy and surgery injury, but also be used as potential preventative and antitumoral agents against carcinomas.
Besides the NF-κB-mediated inflammatory mechanisms, inflammation can induce cancer in other pathways. In chronic inflammatory microenvironment, upregulation of PARP1 will lead to an increase of iNOS, which is an integral unit of ROS/iNOS, subsequently inducing cellular DNA lesions. Thus, PARP1 inhibitors also serve as cell protectors against enhanced ROS/iNOS and inflammation-induced carcinogenesis (Figure 3) [56].
3.4. PARP1 in Cell Cycling Regulation
Carcinomas are characterized by slow metabolism, a low cell death rate, and high proliferation rate. Weakened mitosis, downregulation of cell death factors, evasion of growth repressing factors, or upregulation of anti-death factors may all act as tumor drivers. PARP1 modulates the cancer cellular life cycle via regulating cellular mitosis and cell death pathways, including apoptosis, necrosis, and necroptosis [5]. Mitosis is important for appropriate chromosome segregation. Cells with dysregulated mitosis survive, but cannot conduct appropriate chromosome segregation, subsequently leading to genomic mutants, and even tumorigenesis. This may also be one of the reasons for tumor deterioration [57].
PARP1 also modulates cancer cell cycle via controlling cell death (Figure 3). Initially, PARP1 in cancer cells will activate the apoptosis pathway. Nevertheless, when DNA damage is severe, overactivated PARP1 will lead to exhausting NAD+/ATP-induced necrosis, as described above. Accordingly, inhibition of PARP1 preserves energy, and keeps PARP1 at relatively normal levels, subsequently causing the generation of apoptosis cancer cells which can be cleared by macrophages [4]. In addition, PARP1 also regulates necroptosis of cancer cells through extracellular signal-regulated kinases (ERK), and C-jun N-terminal kinase (JNK) pathways. ERK, which can improve anti-apoptotic protein expression and inhibit activity of the caspase family (apoptosis-inducing enzymes), serves to protect cancer cells from death [58]. PARP1 can stimulate ERK, inducing survival of malignant cells. JNK, which activates PARP1 continuously, will lead to NAD+/ATP depletion-induced necrosis. Interestingly, PARP1 can also simulate JNK, which makes a JNK–PARP1–JNK sustained activating loop and further drives nonapoptotic cell death [59]. PARP1 inhibition, which blocks the JNK–PARP1–JNK loop and ERK-mediated anti-apoptotic protein expression, will result in cancer apoptosis. In general, downregulation of PARP1 could modulate many factors in the cell life cycle and cell death to suppress proliferation of carcinomas, which has been demonstrated in PARP−/− mice models and human breast cancer cells, and so on [49,60].
3.5. PARP1 in Metastasis and Angiogenesis
Multiple research studies have demonstrated the anti-angiogenesis and even anti-metastasis effect of PARP1 inhibitors. ERK, which can inhibit cancer cell apoptosis as mentioned above, was also found to be highly related to tumor metastasis and angiogenesis. Stimulation of ERK by PARP1 will improve the activity of pro-angiogenic factors, such as vascular endothelial growth factor (VEGF), platelet/endothelial cell adhesion molecule (PECAM1/CD31), transmembrane signal protein syndecan-4 (SDC-4), and hypoxia inducible factor (HIF), inducing angiogenesis and metastasis [61,62]. In addition, immunohistochemistry studies on clinical human epithelial ovarian tumor samples found that PARP1 was significantly associated with microvascular density (MVD, CD34), tumor volume, and lymphatic metastasis. In vitro research demonstrated that PARP1 inhibition could repress related human umbilical vein endothelial cell (HUVEC) tubule formation [63]. The study also found that PARP1 was positively related to vimentin (angiogenic intermediary filament) of endothelial cells in malignancies. Inhibition of PARP1 reduced vasculogenic mimicry in melanoma cells, indicating the potential of PARP1 inhibitors on attenuating tumor metastasis [64]. In addition, as mentioned above, the classic tumor-promoting inflammatory factor NF-κB is significantly associated with carcinomas angiogenesis and metastasis. Metastasis may also link to PARP1-mediated chronic inflammation in carcinomas (Figure 3).
Furthermore, PARP1-mediated carcinoma metastasis may be in gene transcription manner. For instance, Choi’s research demonstrated that inhibition of PARP1 blocked lung carcinoma metastasis in a DNA repair-independent mechanism. PARP1 upregulates metastasis of lung cancers by improving PARP1-mediated transcription of S1000A4 and CLDN7. Besides, it also promotes metastasis of lung carcinoma in brain and bone via facilitating cancer cell extravasation, invasion, and self-renewal, resisting apoptosis, as well as changing the brain microenvironment to induce relapse of lung carcinoma cells to the brain [65].
4. PARP1 Inhibitors against Carcinomas
PARP1 inhibitors, as used in monotherapy or maintenance therapy, have clinically presented promising efficiency against ovarian, breast, and other carcinomas [66,67]. Up until now, preclinically studied PARP1 inhibition methodologies are divided into two types: pharmacological inhibition, and genetic knockdown. PARP1 inhibitors include chemical compounds and nucleic acids [65]. In general, the antineoplastic mechanism of currently mature PARP1 inhibitors mainly focuses on DNA repair pathways [67,68].
The earliest discovered PARP1 inhibitors included nicotinamide/ benzamide derivatives, which repress PARP1-induced DNA repair and improve the sensitivity of cancer cells to DNA damaging agents [69,70,71]. Nicotinamide, the catabolite of NAD+ in PARP1-mediated DNA repair pathway, has become the investigating structural model for PARP1 inhibitors. Relevant PARP1 inhibitors have entered late-stage clinical trials or been approved as anticancer drugs, working with other anticancer chemotherapy drugs or being used for monotherapy. Thereinto, olaparib (AZD2281, trade name: Lynparza, given orally), the first single drug for PARP1-mediated carcinoma therapy, clinically showed great inhibition ability against BRCA-mutation carcinomas [72]. Its indications included ovarian carcinoma, breast carcinoma, pancreatic carcinoma, prostate carcinoma, and solid tumors like Ewing’s sarcoma, and so on [1,66] (Table 2). In 2014, the European Medicines Agency (EMA) approved olaparib as a maintenance therapeutic drug for patients with BRCA-mutation ovarian carcinoma following platinum chemotherapy. The United States Food and Drug Administration (FDA) also approved olaparib as a PARP1/2-related drug against BRCA-defective ovarian carcinoma with three or more pro-chemotherapies [73]. Preclinical and clinical investigations of olaparib against other malignancies are in progress. However, as the substrate of the p-glycoprotein efflux pump, olaparib might be resisted by some patients [74]. Studies initially discovered that some subtypes of breast cancer might be resistant to olaparib [75]. In addition, side effects, including bone marrow problems called myelodysplastic syndrome (MDS) or acute myeloid leukemia (AML), are nonnegligible.
Following olaparib, an array of PARP1-based antitumor agents are being discovered, have entered clinical trials, or have been approved to be used as anticancer drugs. Niraparib (MK-4827, given orally) is another well investigated PARP1 inhibitor approved by the FDA as a maintenance therapy drug with platinum-sensitive recurrent ovarian carcinoma [76]. Other adaptation diseases also include breast carcinoma, melanoma, and unspecified solid tumors [77]. Rucaparib (AG-014669, PF-01367338, trade name: Rubraca, given orally), an FDA granted and recently approved PARP1-based drug for BRCA-mutant ovarian carcinoma patients, is indicated for therapy one line earlier than olaparib [78]. It has also been investigated against breast, pancreatic, fallopian tube, and peritoneal carcinomas [79]. Further investigations have significantly found the tumoricidal effect of rucaparib in human non-BRCA-mutant ovarian cancer for both monotherapy and combination therapy [78,80]. Veliparib (ABT-888), a benzimidazole-4-carboxamide derivative, has been preclinically studied against breast, ovarian, lung, pancreatic, prostate, testicular and colorectal cancers, and other unspecified solid tumors. Most of clinical studies of veliparib were for combination therapy (www.clinicaltrials.gov) [81,82,83]. Talazoparib (BMN-673) has been reported to be used as both a mono-therapeutic and combination therapeutic drug with leukemia, Ewing’s sarcoma, BRCA-mutant and proficient ovarian carcinoma, breast carcinoma, and osteosarcoma [84,85,86,87]. It is a promising PARP1-based anticancer drug candidate, because its PARP–DNA complex trapping ability is almost 100-fold stronger than that of olaparib and rucaparib [88] (Table 2). Apart from these well-known PARP1 inhibitors, other novel compounds also demonstrate potential for PARP1 inhibition before clinical trials. For example, one natural product, ZINC67913374, is more stable than olaparib when interacting with PARP1 in molecular dynamics simulations [89]. With pharmacophore and co-crystallization studies, a novel PARP1 inhibitor OL-1 was found to induce cell death and inhibit cell migration in BRAC1 mutant MDA-MB-436 cells [90].
General well-investigated eligibilities of PARP1 inhibitors include ovarian and breast carcinomas, both BRCA-mutant and BRCA-proficient. Additionally, most of the currently discovered PARP1 drugs are taken orally, which could be one of the advantages of PARP1 drugs, offering patients a more comfortable and acceptable therapeutic experience. Due to the DNA repair-related tumoricidal mechanism of currently mature PARP1 anticancer drugs, reduction in pADPr formation has generally been used to evaluate therapeutic efficiency of PARP1 inhibitors as pharmacodynamic (PD) biomarkers. However, clinical results showed that there was no obvious correlation between the clinical tumoricidal efficiency and pADPr-related PD read-out for some PARP1 inhibiting drugs, such as olaparib and rucaparib. As poly (ADPr) is generated not only by PARP1, but also by other PARP isoforms, we speculate that this could be attributed to the selective inhibitory potency of the drugs on PARP1/2, rather than other PARP isoforms. Thus, other PD biomarkers, including the one which indicates PARP1 expression level and basic activity, as well as genomic approaches, should be taken into account in predicting or evaluating the sensitivity of PARP1 inhibitors [91,92].
