
Hydroxypyridinone Chelators: From Iron Scavenging to Radiopharmaceuticals for PET Imaging with Gallium-68

 ,
,
Abstract
:
1. Introduction
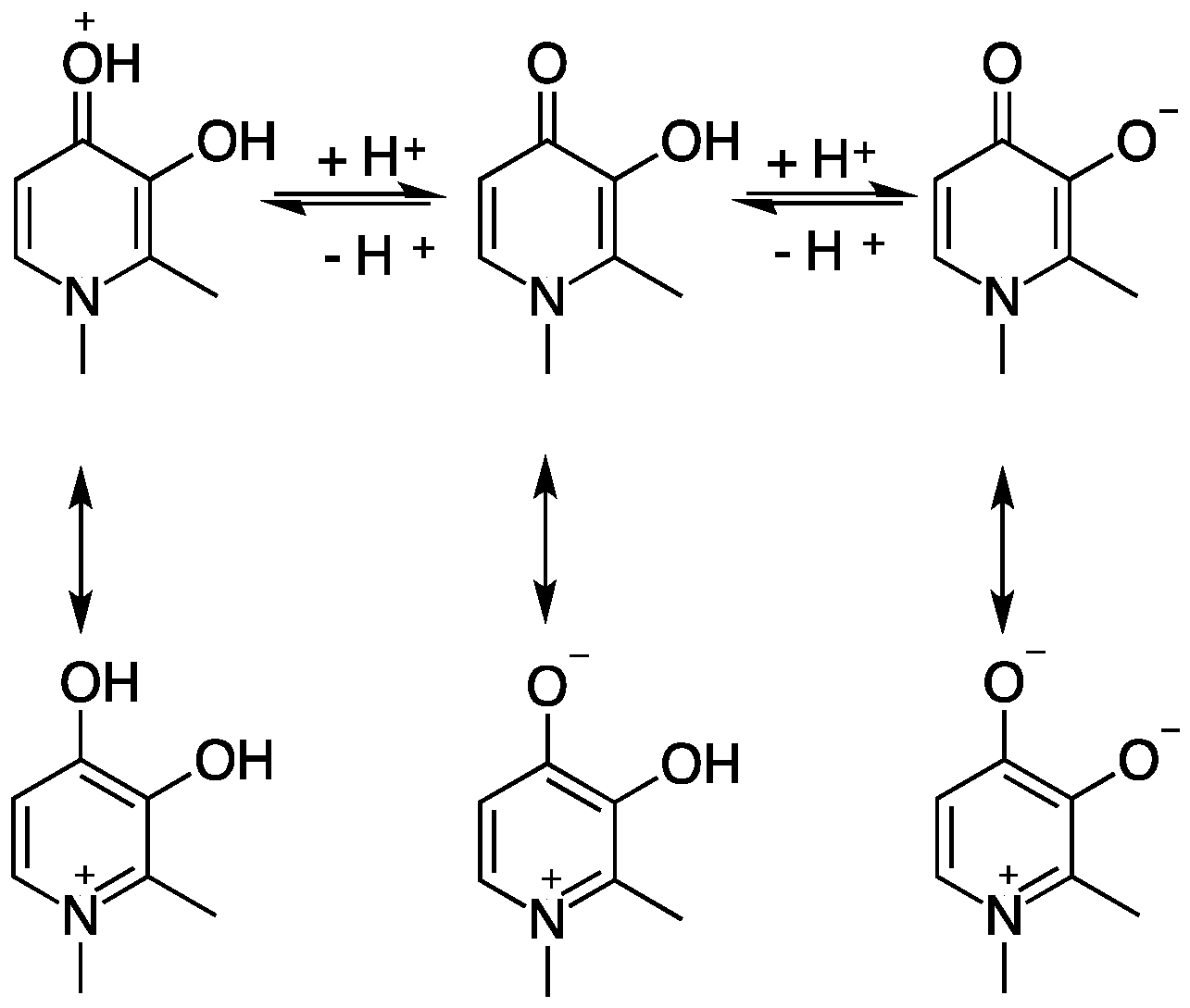