From the perspective of the structural angle, present mature PARP1 inhibitors entered clinical trials always contain nicotinamide-mimic motifs to inhibit PARP1/NAD+ interaction via competing with the nicotinamide pocket of NAD+ [93]. All the PARP1-inhibiting drugs in clinical use contain a benzamide unit in a ring. This ring is essential for activity of PARP1 inhibitors because it holds the amide in the plane of the benzene ring. In olaparib, rucaparib, and talazoparib, this amide-containing ring is covalent. In veliparib and niraparib, the ring is formed from five covalent bonds and a strong intramolecular hydrogen bond, which holds the conformation of the pharmacophore. Nicotinamide/benzamide-mimic pharmacophores have become a classic structural basis for investigating and designing PARP1 inhibitors. However, due to the structural characteristic, many mature PARP1 inhibitors should have the potential to block most of PARP family enzymes which can interact with NAD+ [94]. Nowadays, more and more novel chemical compounds or nucleic acids, which do not imitate nicotinamide and may be PARP1-specific inhibitors, are gradually emerging. Computational methodology, which screens libraries of chemical compounds, proteins, and nucleic acids to find key features of interactions between drug candidates and targets, has already become a vital tool for PARP1-based drug discovery [95].
5. Conclusions and Future Perspectives
In this paper, the structure, expression, and functions of PARP1 in cancers were reviewed. PARP1 has been preclinically or clinically found to be overexpressed in various carcinomas, such as various subtypes of breast cancers, ovarian cancer, and lung cancer. Furthermore, the expression level of PARP1 in these cancers is different. This may be related to the sensitivity of carcinomas to PARP1 inhibitors. This should be considered when precisely choosing therapeutic strategies for patients with carcinomas. Besides recognizing lesion DNA and catalyzing DNA repair, PAPR1 also regulates cancer cells via modulating other cancer-related biological processes, including gene transcription, inflammation, cell life cycle, angiogenesis, and so on. Due to its multiple functions, PARP1 inhibitors can not only cure BRCA-deficient cancers, but also is efficient for BRCA-proficient cancers. PARP1 inhibitors are reported to be effective on blocking cancer metastasis.
As mono-therapeutic or combination therapeutic drugs, the mature PARP1 inhibitors against human malignancies were overviewed. Following the FDA-approved drugs olaparib, niraparib, and rucaparib, other well-investigated PARP1 inhibitors such as veliparib and talazoparib have entered clinical trials. The general well-investigated eligibilities of these PARP1 inhibiting drugs included ovarian and breast carcinomas, both of which presented high sensitivity to these drugs. Currently, mature PARP1 inhibitors mainly focus on DNA repair pathways. From the structural angle, all these PARP1 inhibitors contain amide motif as an active group. Investigating new PARP1-based antineoplastic drugs according to this rule may be easy. However, this also limits the development of PARP1 inhibitors. Thus, further studies on the expression level and location of PARP1 in numerous cancers to assist to find more eligibilities of PARP1 inhibitors may be significant. Investigating and designing chemical compounds and nucleic acids, which can inhibit tumor progression and metastasis based on other PARP1-related functions instead of DNA repair pathways, may expand the indications range of PARP1 inhibitors, and be helpful to break the bottleneck in investigating novel PARP1 drugs.
Acknowledgments
We thank the other academic staff members in Aiping Lu and Ge Zhang’s group at Hong Kong Baptist University. This review was supported by the Natural Science Foundation Council of China (81703049 to F.L.) and the Science and Technology Innovation Commission of Shenzhen Municipality Funds (JCYJ20170307161659648 to F.L.).
Author Contributions
Luyao Wang, Chao Liang, and Fangfei Li wrote the whole manuscript; Daogang Guan, Xiaoqiu Wu, Xuekun Fu contributed the manuscript for literature research; Aiping Lu and Ge Zhang revised and approved the manuscript.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Kaufman, B.; Shapira-Frommer, R.; Schmutzler, R.K.; Audeh, M.W.; Friedlander, M.; Balmana, J.; Mitchell, G.; Fried, G.; Stemmer, S.M.; Hubert, A.; et al. Olaparib monotherapy in patients with advanced cancer and a germline BRCA1/2 mutation. J. Clin. Oncol 2015, 33, 244–250. [Google Scholar] [CrossRef] [PubMed]
- Solimando, D.A., Jr.; Waddell, J.A. Drug Monographs: Olaratumab and Rucaparib. Hosp. Pharm. 2017, 52, 258–263. [Google Scholar] [CrossRef] [PubMed]
- Kummar, S.; Chen, A.; Ji, J.; Zhang, Y.; Reid, J.M.; Ames, M.; Jia, L.; Weil, M.; Speranza, G.; Murgo, A.J.; et al. Phase I study of PARP inhibitor ABT-888 in combination with topotecan in adults with refractory solid tumors and lymphomas. Cancer Res. 2011, 71, 5626–5634. [Google Scholar] [CrossRef] [PubMed]
- Jagtap, P.; Szabó, C. Poly(ADP-ribose) polymerase and the therapeutic effects of its inhibitors. Nat. Rev. Drug Discov. 2005, 4, 421–440. [Google Scholar] [CrossRef] [PubMed]
- Weaver, A.N.; Yang, E.S. Beyond DNA Repair: Additional Functions of PARP-1 in Cancer. Front. Oncol. 2013, 3, 290. [Google Scholar] [CrossRef] [PubMed]
- Espinoza, L.A. Advances in Prostate Cancer. In The Role of PARP Activation in Prostate Cancer; Hamilton, G., Ed.; InTech: Lexington, KY, USA, 2013. [Google Scholar]
- Wielgos, M.; Rajbhandari, R.; Cooper, T.S.; Wei, S.; Nozell, S.; Yang, E.S. Let-7 Status is crucial for PARP1 expression in HER2-over-expressing breast tumors. Mol. Cancer Res. 2017, 15, 340–347. [Google Scholar] [CrossRef] [PubMed]
- Satoh, M.S.; Lindahl, T. Role of poly(ADP-ribose) formation in DNA repair. Nature 1992, 356, 356–358. [Google Scholar] [CrossRef] [PubMed]
- Beck, C.; Robert, I.; Reina-San-Martin, B.; Schreiber, V.; Dantzer, F. Poly(ADP-ribose) polymerases in double-strand break repair: focus on PARP1, PARP2 and PARP3. Exp. Cell Res. 2014, 329, 18–25. [Google Scholar] [CrossRef] [PubMed]
- Bryant, H.E.; Schultz, N.; Thomas, H.D.; Parker, K.M.; Flower, D.; Lopez, E.; Kyle, S.; Meuth, M.; Curtin, N.J.; Helleday, T. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature 2005, 434, 913–920. [Google Scholar] [CrossRef] [PubMed]
- Farmer, H.; McCabe, N.; Lord, C.J.; Tutt, A.N.J.; Johnson, D.A.; Richardson, T.B.; Santarosa, M.; Dillon, K.J.; Hickson, I.; Knights, C.; et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature 2005, 434, 917–921. [Google Scholar] [CrossRef] [PubMed]
- Ashworth, A. A synthetic lethal therapeutic approach: poly(ADP) ribose polymerase inhibitors for the treatment of cancers deficient in DNA double-strand break repair. J. Clin. Oncol. 2008, 26, 3785–3790. [Google Scholar] [CrossRef] [PubMed]
- Ledermann, J.A. PARP inhibitors in ovarian cancer. Ann. Oncol. 2016, 27, i40–i44. [Google Scholar] [CrossRef] [PubMed]
- Swindall, A.F.; Stanley, J.A.; Yang, E.S. PARP-1: Friend or Foe of DNA Damage and Repair in Tumorigenesis? Cancers 2013, 5, 943–958. [Google Scholar] [CrossRef] [PubMed]
- Brenner, J.C.; Feng, F.Y.; Han, S.; Patel, S.; Goyal, S.V.; Bou-Maroun, L.M.; Liu, M.; Lonigro, R.; Prensner, J.R.; Tomlins, S.A.; et al. PARP-1 inhibition as a targeted strategy to treat Ewing’s sarcoma. Cancer Res. 2012, 72, 1608–1613. [Google Scholar] [CrossRef] [PubMed]