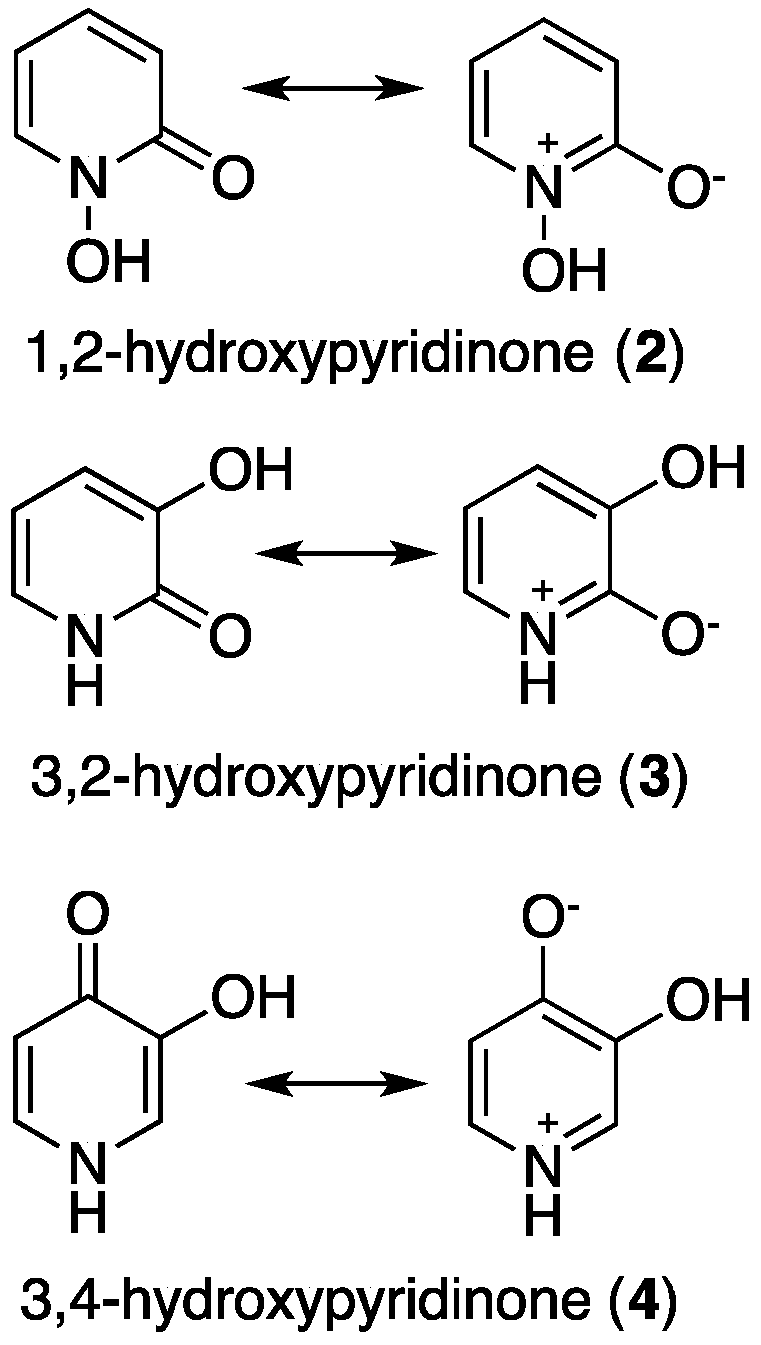
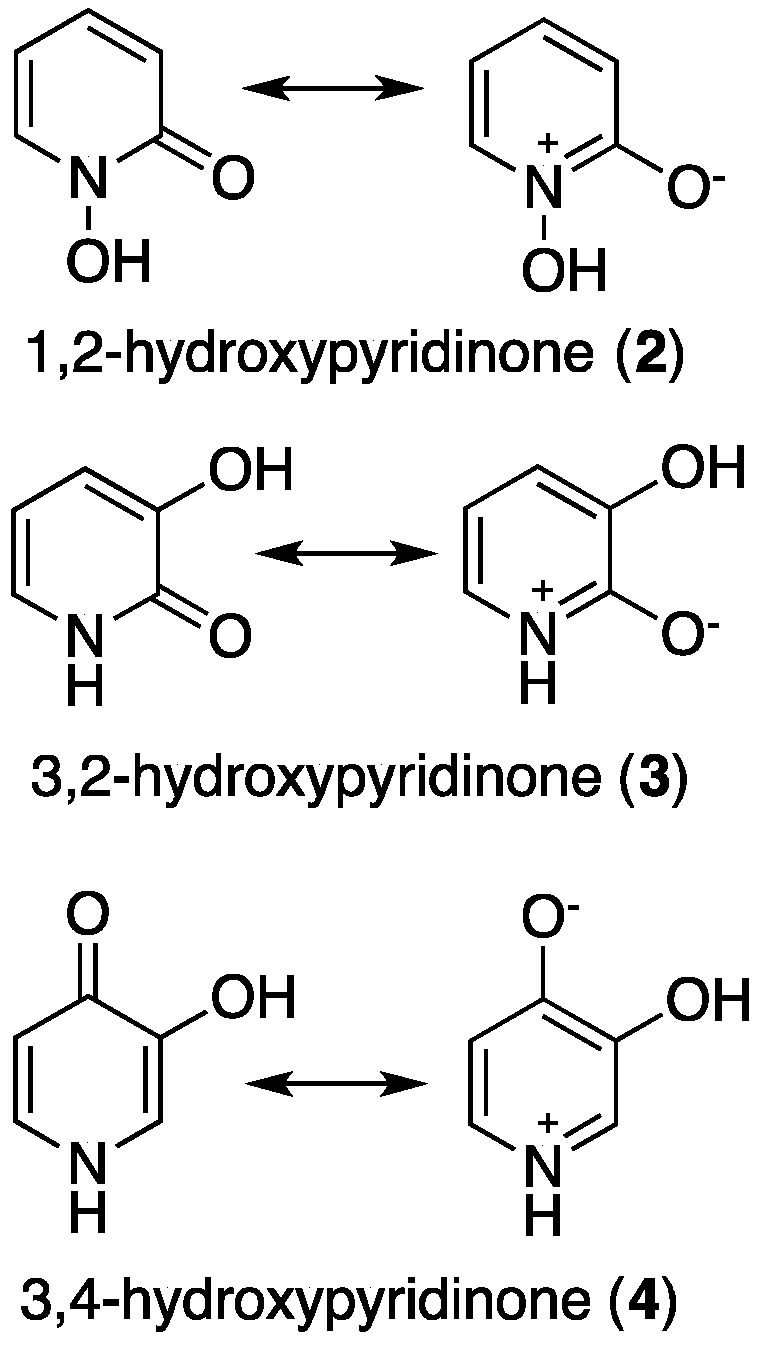
2. Hydroxypyridinones
3. 3,4-Hydroxypyridinones for Fe3+ Complexation
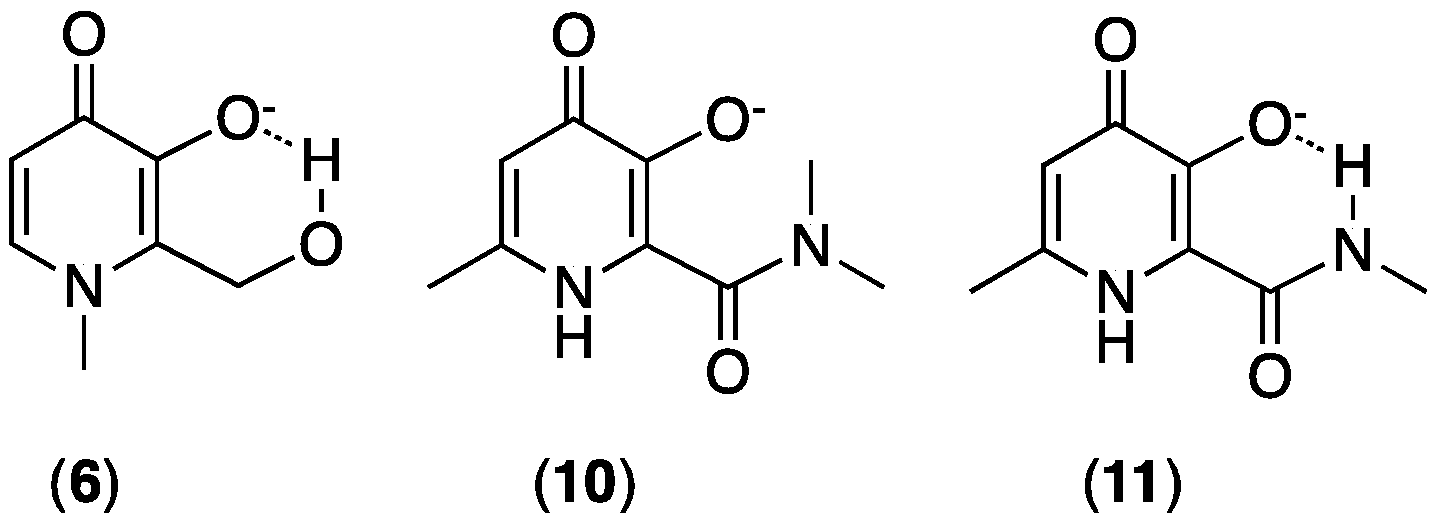
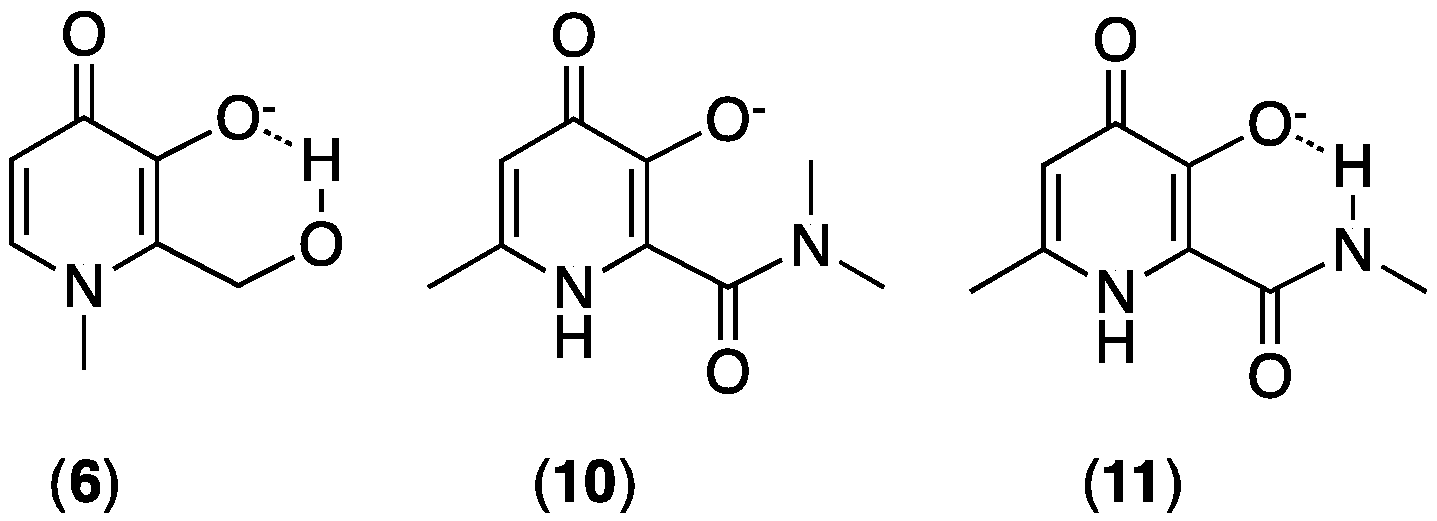
3.1. Tailoring 3,4-HP Properties by Ring Substitution
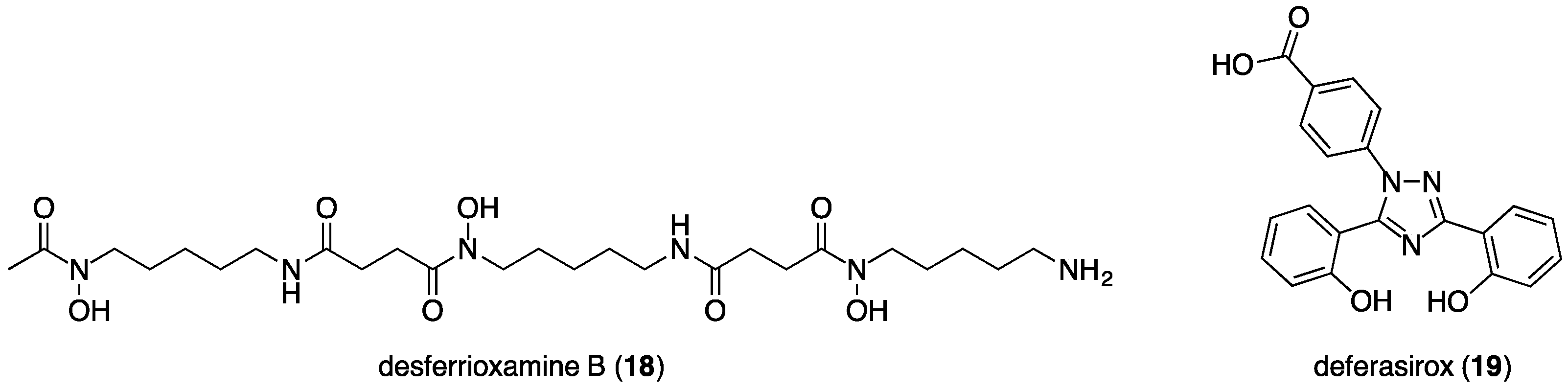
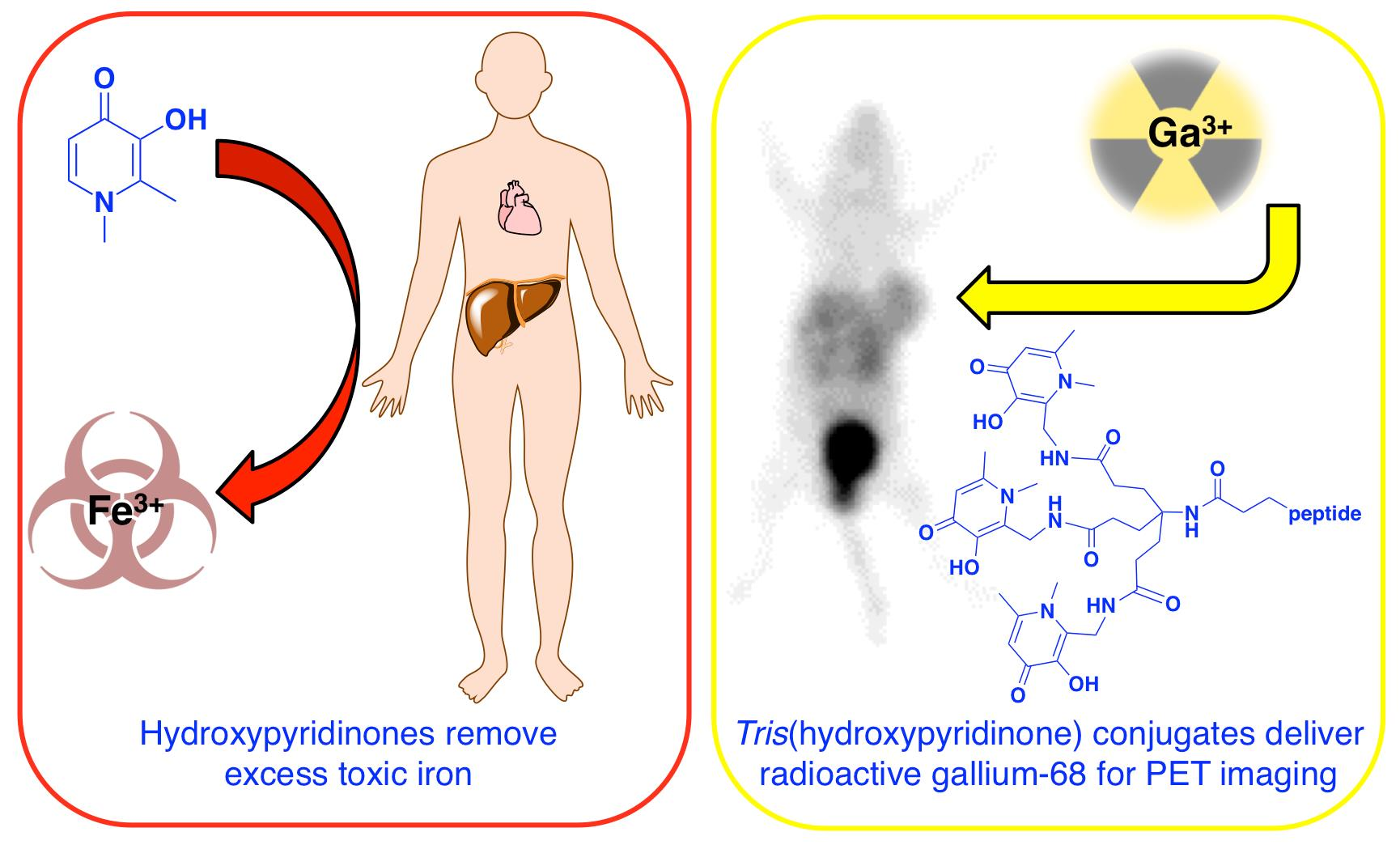
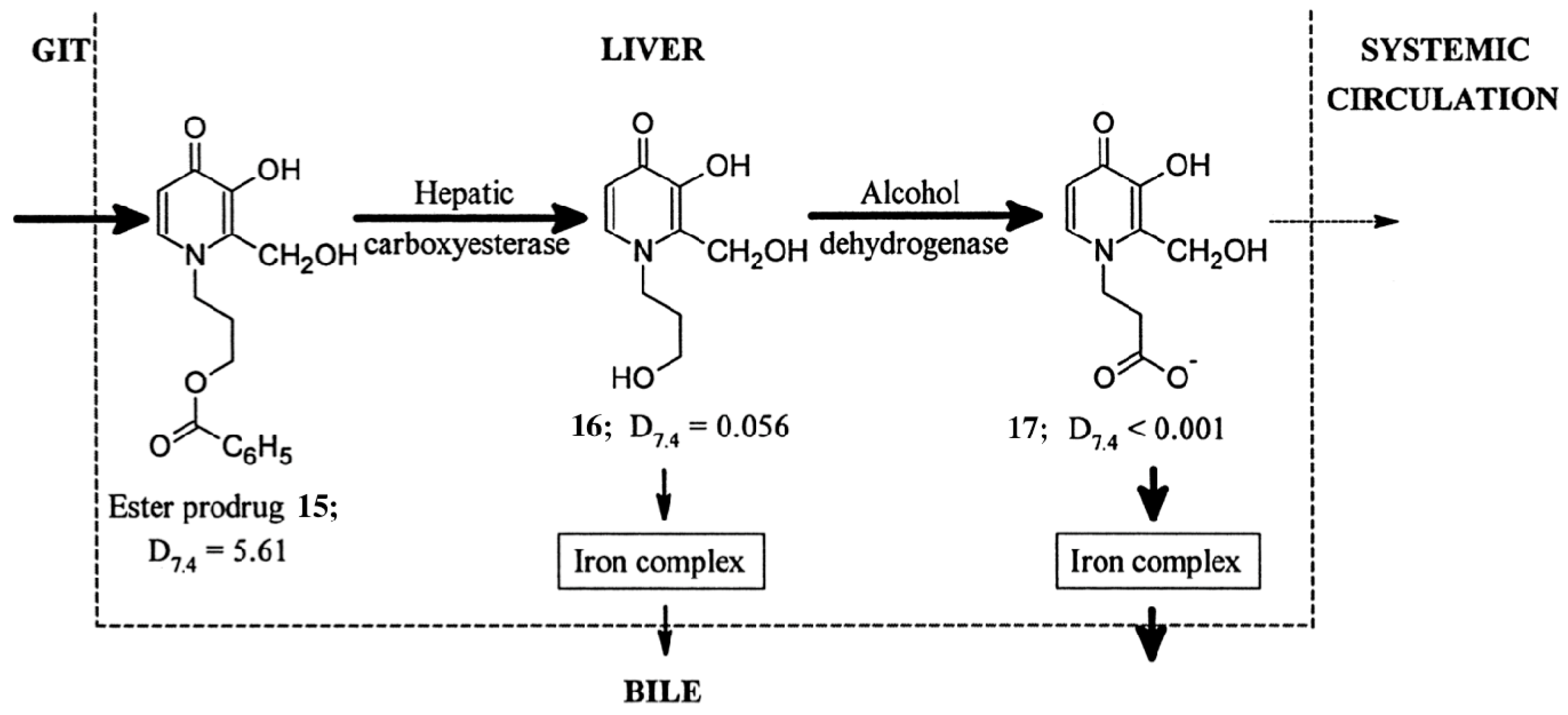