- Nowsheen, S.; Cooper, T.; Bonner, J.A.; LoBuglio, A.F.; Yang, E.S. HER2 overexpression renders human breast cancers sensitive to PARP inhibition independently of any defect in homologous recombination DNA repair. Cancer Res. 2012, 72, 4796–4806. [Google Scholar] [CrossRef] [PubMed]
- Malyuchenko, N.V.; Kotova, E.Y.; Kulaeva, O.I.; Kirpichnikov, M.P.; Studitskiy, V.M. PARP1 Inhibitors: antitumor drug design. Acta Naturae. 2015, 7, 27–37. [Google Scholar] [PubMed]
- Rouleau, M.; Patel, A.; Hendzel, M.J.; Kaufmann, S.H.; Poirier, G.G. PARP inhibition: PARP1 and beyond. Nat. Rev. Cancer 2010, 10, 293–301. [Google Scholar] [CrossRef] [PubMed]
- Langelier, M.F.; Planck, J.L.; Roy, S.; Pascal, J.M. Structural basis for DNA damage-dependent poly(ADP-ribosyl)ation by human PARP-1. Science 2012, 336, 728–732. [Google Scholar] [CrossRef] [PubMed]
- Ali, A.A.E.; Timinszky, G.; Arribas-Bosacoma, R.; Kozlowski, M.; Hassa, P.O.; Hassler, M.; Ladurner, A.G.; Pearl, L.H.; Oliver, A.W. The zinc-finger domains of PARP1 cooperate to recognize DNA strand breaks. Nat. Struct. Mol. Biol. 2012, 19, 685–692. [Google Scholar] [CrossRef] [PubMed]
- Tao, Z.; Gao, P.; Hoffman, D.W.; Liu, H.-W. Domain C of Human Poly(ADP-ribose) Polymerase-1 Is Important for Enzyme Activity and Contains a Novel Zinc-Ribbon Motif. Biochemistry 2008, 47, 5804–5813. [Google Scholar] [CrossRef] [PubMed]
- Loeffler, P.A.; Cuneo, M.J.; Mueller, G.A.; DeRose, E.F.; Gabel, S.A.; London, R.E. Structural studies of the PARP-1 BRCT domain. BMC Struct. Biol. 2011, 11, 37. [Google Scholar] [CrossRef] [PubMed]
- Ray Chaudhuri, A.; Nussenzweig, A. The multifaceted roles of PARP1 in DNA repair and chromatin remodelling. Nat. Rev. Mol. Cell Biol. 2017, 18, 610–621. [Google Scholar] [CrossRef] [PubMed]
- Eustermann, S.; Wu, W.-F.; Langelier, M.-F.; Yang, J.-C.; Easton, L.E.; Riccio, A.A.; Pascal, J.M.; Neuhaus, D. Structural Basis of Detection and Signaling of DNA Single-Strand Breaks by Human PARP-1. Mol. Cell 2015, 60, 742–754. [Google Scholar] [CrossRef] [PubMed]
- Ossovskaya, V.; Koo, I.C.; Kaldjian, E.P.; Alvares, C.; Sherman, B.M. Upregulation of Poly (ADP-Ribose) Polymerase-1 (PARP1) in Triple-Negative Breast Cancer and Other Primary Human Tumor Types. Genes Cancer 2010, 1, 812–821. [Google Scholar] [CrossRef] [PubMed]
- Domagala, P.; Huzarski, T.; Lubinski, J.; Gugala, K.; Domagala, W. PARP-1 expression in breast cancer including BRCA1-associated, triple negative and basal-like tumors: possible implications for PARP-1 inhibitor therapy. Breast Cancer Res. Treat 2011, 127, 861–869. [Google Scholar] [CrossRef] [PubMed]
- Donizy, P.; Pietrzyk, G.; Halon, A.; Kozyra, C.; Gansukh, T.; Lage, H.; Surowiak, P.; Matkowski, R. Nuclear-cytoplasmic PARP-1 expression as an unfavorable prognostic marker in lymph nodenegative early breast cancer: 15-year follow-up. Oncol. Rep. 2014, 31, 1777–1787. [Google Scholar] [PubMed]
- Mazzotta, A.; Partipilo, G.; De Summa, S.; Giotta, F.; Simone, G.; Mangia, A. Nuclear PARP1 expression and its prognostic significance in breast cancer patients. Tumor Biol. 2016, 37, 6143–6153. [Google Scholar] [CrossRef] [PubMed]
- Byers, L.A.; Wang, J.; Nilsson, M.B.; Fujimoto, J.; Saintigny, P.; Yordy, J.; Giri, U.; Peyton, M.; Fan, Y.H.; Diao, L.; et al. Proteomic profiling identifies dysregulated pathways in small cell lung cancer and novel therapeutic targets including PARP1. Cancer Discov. 2012, 2, 798–811. [Google Scholar] [CrossRef] [PubMed]
- Galia, A.; Calogero, A.E.; Condorelli, R.; Fraggetta, F.; La Corte, A.; Ridolfo, F.; Bosco, P.; Castiglione, R.; Salemi, M. PARP-1 protein expression in glioblastoma multiforme. Eur. J. Histochem. 2012, 56, e9. [Google Scholar] [CrossRef] [PubMed]
- Schiewer, M.J.; Goodwin, J.F.; Han, S.; Brenner, J.C.; Augello, M.A.; Dean, J.L.; Liu, F.; Planck, J.L.; Ravindranathan, P.; Chinnaiyan, A.M.; et al. Dual roles of PARP-1 promote cancer growth and progression. Cancer Discov. 2012, 2, 1134–1149. [Google Scholar] [CrossRef] [PubMed]
- Nosho, K.; Yamamoto, H.; Mikami, M.; Taniguchi, H.; Takahashi, T.; Adachi, Y.; Imamura, A.; Imai, K.; Shinomura, Y. Overexpression of poly(ADP-ribose) polymerase-1 (PARP-1) in the early stage of colorectal carcinogenesis. Eur. J. Cancer 2006, 42, 2374–2381. [Google Scholar] [CrossRef] [PubMed]
- Barton, V.N.; Donson, A.M.; Kleinschmidt-DeMasters, B.K.; Gore, L.; Liu, A.K.; Foreman, N.K. PARP1 Expression in Pediatric Central Nervous System Tumors. Pediatric. Blood Cancer 2009, 53, 1227–1230. [Google Scholar] [CrossRef] [PubMed]
- Mego, M.; Cierna, Z.; Svetlovska, D.; Macak, D.; Machalekova, K.; Miskovska, V.; Chovanec, M.; Usakova, V.; Obertova, J.; Babal, P.; Mardiak, J. PARP expression in germ cell tumours. J Clin Pathol 2013, 66, 607–612. [Google Scholar] [CrossRef] [PubMed]
- Wei, H.; Yu, X. Functions of PARylation in DNA Damage Repair Pathways. Genomics Proteomics Bioinformatics 2016, 14, 131–139. [Google Scholar] [CrossRef] [PubMed]
- Gibson, B.A.; Zhang, Y.; Jiang, H.; Hussey, K.M.; Shrimp, J.H.; Lin, H.; Schwede, F.; Yu, Y.; Kraus, W.L. Chemical genetic discovery of PARP targets reveals a role for PARP-1 in transcription elongation. Science 2016, 353, 45–50. [Google Scholar] [CrossRef] [PubMed]
- Kraus, W.L. Transcriptional control by PARP-1: chromatin modulation, enhancer-binding, coregulation, and insulation. Curr. Opin. Cell Biol. 2008, 20, 294–302. [Google Scholar] [CrossRef] [PubMed]
- Quiles-Perez, R.; Muñoz-Gámez, J.A.; Ruiz-Extremera, A.; O’Valle, F.; Sanjuán-Nuñez, A.; Martín-Álvarez, A.B.; Martín-Oliva, D.; Caballero, T.; Muñoz de Rueda, P.; León, J.; et al. Inhibition of poly adenosine diphosphate-ribose polymerase decreases hepatocellular carcinoma growth by modulation of tumor-related gene expression. Hepatology 2010, 51, 255–266. [Google Scholar] [CrossRef] [PubMed]
- Martin-Oliva, D.; Aguilar-Quesada, R.; O’Valle, F.; Muñoz-Gámez, F.A.; Martínez-Romero, R.; García del Moral, R.; Ruiz de Almodóvar, J.M.; Villuendas, R.; Piris, M.A.; Oliver, F.J. Inhibition of poly(ADP-ribose) polymerase modulates tumor-related gene expression, including hypoxia-inducible factor-1 activation, during skin carcinogenesis. Cancer Res. 2006, 66, 5744–5756. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.; Xu, M.; Makowski, M.M.; Zhang, T.; Law, M.H.; Kovacs, M.A.; Granzhan, A.; Kim, W.J.; Parikh, H.; Gartside, M.; et al. A common intronic variant of PARP1 confers melanoma risk and mediates melanocyte growth via regulation of MITF. Nature Genetics 2017, 49, 1326–1335. [Google Scholar] [CrossRef] [PubMed]
- Nirodi, C.; NagDas, S.; Gygi, S.P.; Olson, G.; Aebersold, R.; Richmond, A. A role for poly(ADP-ribose) polymerase in the transcriptional regulation of the melanoma growth stimulatory activity (CXCL1) gene expression. J. Biol. Chem. 2001, 276, 9366–9374. [Google Scholar] [CrossRef] [PubMed]
- Schiewer, M.J.; Knudsen, K.E. Transcriptional roles of PARP1 in cancer. Mol Cancer Res. 2014, 12, 1069–1080. [Google Scholar] [CrossRef] [PubMed]
- Wieler, S.; Gagné, J.-P.; Vaziri, H.; Poirier, G.G.; Benchimol, S. Poly(ADP-ribose) polymerase-1 is a positive regulator of the p53-mediated G1 arrest response following ionizing radiation. J. Biol. Chem. 2003, 278, 18914–18921. [Google Scholar] [CrossRef] [PubMed]
- Feng, F.Y.; de Bono, J.S.; Rubin, M.A.; Knudsen, K.E. Chromatin to Clinic: The Molecular Rationale for PARP1 Inhibitor Function. Mol. Cell 2015, 58, 925–934. [Google Scholar] [CrossRef] [PubMed]
- Tulin, A.; Spradling, A. Chromatin Loosening by Poly(ADP)-Ribose Polymerase (PARP) at Drosophila Puff Loci. Science reports 2003, 299, 560–564. [Google Scholar] [CrossRef] [PubMed]
- Bouchard, V.J.; Rouleau, M.; Poirier, G.G. PARP-1, a determinant of cell survival in response to DNA damage. Exp. Hematol. 2003, 31, 446–454. [Google Scholar] [CrossRef]
- D’Amours, D.; Desnoyers, S.; D’Silva, I.; Poirier, G.G. Poly(ADP-ribosyl)ation reactions in the regulation of nuclear functions. Biochem. J. 1999, 342, 249–268. [Google Scholar] [CrossRef] [PubMed]
- Kraus, W.L.; Hottiger, M.O. PARP-1 and gene regulation: progress and puzzles. Mol. Aspects Med. 2013, 34, 1109–1123. [Google Scholar] [CrossRef] [PubMed]
- Frizzell, K.M.; Gamble, M.J.; Berrocal, J.G.; Zhang, T.; Krishnakumar, R.; Cen, Y.; Sauve, A.A.; Kraus, W.L. Global analysis of transcriptional regulation by poly(ADP-ribose) polymerase-1 and poly(ADP-ribose) glycohydrolase in MCF-7 human breast cancer cells. J. Biol. Chem. 2009, 284, 33926–33938. [Google Scholar] [CrossRef] [PubMed]
- Reale, A.; De Matteis, G.; Galleazzi, G.; Zampieri, M.; Caiafa, P. Modulation of DNMT1 activity by ADP-ribose polymers. Oncogene 2005, 24, 13–19. [Google Scholar] [CrossRef] [PubMed]
- Hoesel, B.; Schmid, J.A. The complexity of NF-κB signaling in inflammation and cancer. Mol. Cancer 2013, 12, 86. [Google Scholar] [CrossRef] [PubMed]
- Haddad, M.; Rhinn, H.; Bloquel, C.; Coqueran, B.; Szabó, C.; Plotkine, M.; Scherman, D.; Margaill, I. Anti-inflammatory effects of PJ34, a poly(ADP-ribose) polymerase inhibitor, in transient focal cerebral ischemia in mice. Br. J. Pharmacol. 2006, 149, 23–30. [Google Scholar] [CrossRef] [PubMed]
- Smyth, M.J.; Dunn, G.P.; Schreiber, R.D. Cancer Immunosurveillance and Immunoediting: The Roles of Immunity in Suppressing Tumor Development and Shaping Tumor Immunogenicity. Adv. Immunol. 2006, 90, 1–50. [Google Scholar] [PubMed]
- Santini, D.; Perrone, G.; Roato, I.; Godio, L.; Pantano, F.; Grasso, D.; Russo, A.; Vincenzi, B.; Fratto, M.E.; Sabbatini, R.; et al. Expression pattern of receptor activator of NFkappaB (RANK) in a series of primary solid tumors and related bone metastases. J. Cell Physiol. 2011, 226, 780–784. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, Y.; Luo, J.L.; Karin, M. IκB kinase α kinase activity is required for self-renewal of ErbB2/Her2-transformed mammary tumor-initiating cells. Proc. Natl. Acad. Sci. USA 2007, 104, 15852–15857. [Google Scholar] [CrossRef] [PubMed]
- Dalaklioglu, S.; Sahin, P.; Ordueri, E.G.; Celik-Ozenci, C.; Tasatargil, A. Potential role of poly(ADP-ribose) polymerase (PARP) activation in methotrexate-induced nephrotoxicity and tubular apoptosis. Int. J. Toxicol. 2012, 31, 430–440. [Google Scholar] [CrossRef] [PubMed]
- Chang, P.; Jacobson, M.K.; Mitchison, T.J. Poly(ADP-ribose) is required for spindle assembly and structure. Nature 2004, 432, 645–650. [Google Scholar] [CrossRef] [PubMed]
- Boucher, M.-J.; Morisset, J.; Vachon, P.H.; Reed, J.C.; Lainé, J.; Rivard, N. MEK/ERK Signaling Pathway Regulates the Expression of Bcl-2, Bcl-XL, and Mcl-1 and Promotes Survival of Human Pancreatic Cancer Cells. J. Cell. Biochem. 2000, 79, 355–369. [Google Scholar] [CrossRef]
- Zhang, S.; Lin, Y.; Kim, Y.S.; Hande, M.P.; Liu, Z.G.; Shen, H.M. c-Jun N-terminal kinase mediates hydrogen peroxide-induced cell death via sustained poly(ADP-ribose) polymerase-1 activation. Cell. Death. Differ. 2007, 14, 1001. [Google Scholar] [CrossRef] [PubMed]
- Simbulan-Rosenthal, C.M.; Ly, D.H.; Rosenthal, D.S.; Konopka, G.; Luo, R.; Wang, Z.Q.; Schultz, P.G.; Smulson, M.E. Misregulation of gene expression in primary fibroblasts lacking poly(ADP-ribose) polymerase. Proc. Natl. Acad. Sci. USA 2000, 97, 11274–11279. [Google Scholar] [CrossRef] [PubMed]
- Lacal, P.M.; Tentori, L.; Muzi, A.; Ruffini, F.; Dorio, A.S.; Xu, W.; Arcelli, D.; Zhang, J.; Graziani, G. Pharmacological inhibition of poly(ADP-ribose) polymerase activity down-regulates the expression of syndecan-4 and Id-1 in endothelial cells. Int. J. Oncol. 2009, 34, 861–872. [Google Scholar] [PubMed]
- Rajesh, M.; Mukhopadhyay, P.; Godlewski, G.; Bátkai, S.; Haskó, G.; Liaudet, L.; Pacher, P. Poly(ADP-ribose)polymerase inhibition decreases angiogenesis. Biochem. Biophys. Res. Commun. 2006, 350, 1056–1062. [Google Scholar] [CrossRef] [PubMed]
- Wei, W.; Li, Y.; Lv, S.; Zhang, C.; Tian, Y. PARP-1 may be involved in angiogenesis in epithelial ovarian cancer. Oncol. Lett. 2016, 12, 4561–4567. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, M.I.; Peralta-Leal, A.; O’Valle, F.; Rodriguez-Vargas, J.M.; Gonzalez-Flores, A.; Majuelos-Melguizo, J.; López, L.; Serrano, S.; de Herreros, A.G.; Rodríguez-Manzaneque, J.-C.; et al. PARP-1 regulates metastatic melanoma through modulation of vimentin-induced malignant transformation. PLoS Genet. 2013, 9, e1003531. [Google Scholar] [CrossRef] [PubMed]
- Choi, E.B.; Yang, A.Y.; Kim, S.C.; Lee, J.; Choi, J.K.; Choi, C.; Kim, M.Y. PARP1 enhances lung adenocarcinoma metastasis by novel mechanisms independent of DNA repair. Oncogene 2016, 35, 4569–4579. [Google Scholar] [CrossRef] [PubMed]
- Curtin, N.J.; Szabo, C. Therapeutic applications of PARP inhibitors: anticancer therapy and beyond. Mol. Aspects Med. 2013, 34, 1217–1256. [Google Scholar] [CrossRef] [PubMed]
- Murai, J.; Huang, S.Y.; Das, B.B.; Renaud, A.; Zhang, Y.; Doroshow, J.H.; Ji, J.; Takeda, S.; Pommier, Y. Trapping of PARP1 and PARP2 by Clinical PARP Inhibitors. Cancer Res. 2012, 72, 5588–5599. [Google Scholar] [CrossRef] [PubMed]
- Helleday, T. The underlying mechanism for the PARP and BRCA synthetic lethality: clearing up the misunderstandings. Mol. Oncol. 2011, 5, 387–393. [Google Scholar] [CrossRef] [PubMed]
- Purnell, M.R.; Whish, W.J.D. Novel Inhibitors of Poly(ADP-Ribose) Synthetase. Biochem. J. 1980, 185, 775–777. [Google Scholar] [CrossRef] [PubMed]
- Papeo, G.; Forte, B.; Orsini, P.; Perrera, C.; Posteri, H.; Scolaro, A.; Montagnoli, A. Poly(ADP-ribose) polymerase inhibition in cancer therapy: Are we close to maturity? Expert. Opin. Ther. Pat. 2009, 19, 1377–1400. [Google Scholar] [CrossRef] [PubMed]
- Durrant, L.G.; Boyle, J.M. Potentiation of cell killing by inhibitors of poly (ADP-ribose) polymerase in four rodent cell lines exposed to N-methyl-N-nitrosourea or UV light. Chem. Biol. Inter. 1982, 38, 325–338. [Google Scholar] [CrossRef]
- Fong, P.C.; Boss, D.S.; Yap, T.A.; Tutt, A.; Wu, P.; Mergui-Roelvink, M.; Mortimer, P.; Swaisland, H.; Lau, A.; O’Connor, M.J.; et al. Inhibition of poly(ADP-ribose) polymerase in tumors from BRCA mutation carriers. N. Engl. J. Med. 2009, 361, 123–134. [Google Scholar] [CrossRef] [PubMed]
- Kim, G.; Ison, G.; McKee, A.E.; Zhang, H.; Tang, S.; Gwise, T.; Sridhara, R.; Lee, E.; Tzou, A.; Philip, R.; et al. FDA Approval Summary: Olaparib Monotherapy in Patients with Deleterious Germline BRCA-Mutated Advanced Ovarian Cancer Treated with Three or More Lines of Chemotherapy. Clin. Cancer Res. 2015, 21, 4257–4261. [Google Scholar] [CrossRef] [PubMed]
- Lawlor, D.; Martin, P.; Busschots, S.; Thery, J.; O’leary, J.J.; Hennessy, B.T.; Stordal, B. PARP Inhibitors as P-glyoprotein Substrates. J. Pharm. Sci. 2014, 103, 1913–1920. [Google Scholar] [CrossRef] [PubMed]