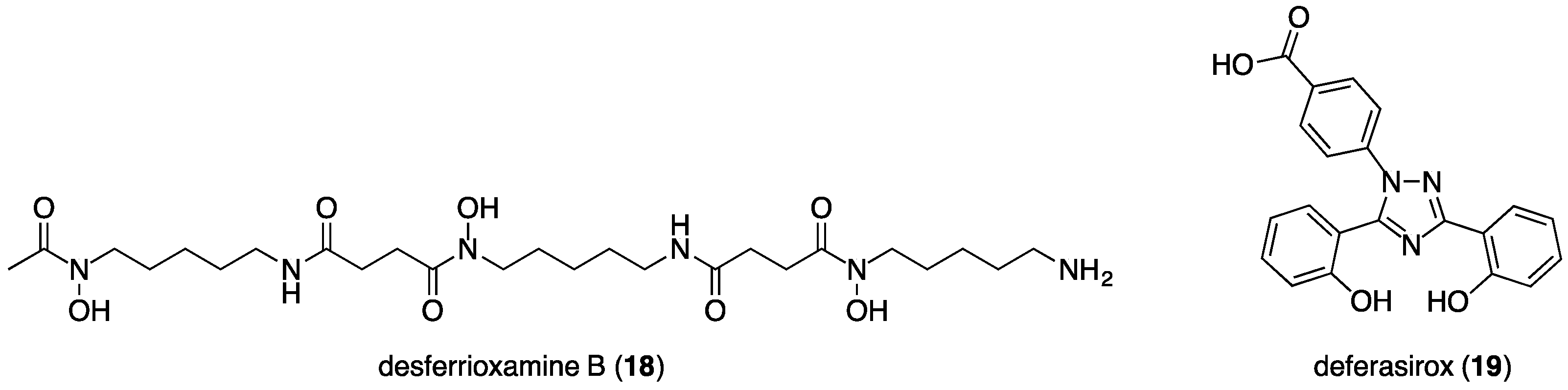
3.2. Deferiprone, Desferrioxamine and Deferasirox
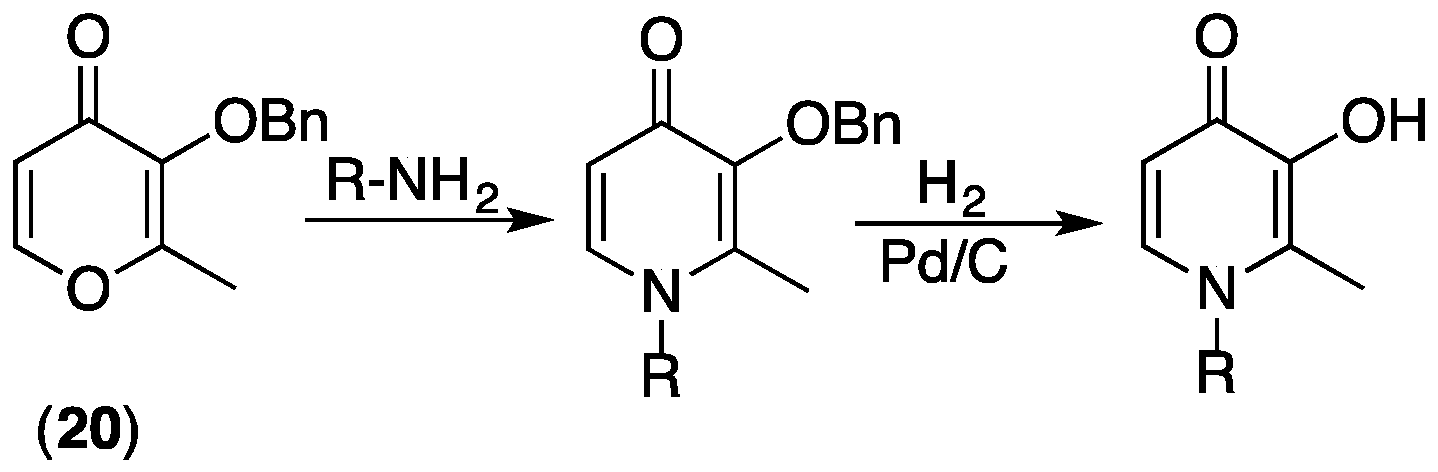
3.3. Synthesis of 3,4-HPs
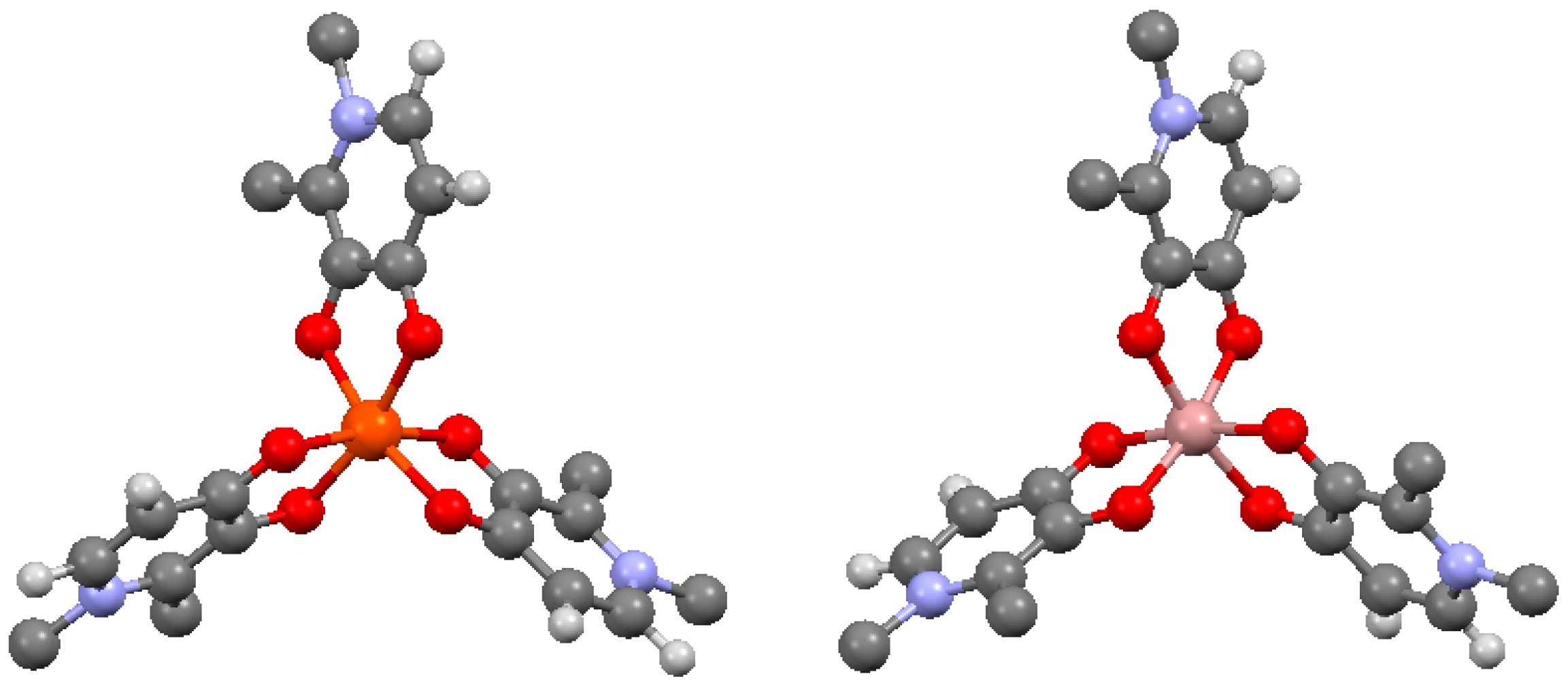
4. Hexadentate Tris(hydroxypyridinone) Ligands
4.1. Topology and Fe3+ Affinity
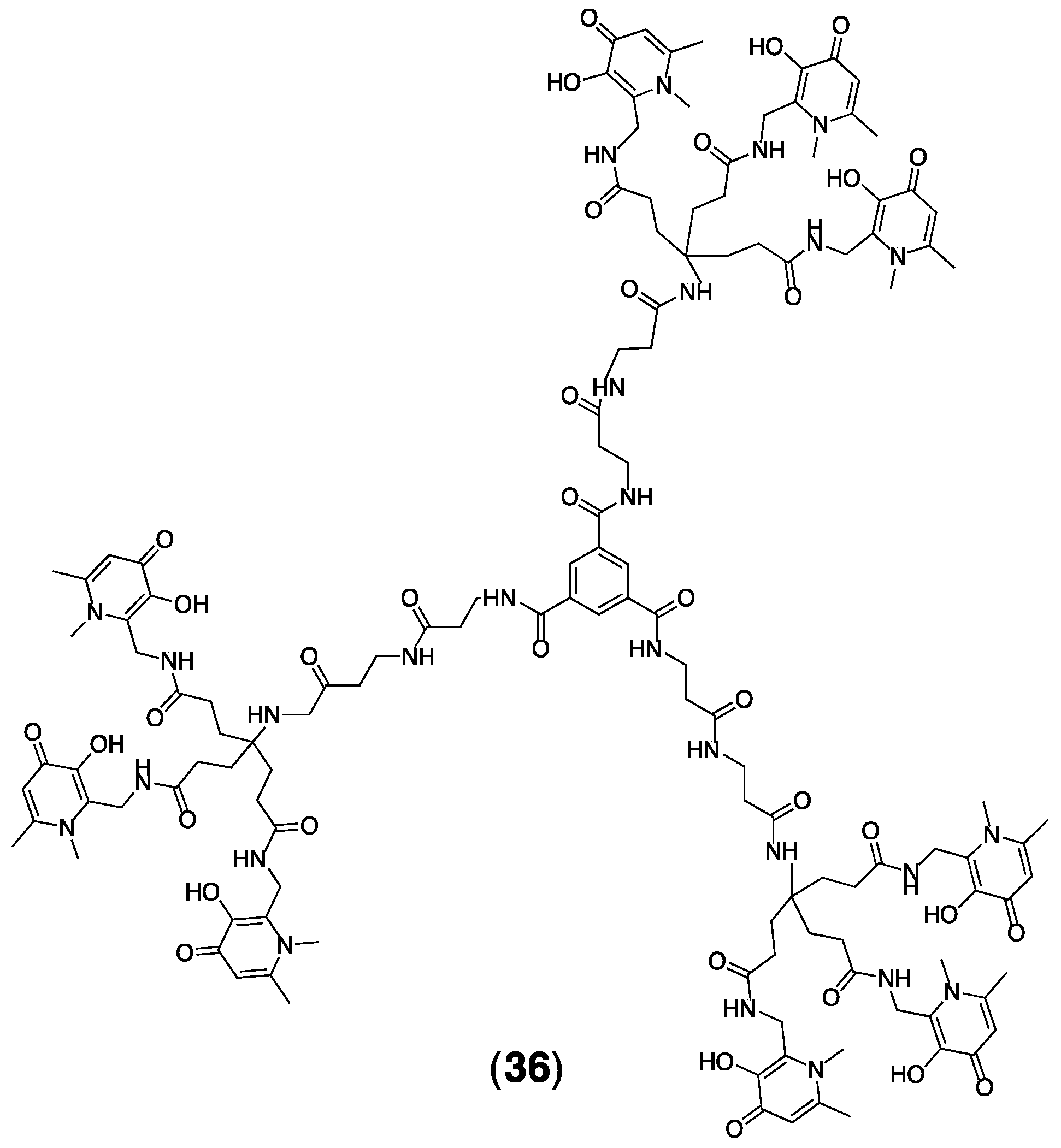
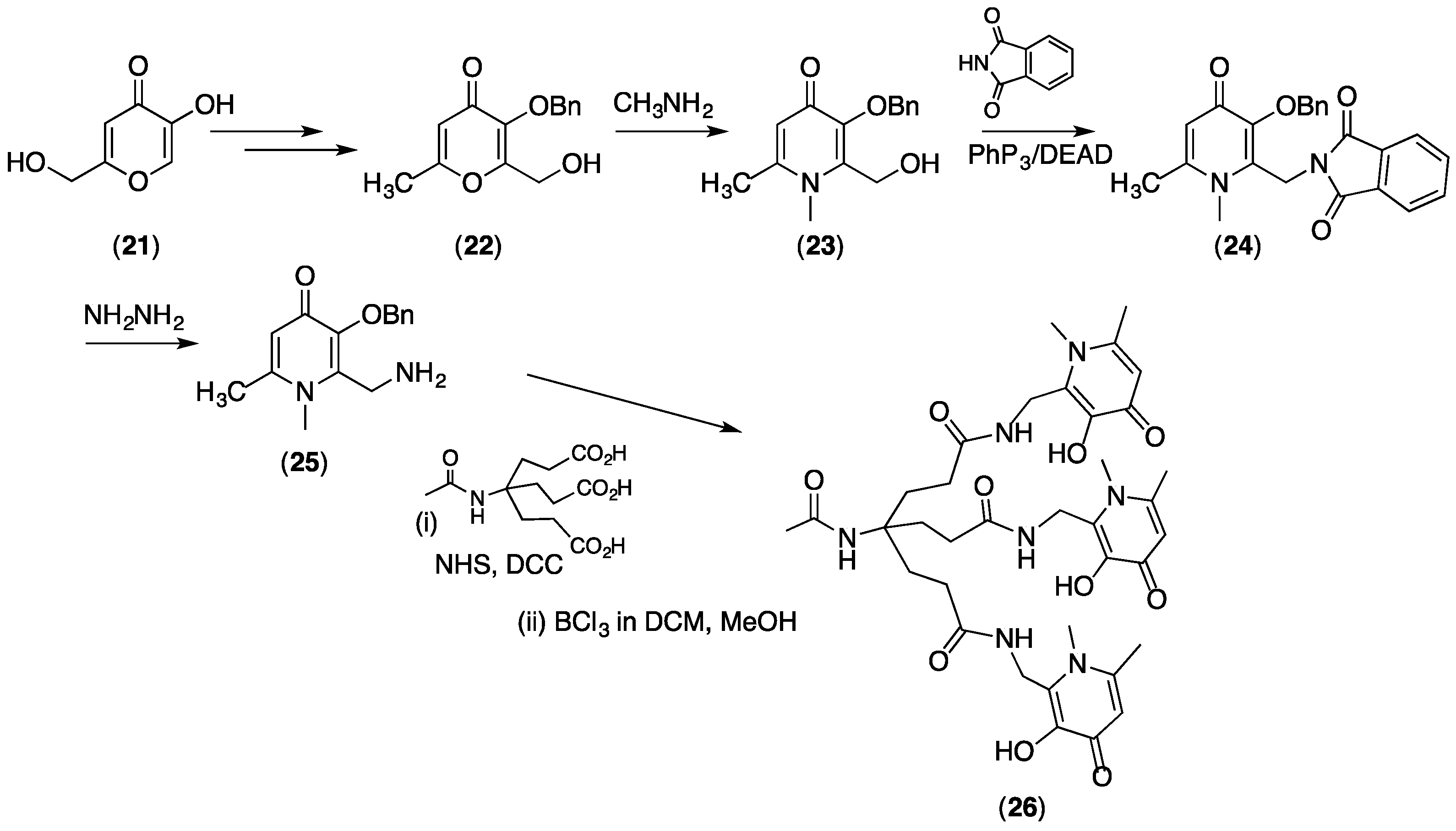
4.2. Dendrimers Based on THP Units