- Gilabert, M.; Launay, S.; Ginestier, C.; Bertucci, F.; Audebert, S.; Pophillat, M.; Toiron, Y.; Baudelet, E.; Finetti, P.; Noguchi, T.; Sobol, H.; et al. Poly(ADP-Ribose) Polymerase 1 (PARP1) Overexpression in Human Breast Cancer Stem Cells and Resistance to Olaparib. PLoS ONE 2014, 9, e104302. [Google Scholar] [CrossRef] [PubMed]
- Mirza, M.R.; Monk, B.J.; Herrstedt, J.; Oza, A.M.; Mahner, S.; Redondo, A.; Fabbro, M.; Ledermann, J.A.; Lorusso, D.; Vergote, I.; et al. Niraparib Maintenance Therapy in Platinum-Sensitive, Recurrent Ovarian Cancer. N. Engl. J. Med. 2016, 375, 2154–2164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lupo, B.; Trusolino, L. Inhibition of poly(ADP-ribosyl)ation in cancer: old and new paradigms revisited. Biochim. Biophys. Acta. 2014, 1846, 201–215. [Google Scholar] [CrossRef] [PubMed]
- Dockery, L.E.; Gunderson, C.C.; Moore, K.N. Rucaparib: the past, present, and future of a newly approved PARP inhibitor for ovarian cancer. Onco Targets Ther. 2017, 10, 3029–3037. [Google Scholar] [CrossRef] [PubMed]
- Lin, K.; Sun, J.; Maloney, L.; Goble, S.; Oza, A.; Coleman, R.; Scott, C.; Robillard, L.; Mann, E.; Isaacson, J.; et al. Quantification of genomic loss of heterozygosity enables prospective selection of ovarian cancer patients who may derive benefit from the PARP inhibitor rucaparib. Europ. Cancer Congr. 2015, 51, S531. [Google Scholar] [CrossRef]
- Ihnen, M.; zu Eulenburg, C.; Kolarova, T.; Qi, J.W.; Manivong, K.; Chalukya, M.; Dering, J.; Anderson, L.; Ginther, C.; Meuter, A.; et al. Therapeutic potential of the poly(ADP-ribose) polymerase inhibitor rucaparib for the treatment of sporadic human ovarian cancer. Mol. Cancer Ther. 2013, 12, 1002–1015. [Google Scholar] [CrossRef] [PubMed]
- Soumerai, J.D.; Zelenetz, A.D.; Moskowitz, C.H.; Palomba, M.L.; Hamlin, P.A., Jr.; Noy, A.; Straus, D.J.; Moskowitz, A.J.; Younes, A.; Matasar, M.J.; et al. The PARP Inhibitor Veliparib Can Be Safely Added to Bendamustine and Rituximab and Has Preliminary Evidence of Activity in B-Cell Lymphoma. Clin. Cancer Res. 2017, 23, 4119–4126. [Google Scholar] [CrossRef] [PubMed]
- Somlo, G.; Frankel, P.H.; Arun, B.K.; Ma, C.X.; Garcia, A.A.; Cigler, T.; Cream, L.V.; Harvey, H.A.; Sparano, J.A.; Nanda, R.; et al. Efficacy of the PARP Inhibitor Veliparib with Carboplatin or as a Single Agent in Patients with Germline BRCA1- or BRCA2-Associated Metastatic Breast Cancer: California Cancer Consortium Trial NCT01149083. Clin. Cancer Res. 2017, 23, 4066–4076. [Google Scholar] [CrossRef] [PubMed]
- Reiss, K.A.; Herman, J.M.; Zahurak, M.; Brade, A.; Dawson, L.A.; Scardina, A.; Joffe, C.; Petito, E.; Hacker-Prietz, A.; Kinders, R.J.; et al. A Phase I study of veliparib (ABT-888) in combination with low-dose fractionated whole abdominal radiation therapy in patients with advanced solid malignancies and peritoneal carcinomatosis. Clin. Cancer Res. 2015, 21, 68–76. [Google Scholar] [CrossRef] [PubMed]
- Smith, M.A.; Reynolds, C.P.; Kang, M.H.; Kolb, E.A.; Gorlick, R.; Carol, H.; Lock, R.B.; Keir, S.T.; Maris, J.M.; Billups, C.A.; et al. Synergistic activity of PARP inhibition by talazoparib (BMN 673) with temozolomide in pediatric cancer models in the pediatric preclinical testing program. Clin. Cancer Res. 2015, 21, 819–832. [Google Scholar] [CrossRef] [PubMed]
- Litton, J.K.; Scoggins, M.; Ramirez, D.L.; Murthy, R.K.; Whitman, G.J.; Hess, K.R.; Adrada, B.E.; Moulder, S.L.; Barcenas, C.H.; Valero, V.; et al. A pilot study of neoadjuvant talazoparib for early-stage breast cancer patients with a BRCA mutation. Ann. Oncol. 2016. [Google Scholar] [CrossRef]
- Ashleigh, H.; Tudhope, S.J.; Junge, G.; Rodrigues, N.; Patterson, M.J.; Woodhouse, L.; Lunec, J.; Hunter, J.E.; Mulligan, E.A.; Cole, M.; et al. PARP1 expression, activity and ex vivo sensitivity to the PARP inhibitor, talazoparib (BMN 673), in chronic lymphocytic leukaemia. Oncotarget 2015, 6, 43978–43991. [Google Scholar]
- Engert, F.; Kovac, M.; Baumhoer, D.; Nathrath, M.; Fulda, S. Osteosarcoma cells with genetic signatures of BRCAness are susceptible to the PARP inhibitor talazoparib alone or in combination with chemotherapeutics. Oncotarget 2017, 8, 48794–48806. [Google Scholar] [CrossRef] [PubMed]
- Murai, J.; Huang, S.Y.; Renaud, A.; Zhang, Y.; Ji, J.; Takeda, S.; Morris, J.; Teicher, B.; Doroshow, J.H.; Pommier, Y. Stereospecific PARP trapping by BMN 673 and comparison with olaparib and rucaparib. Mol. Cancer Ther. 2014, 13, 433–443. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Zhou, N.; Cai, P.; Bao, J. In Silico Screening Identifies a Novel Potential PARP1 Inhibitor Targeting Synthetic Lethality in Cancer Treatment. Int. J. Mol. Sci. 2016, 17, 258. [Google Scholar] [CrossRef] [PubMed]
- Fu, L.; Wang, S.; Wang, X.; Wang, P.; Zheng, Y.; Yao, D.; Guo, M.; Zhang, L.; Ouyang, L. Crystal structure-based discovery of a novel synthesized PARP1 inhibitor (OL-1) with apoptosis-inducing mechanisms in triple-negative breast cancer. Sci. Rep. 2016, 6, 3. [Google Scholar] [CrossRef] [PubMed]
- Brown, J.S.; Kaye, S.B.; Yap, T.A. PARP inhibitors: The race is on. Br. J. Cancer 2016, 114, 713–715. [Google Scholar] [CrossRef] [PubMed]
- Konstantinopoulos, P.A.; Spentzos, D.; Karlan, B.Y.; Taniguchi, T.; Fountzilas, E.; Francoeur, N.; Levine, D.A.; Cannistra, S.A. Gene expression profile of BRCAness that correlates with responsiveness to chemotherapy and with outcome in patients with epithelial ovarian cancer. J. Clin. Oncol. 2010, 28, 3555–3561. [Google Scholar] [CrossRef] [PubMed]
- Ekblad, T.; Schüler, H. Sirtuins are Unaffected by PARP Inhibitors Containing Planar Nicotinamide Bioisosteres. Chem. Biol. Drug Des. 2016, 87, 478–482. [Google Scholar] [CrossRef] [PubMed]
- Zaremba, T.; Curtin, N.J. PARP Inhibitor Development for Systemic Cancer Targeting. AntiCancer Agents Med. Chem. 2007, 7, 515–523. [Google Scholar] [CrossRef] [PubMed]
- Baptista, S.J.; Silva, M.M.C.; Moroni, E.; Meli, M.; Colombo, G.; Dinis, T.C.P.; Salvador, J.A.R. Novel PARP-1 Inhibitor Scaffolds Disclosed by a Dynamic Structure-Based Pharmacophore Approach. PLoS ONE 2017, 12, e0170846. [Google Scholar] [CrossRef] [PubMed]
- Elmongy, E.I.; Khedr, M.A.; Taleb, N.A.; Awad, H.M.; Abbas, S.E.S. Design, Synthesis, and Biological Evaluation of Some Cyclohepta[b]Thiophene and Substituted Pentahydrocycloheptathieno[2,3-d]Pyrimidine Derivatives. J. Heter. Chem. 2017, 54, 1084–1093. [Google Scholar] [CrossRef]
- Zhu, Q.; Wang, X.; Chu, Z.; He, G.; Dong, G.; Xu, Y. Design, synthesis and biological evaluation of novel imidazo[4,5-c]pyridinecarboxamide derivatives as PARP-1 inhibitors. Bioorg. Med. Chem. Lett. 2013, 23, 1993–1996. [Google Scholar] [CrossRef] [PubMed]
- Wagner, L.M. Profile of veliparib and its potential in the treatment of solid tumors. Onco. Targets Ther. 2015, 8, 1931–1939. [Google Scholar] [CrossRef] [PubMed]
- Gavande, N.S.; VanderVere-Carozza, P.S.; Hinshaw, H.D.; Jalal, S.I.; Sears, C.R.; Pawelczak, K.S.; Turchi, J.J. DNA repair targeted therapy: The past or future of cancer treatment? Pharmacol. Ther. 2016, 160, 65–83. [Google Scholar] [CrossRef] [PubMed]
- Steffen, J.D.; Tholey, R.M.; Langelier, M.F.; Planck, J.L.; Schiewer, M.J.; Lal, S.; Bildzukewicz, N.A.; Yeo, C.J.; Knudsen, K.E.; Brody, J.R.; et al. Targeting PARP-1 allosteric regulation offers therapeutic potential against cancer. Cancer Res. 2014, 74, 31–37. [Google Scholar] [CrossRef] [PubMed]
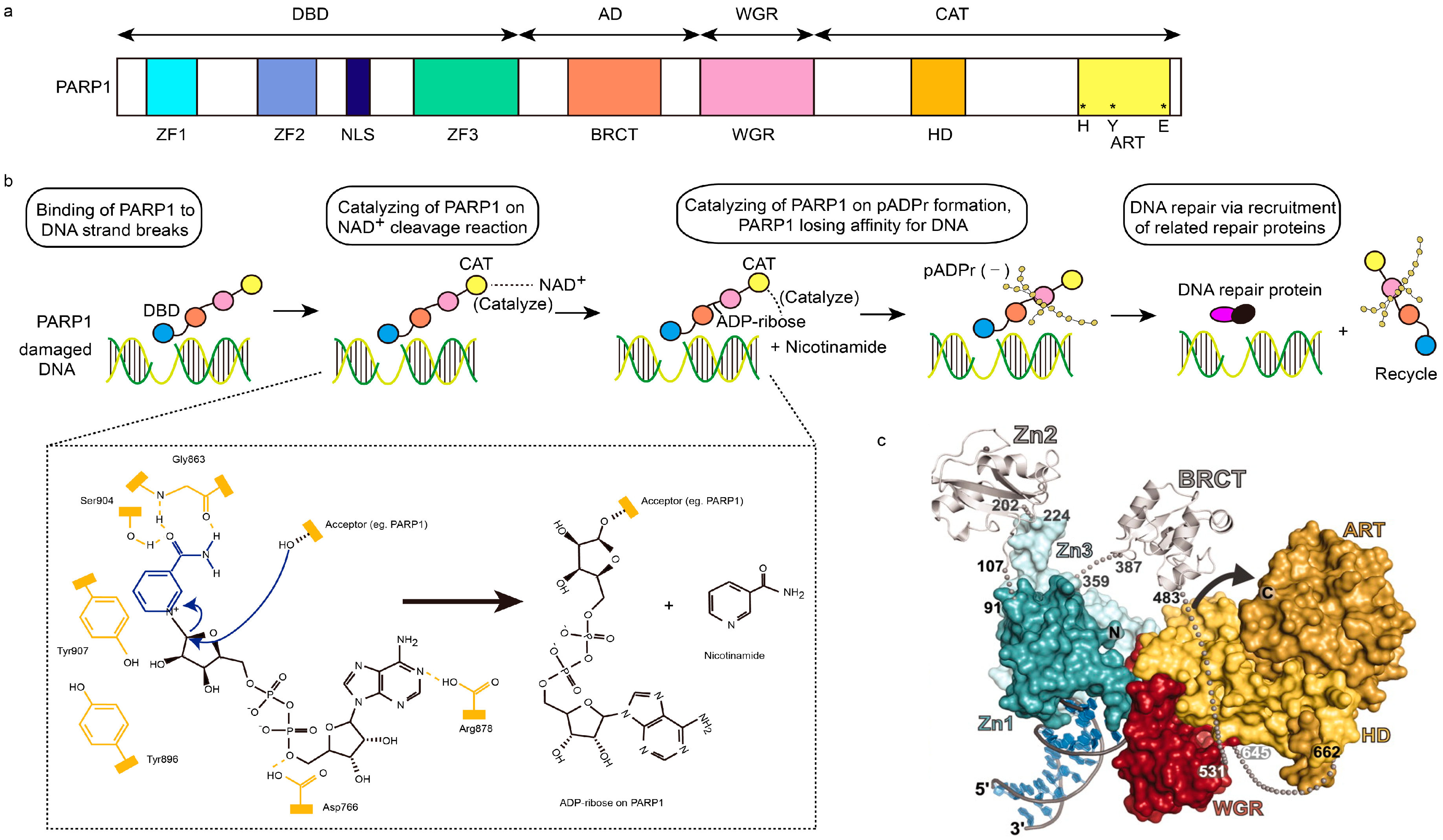
Figure 1.