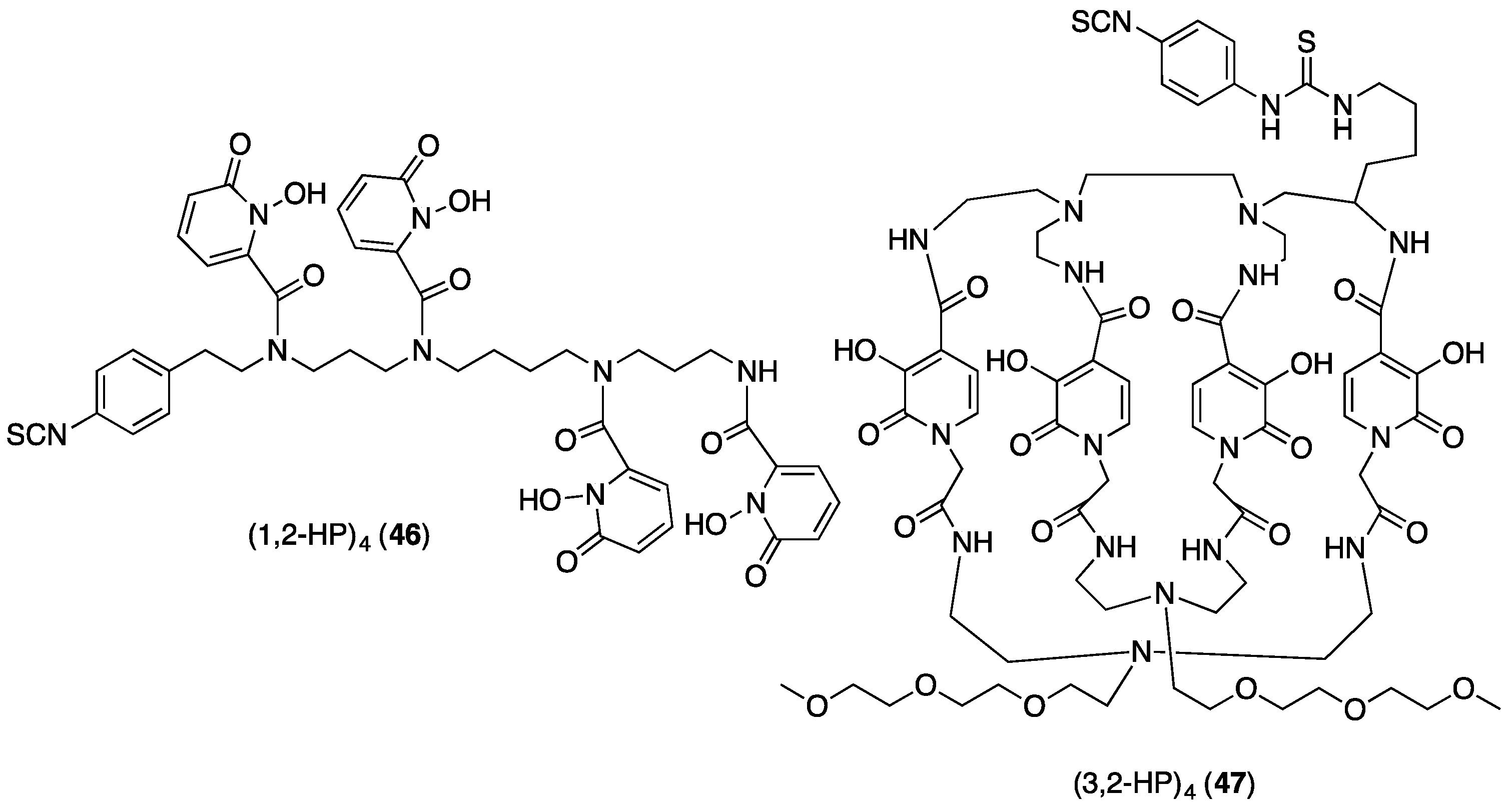
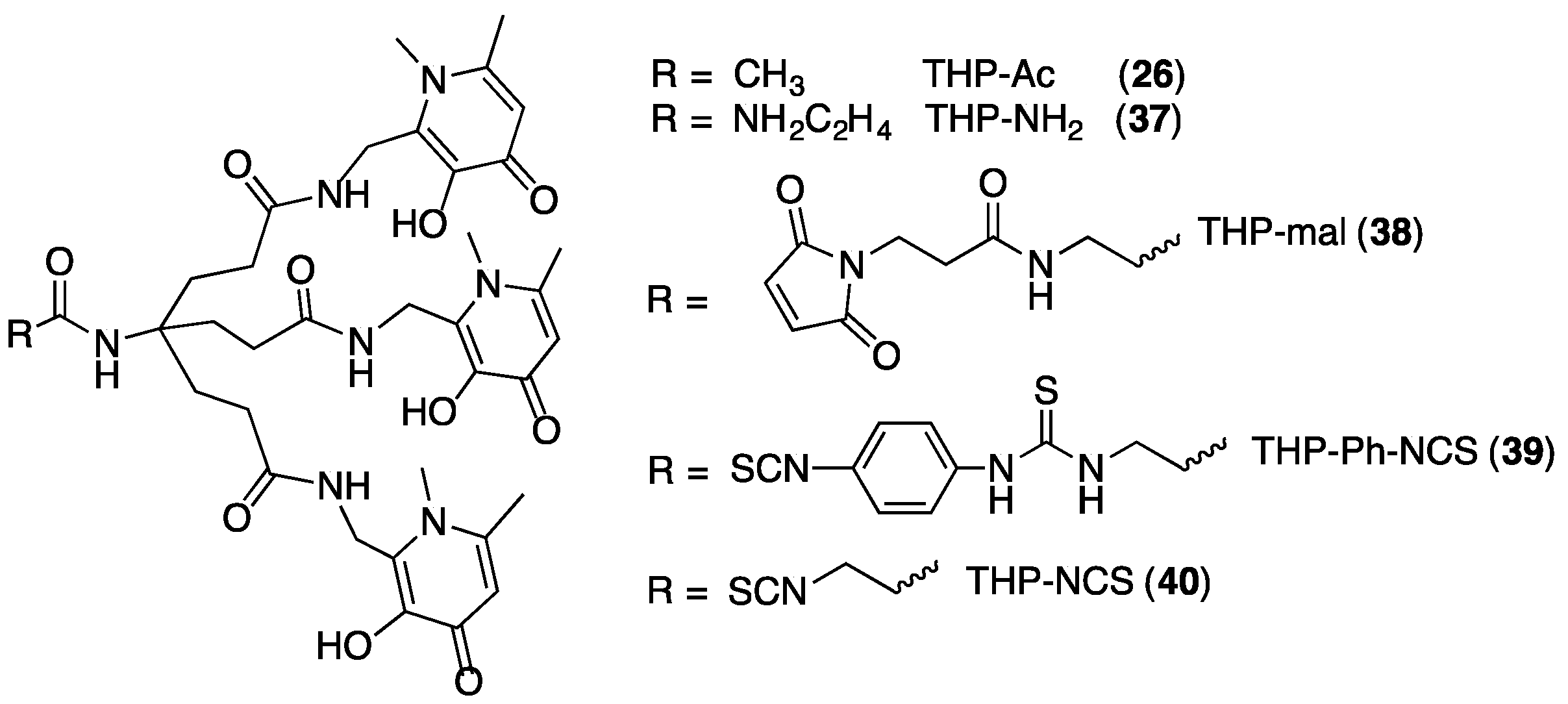
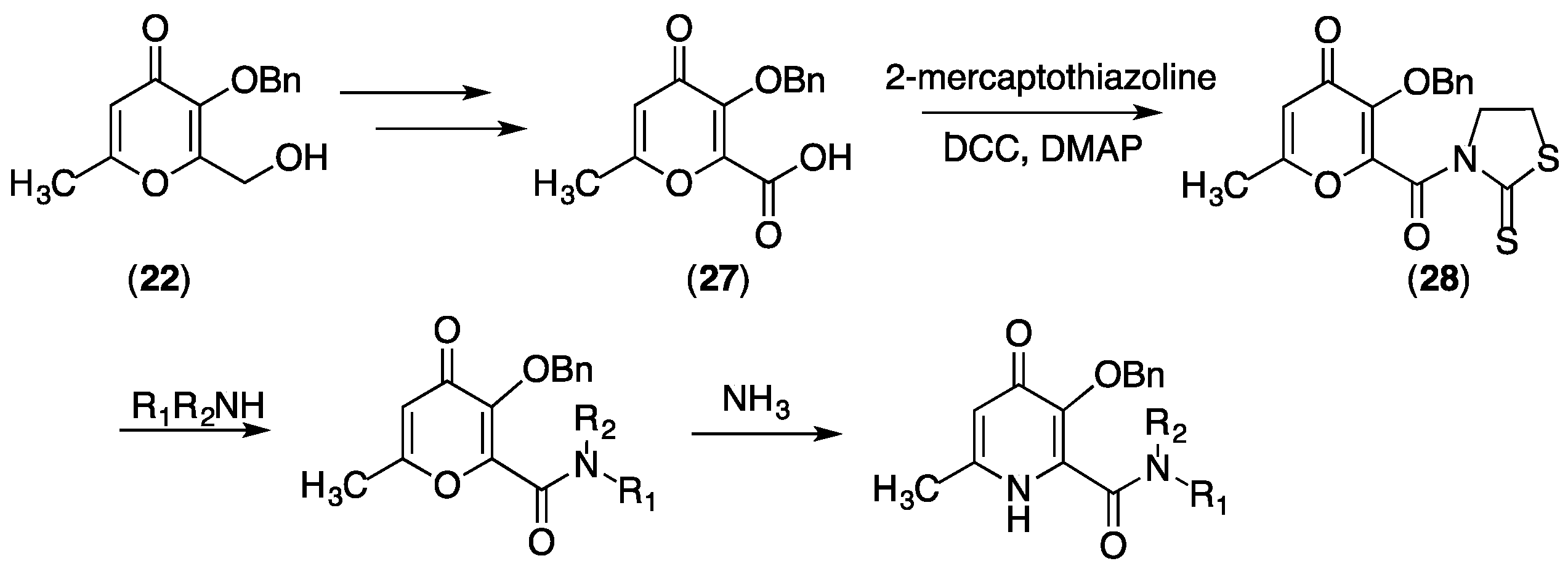
4.3. Derivatising THP Ligands
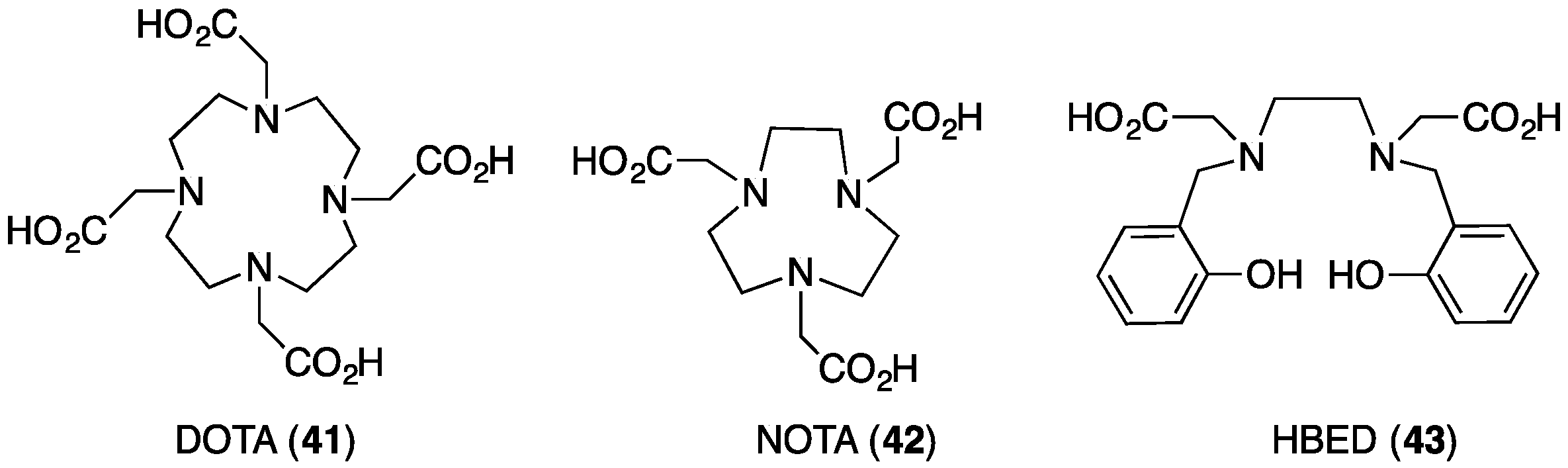
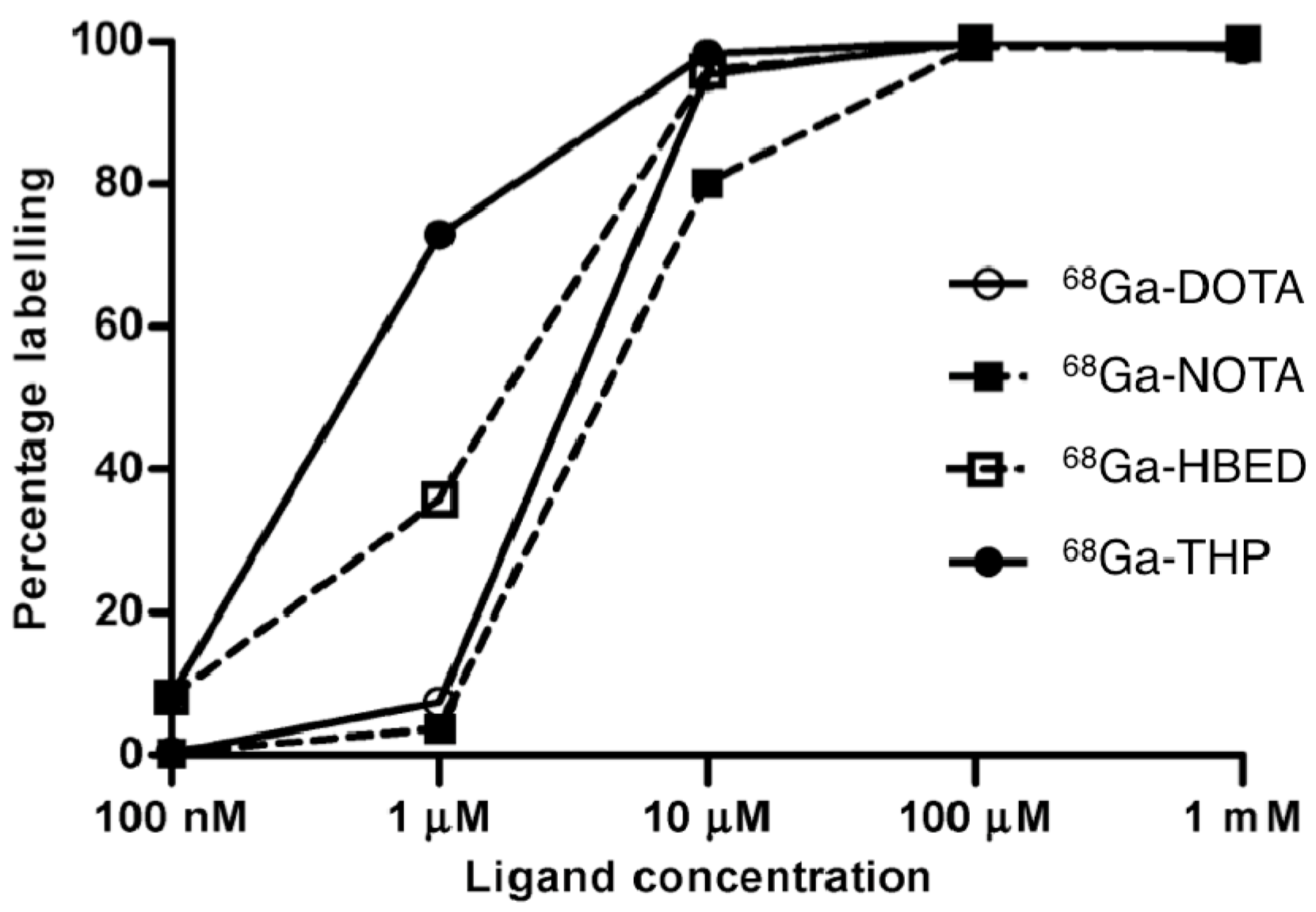
5. Tris(hydroxypyridinone) Ligands for Radiolabelling with 68Ga3+
5.1. Radiolabelling Peptides with 68Ga for PET Imaging: The Case for Tris(hydroxypyridinone) Derivatives