Structural characteristics of PARP1 and PARP1-based DNA repair. (a) Schematic representation of human PARP1 molecular architecture; (b) Structural process of PARP1-mediated DNA repair. After DNA-binding domain (DBD, blue ball) of PARP1 senses and binds damaged DNA, the NAD+ will be cleaved into ADP-ribose and nicotinamide, which are catalyzed by the catalytic domain (CAT, yellow ball) of PARP1. Then, poly (ADP-ribose) (pADPr) is synthesized on the acceptor PARP1 through the combination of ADP-ribose, assisted by the catalysis of CAT too. Subsequently, PARP1 leaves damaged DNA, due to the dense negative charge of pADPr, allowing the recruitment of related repair proteins and replication. In chemical formulae, domains in yellow represents active domains of PARP1, while blue and black parts represent NAD+. Hydroxyl of Ser904 and carbonyl and NH group of Gly863 in PARP1 interact with the amide moiety of NAD+ via hydrogen bonding interaction, while Tyr907 of PARP1 via π–π stacking interaction. The curved arrows in blue represent the nucleophilic attack by the 2′-hydroxyl of the acceptor site in PARP1, and the release of the nicotinamide procedures; AD, orange ball; WGR, red ball. (c) The PARP1/damaged DNA crystal structure. Zn1, dark green; Zn2, silver (left); Zn3, light green; BRCT, silver (right); ART, dark yellow; HD, light yellow; red, WGR [4,18,19].
Figure 1.
Structural characteristics of PARP1 and PARP1-based DNA repair. (a) Schematic representation of human PARP1 molecular architecture; (b) Structural process of PARP1-mediated DNA repair. After DNA-binding domain (DBD, blue ball) of PARP1 senses and binds damaged DNA, the NAD+ will be cleaved into ADP-ribose and nicotinamide, which are catalyzed by the catalytic domain (CAT, yellow ball) of PARP1. Then, poly (ADP-ribose) (pADPr) is synthesized on the acceptor PARP1 through the combination of ADP-ribose, assisted by the catalysis of CAT too. Subsequently, PARP1 leaves damaged DNA, due to the dense negative charge of pADPr, allowing the recruitment of related repair proteins and replication. In chemical formulae, domains in yellow represents active domains of PARP1, while blue and black parts represent NAD+. Hydroxyl of Ser904 and carbonyl and NH group of Gly863 in PARP1 interact with the amide moiety of NAD+ via hydrogen bonding interaction, while Tyr907 of PARP1 via π–π stacking interaction. The curved arrows in blue represent the nucleophilic attack by the 2′-hydroxyl of the acceptor site in PARP1, and the release of the nicotinamide procedures; AD, orange ball; WGR, red ball. (c) The PARP1/damaged DNA crystal structure. Zn1, dark green; Zn2, silver (left); Zn3, light green; BRCT, silver (right); ART, dark yellow; HD, light yellow; red, WGR [4,18,19].
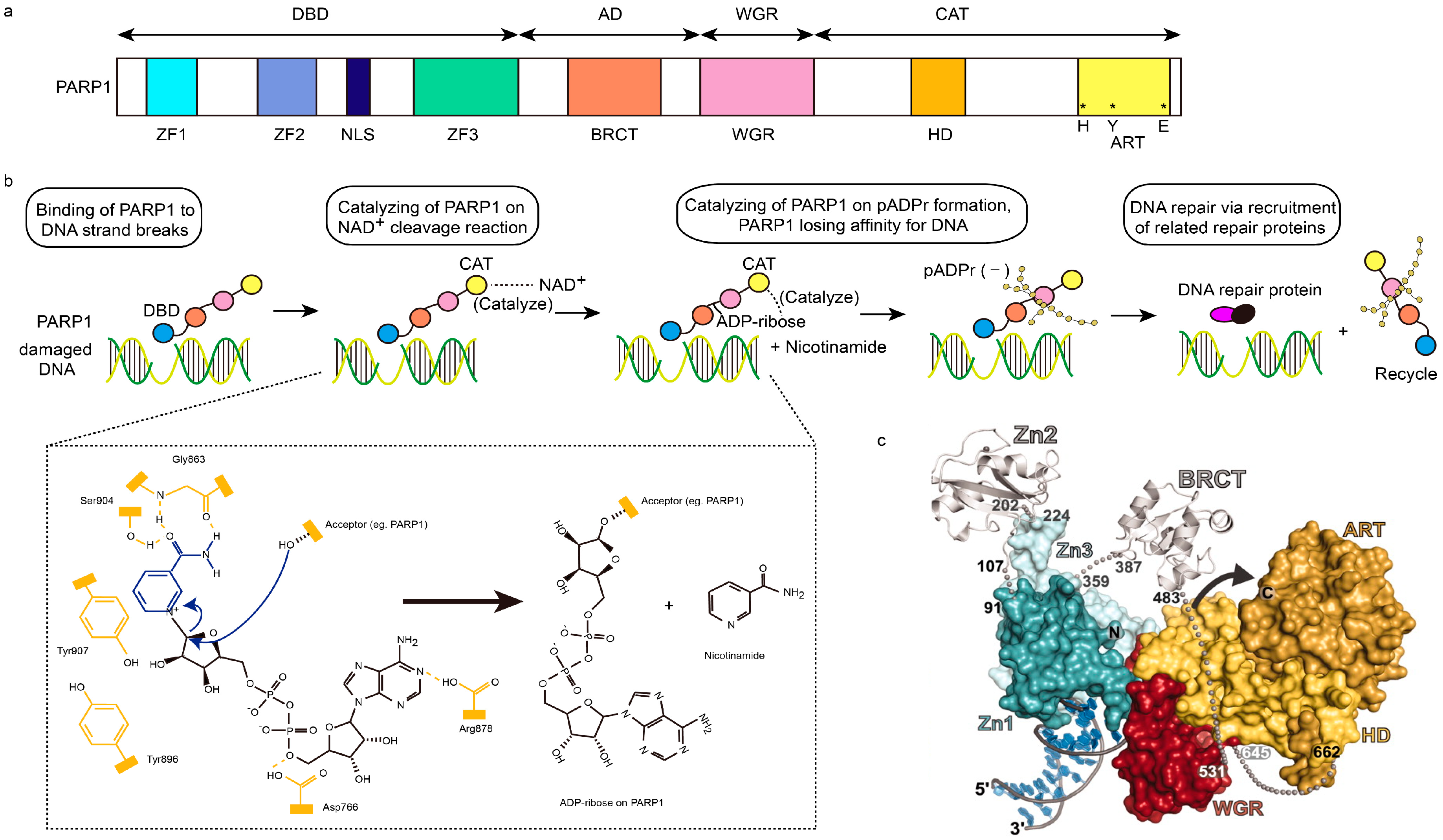
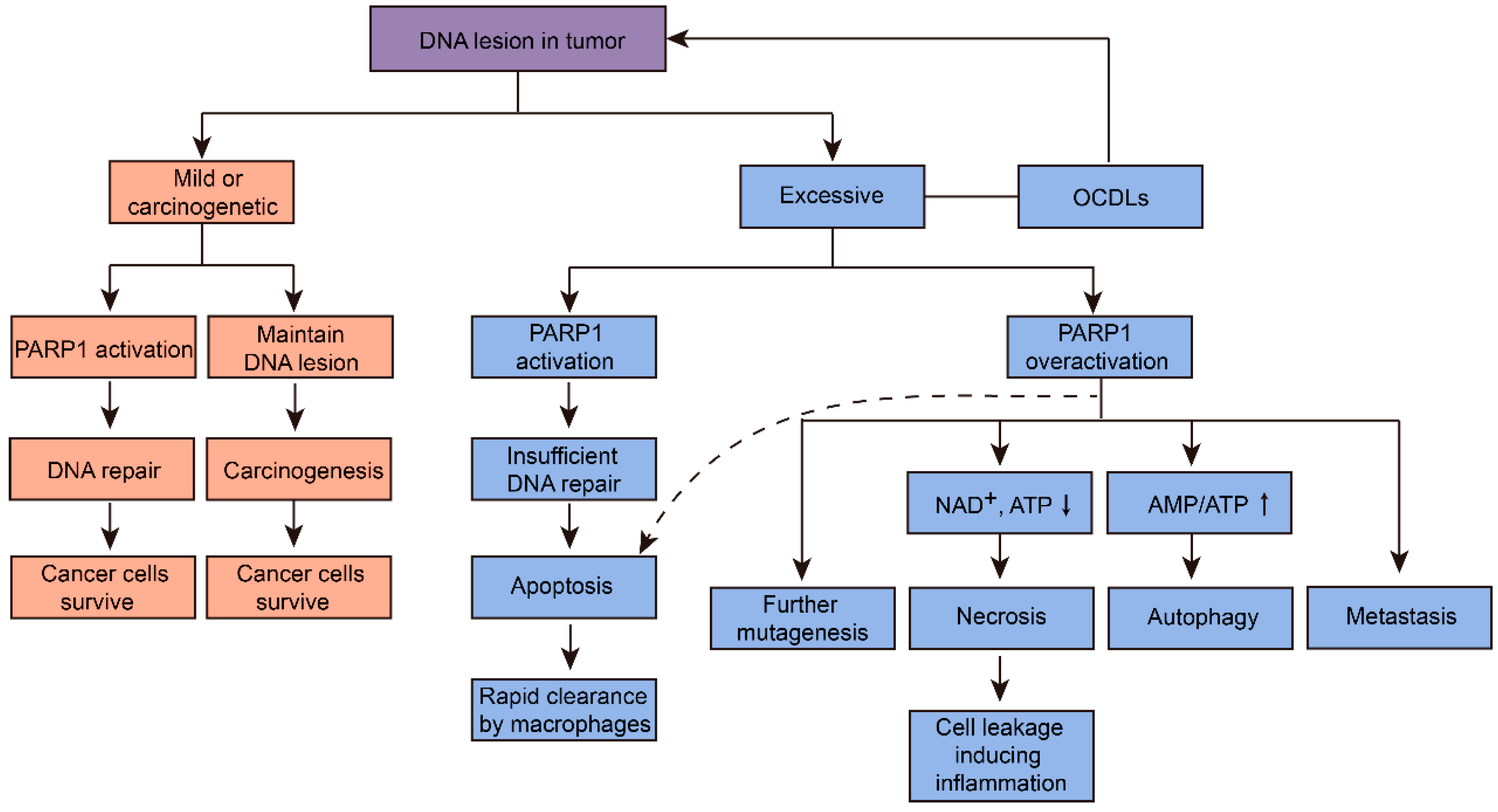
Figure 2.
Cancer cells fate induced by different levels of PARP1-related DNA repair in tumor. PARP1 regulated various pathways due to the degree of DNA lesion in tumor. To survive, cancer cells will activate PARP1 to repair mildly damaged DNA, and harbor carcinogenetic genetic mutations via maintaining DNA lesions. In case of excessive DNA lesions, normal PARP1 activation will cause insufficient DNA repair, subsequently leading to cancer cell apoptosis. This may be a more desirable result of cancer cells in anticancer therapy, because apoptosis cells will then be cleared by macrophages rapidly. On the other hand, overactivation of PARP1 will lead to further mutagenesis, metastasis, and energy-depleted necrosis and autophagy of tumors. Necrosis will then further induce inflammation, which is another important carcinogenetic factor. Inhibition of PARP1 overactivation by PARP1 inhibitors will transform cells’ fate to apoptosis, subsequently reduce further mutagenesis, metastasis, autophagy, necrosis, and tumor-promoting inflammation (dashed arrow) [4,14,18].
Figure 2.
Cancer cells fate induced by different levels of PARP1-related DNA repair in tumor. PARP1 regulated various pathways due to the degree of DNA lesion in tumor. To survive, cancer cells will activate PARP1 to repair mildly damaged DNA, and harbor carcinogenetic genetic mutations via maintaining DNA lesions. In case of excessive DNA lesions, normal PARP1 activation will cause insufficient DNA repair, subsequently leading to cancer cell apoptosis. This may be a more desirable result of cancer cells in anticancer therapy, because apoptosis cells will then be cleared by macrophages rapidly. On the other hand, overactivation of PARP1 will lead to further mutagenesis, metastasis, and energy-depleted necrosis and autophagy of tumors. Necrosis will then further induce inflammation, which is another important carcinogenetic factor. Inhibition of PARP1 overactivation by PARP1 inhibitors will transform cells’ fate to apoptosis, subsequently reduce further mutagenesis, metastasis, autophagy, necrosis, and tumor-promoting inflammation (dashed arrow) [4,14,18].
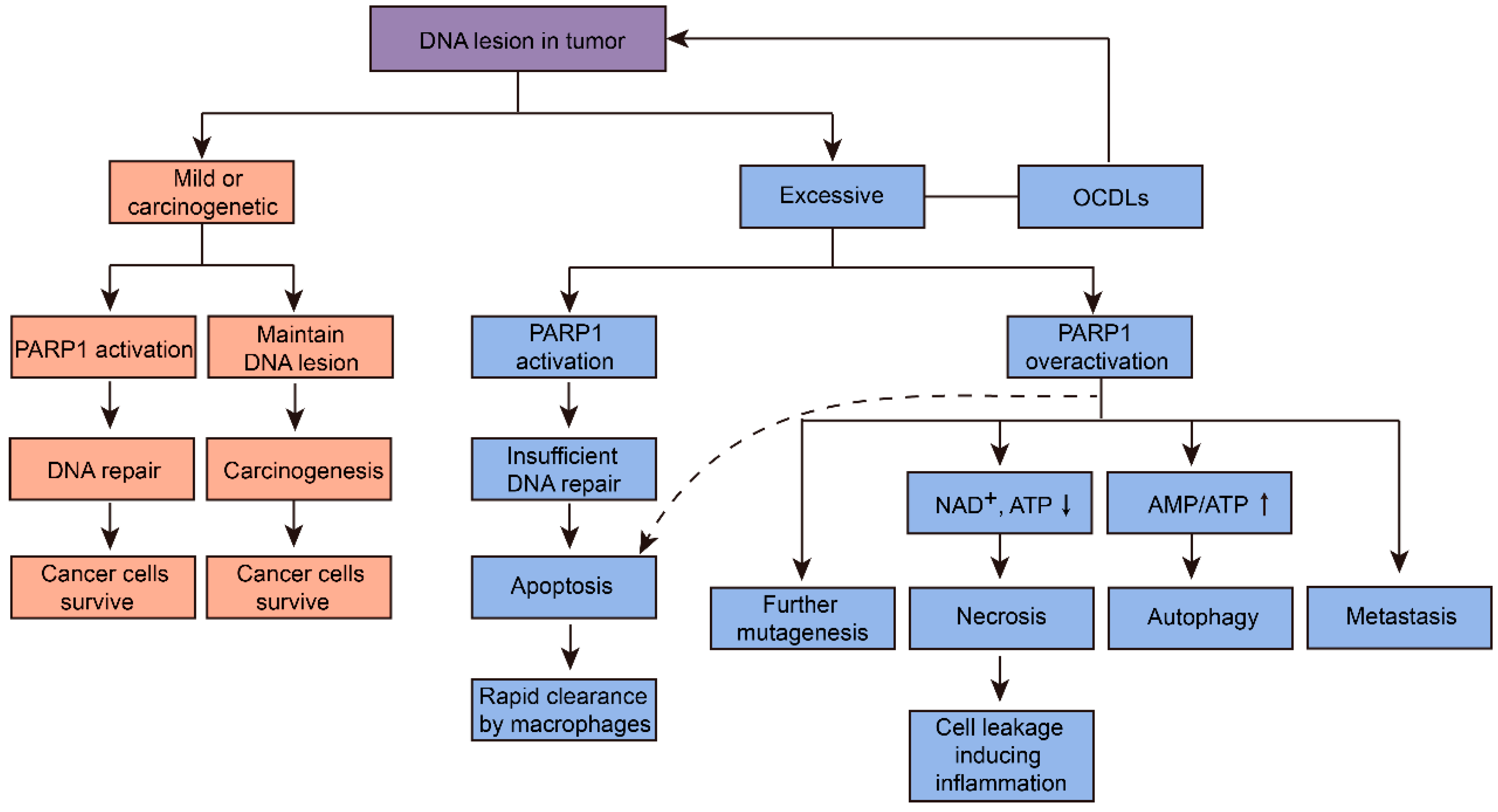
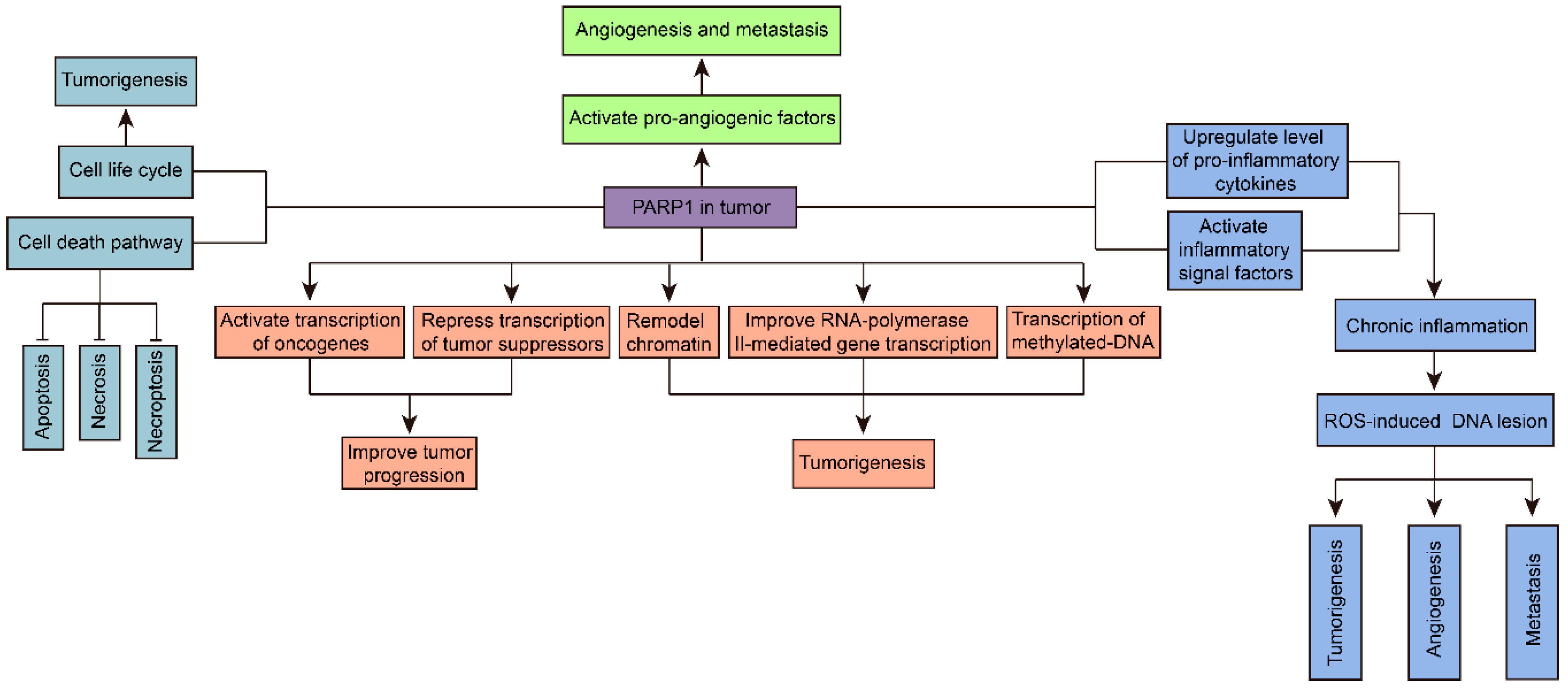
Figure 3.
The multifunction of PARP1 in tumorigenesis. PARP1 is considered to promote tumor development potentially through many pathways. Briefly, PARP1 regulates gene transcription through interacting with transcription factors, transcription machinery, and chromatin modulators. Hyperactivated PARP1 upregulates inflammatory signal factors in tumors. Also, PARP1 modulates cancer cellular life cycle via regulating cellular mitosis and cell death pathways, including apoptosis, necrosis, and necroptosis. PARP1 activates pro-angiogenic factors and induces angiogenesis and metastasis in tumors.
Figure 3.
The multifunction of PARP1 in tumorigenesis. PARP1 is considered to promote tumor development potentially through many pathways. Briefly, PARP1 regulates gene transcription through interacting with transcription factors, transcription machinery, and chromatin modulators. Hyperactivated PARP1 upregulates inflammatory signal factors in tumors. Also, PARP1 modulates cancer cellular life cycle via regulating cellular mitosis and cell death pathways, including apoptosis, necrosis, and necroptosis. PARP1 activates pro-angiogenic factors and induces angiogenesis and metastasis in tumors.


{kind=link}
{kind=link}
{kind=link}
Table 1.
Expression level of PARP1 in different carcinomas.
| Cancer Types | Expression of PARP1 |
|---|---|
| Breast Cancer | Up-regulated |
| Ovarian Cancer | Up-regulated |
| Uterine Cancer | Up-regulated |
| Lung Cancer | Up-regulated |
| Skin Cancer | Up-regulated |
| Non-Hodgkin’s Lymphoma | Up-regulated |
| Glioblastoma Multiforme | Up-regulated |
| Prostate Cancer | Up-regulated |
| Ewing’s Sarcoma | Up-regulated |
| Colorectal Cancer | Up-regulated |
| Pediatric Central Nervous System Cancer | Up-regulated |
| Testicular Germ Cell Tumor | Up-regulated |
Table 2.
Structure and indications of mature anticancer PARP1-based drugs entered clinical trials.
| Drug | Structure | Active Structural Group | Biophysical Parameters (PARP1) | Well Studied Cancer Types | Typical Clinical Trials No. (The Latest Stage) | Phase | REF |
|---|---|---|---|---|---|---|---|
| Olaparib (AZD2281) |  | Amide motif enclosed in cyclic ring | IC50 = 5 nM | Ovarian carcinoma | NCT02476968 (Phase 4) | 1–4 | [1,73] |
| Breast carcinoma | NCT02032823 (Phase 3) | 1–3 | |||||
| Prostate carcinoma | NCT02987543 (Phase 3) | 1–3 | |||||
| Pancreatic carcinoma | NCT02184195 (Phase 3) | 1–3 | |||||
| Ewing’s sarcoma | NCT01583543 (Phase 2) | 1–2 | |||||
| Gastric carcinoma | NCT01924533 (Phase 3) | 1–3 | |||||
| Lung carcinoma | NCT03009682 (Phase 2) | 1–2 | |||||
| Germ Cell Tumor | NCT02533765 (Phase 2) | 1–2 | |||||
| Colorectal carcinoma | NCT00912743 (Phase 2) | 1–2 | |||||
| Unknown solid tumors | NCT03233204 (Phase 2) | 1–2 | |||||
| Niraparib (MK-4827) |  | Conventional embedded primary amide | IC50 = 3.2 nM | Ovarian carcinoma | NCT01847274 (Phase 3) | 1–3 | [76,93] |
| Breast carcinoma | NCT01905592 (Phase 3) | 1–3 | |||||
| Endometrial carcinoma | NCT03016338 (Phase 2) | 1–2 | |||||
| Uveal melanoma | NCT03207347 (Phase 2) | 1–2 | |||||
| Fallopian tube carcinoma | NCT02657889 (Phase 2) | 1–2 | |||||
| Peritoneal carcinoma | NCT02657889 (Phase 2) | 1–2 | |||||
| Rucaparib (AG-014669, PF-01367338) |  | Amide motif enclosed in cyclic ring | IC50 = 1.4 nM | Ovarian carcinoma | NCT02855944 (Phase 3) | 1–3 | [78,80,96] |
| Fallopian tube carcinoma | NCT02855944 (Phase 3) | 1–3 | |||||
| Peritoneal carcinoma | NCT01968213 (Phase 3) | 1–3 | |||||
| Prostate carcinoma | NCT02975934 (Phase 3) | 1–3 | |||||
| Pancreatic carcinoma | NCT02042378 (Phase 2) | 1–2 | |||||
| Breast carcinoma | NCT02505048 (Phase 2) | 1–2 | |||||
| Veliparib (ABT-888) |  | Conventional embedded primary amide | Ki = 5.2 nM | Breast carcinoma | NCT02032277 (Phase 3) | 1–3 | [82,97,98] |
| Ovarian carcinoma | NCT02470585 (Phase 3) | 1–3 | |||||
| Non-small cell lung carcinoma | NCT02106546 (Phase 3) | 1–3 | |||||
| Solid tumors | NCT01193140 (Phase 2) | 1–2 | |||||
| Testicular carcinoma | NCT02860819 (Phase 2) | 1–2 | |||||
| Talazoparib (BMN-673) |  | Amide motif enclosed in cyclic ring | IC50 = 1.2 nM | Breast carcinoma | NCT01945775 (Phase 3) | 1–3 | [85,86,99,100] |
| Prostate carcinoma | NCT03148795 (Phase 2) | 1–2 | |||||
| Ovarian carcinoma | NCT02326844 (Phase 2) | 1–2 | |||||
| Endometrial carcinoma | NCT02127151 (Phase 2) | 1–2 | |||||
| Acute Myeloid Leukemia | NCT02878785 (Phase 2) | 1–2 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Wang, L.; Liang, C.; Li, F.; Guan, D.; Wu, X.; Fu, X.; Lu, A.; Zhang, G. PARP1 in Carcinomas and PARP1 Inhibitors as Antineoplastic Drugs. Int. J. Mol. Sci. 2017, 18, 2111. https://doi.org/10.3390/ijms18102111
AMA Style
Wang L, Liang C, Li F, Guan D, Wu X, Fu X, Lu A, Zhang G. PARP1 in Carcinomas and PARP1 Inhibitors as Antineoplastic Drugs. International Journal of Molecular Sciences. 2017; 18(10):2111. https://doi.org/10.3390/ijms18102111
Chicago/Turabian StyleWang, Luyao, Chao Liang, Fangfei Li, Daogang Guan, Xiaoqiu Wu, Xuekun Fu, Aiping Lu, and Ge Zhang. 2017. "PARP1 in Carcinomas and PARP1 Inhibitors as Antineoplastic Drugs" International Journal of Molecular Sciences 18, no. 10: 2111. https://doi.org/10.3390/ijms18102111
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.