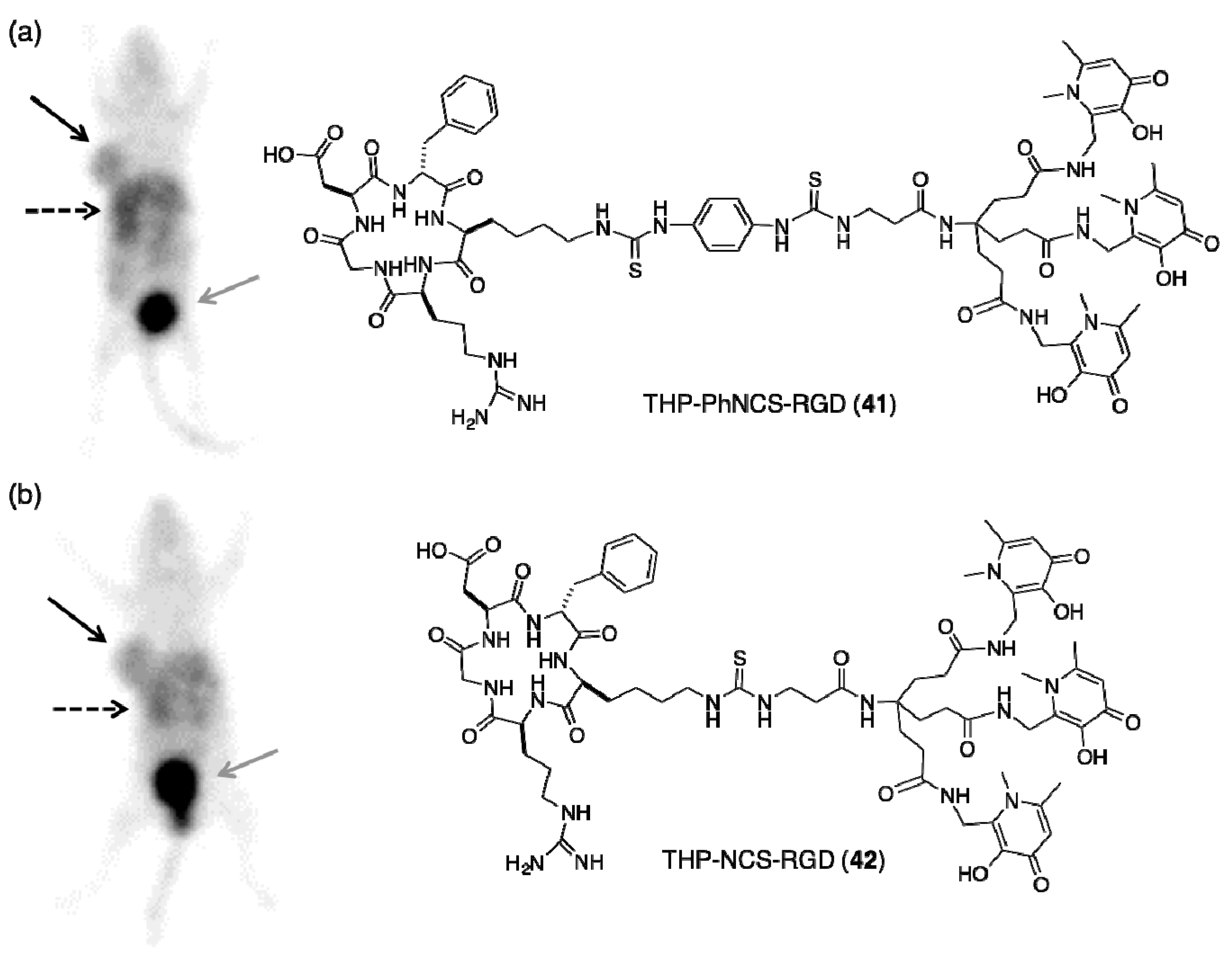
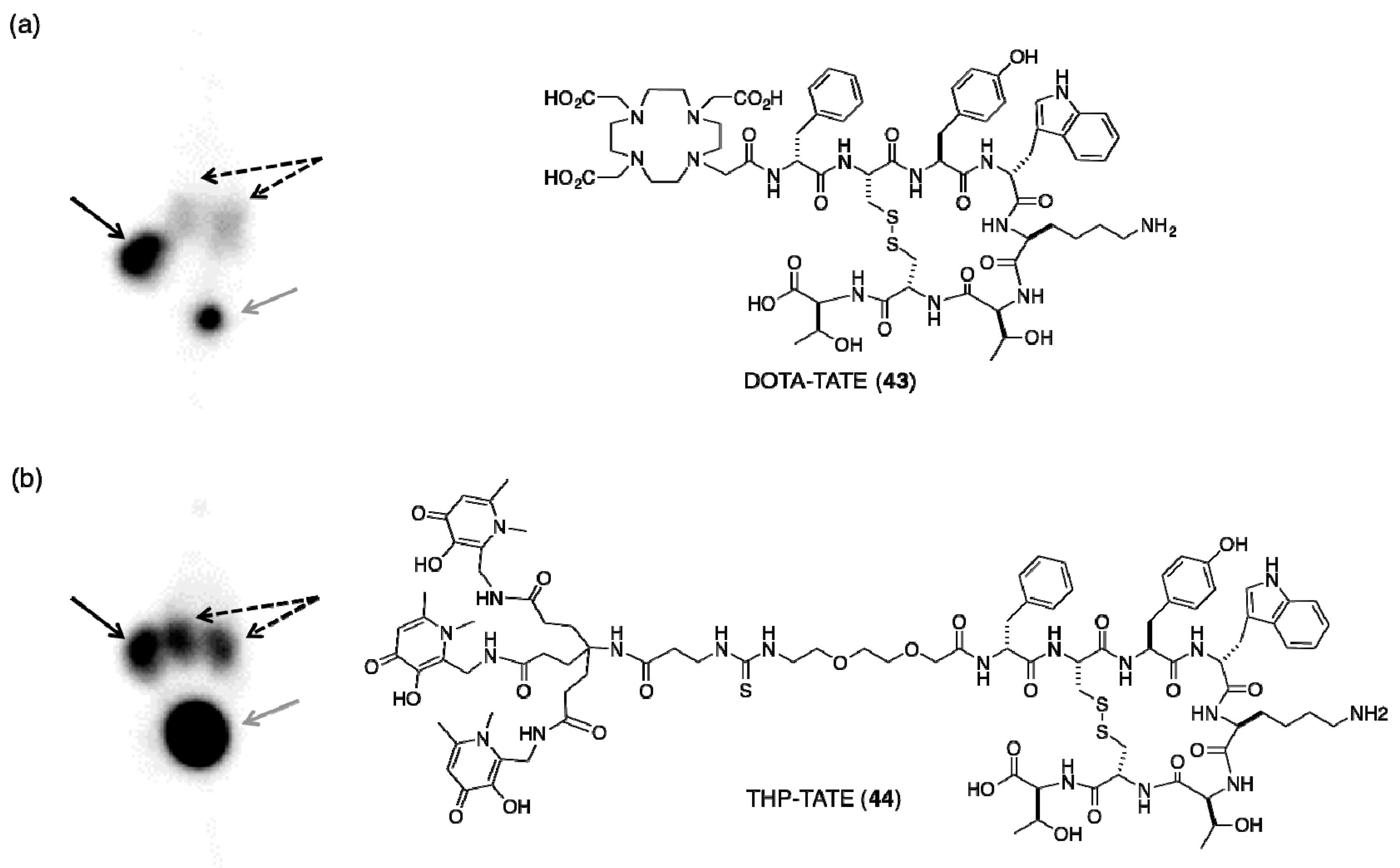
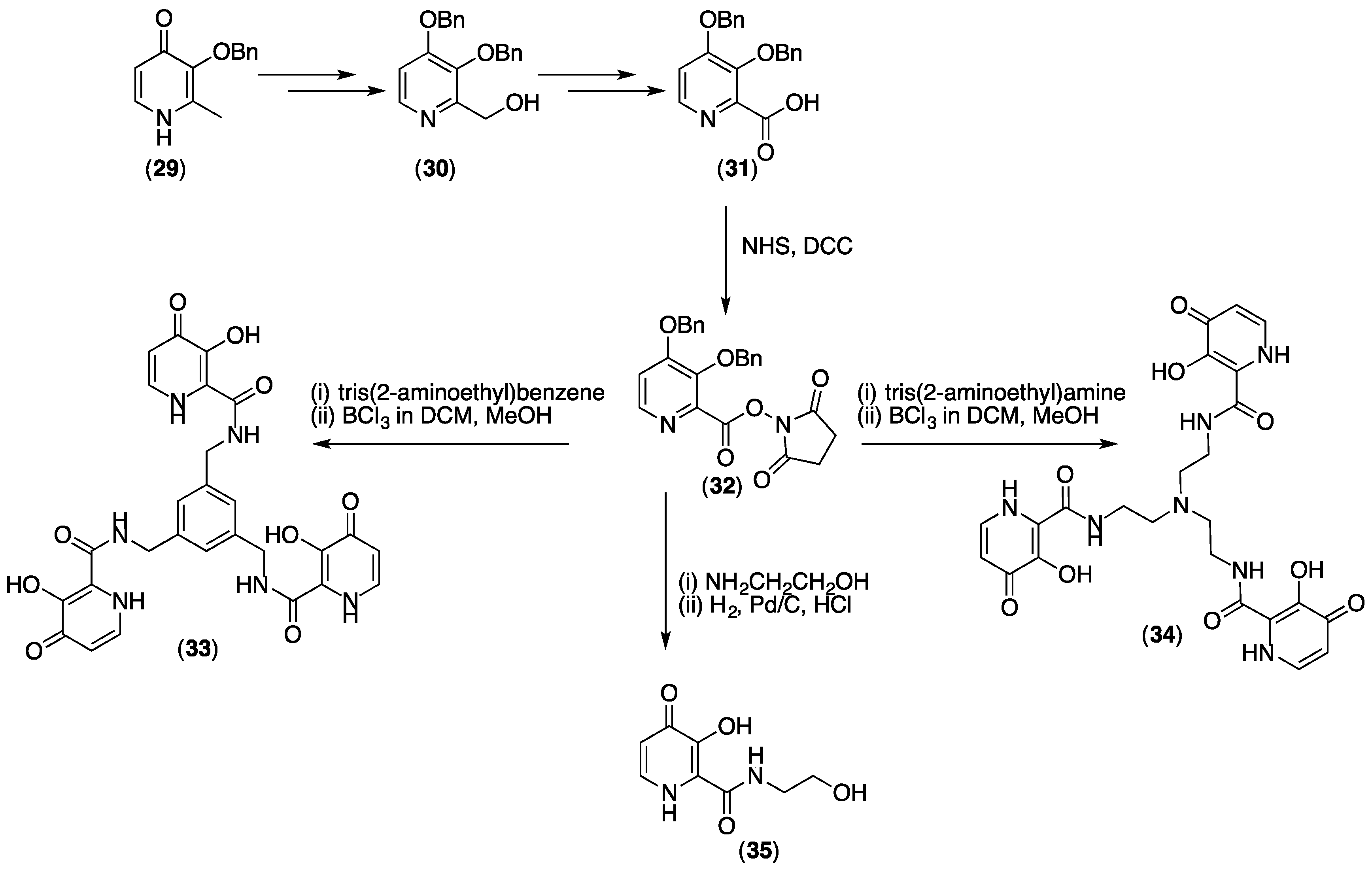
5.2. Tris(hydroxypyridinone) Bioconjugates
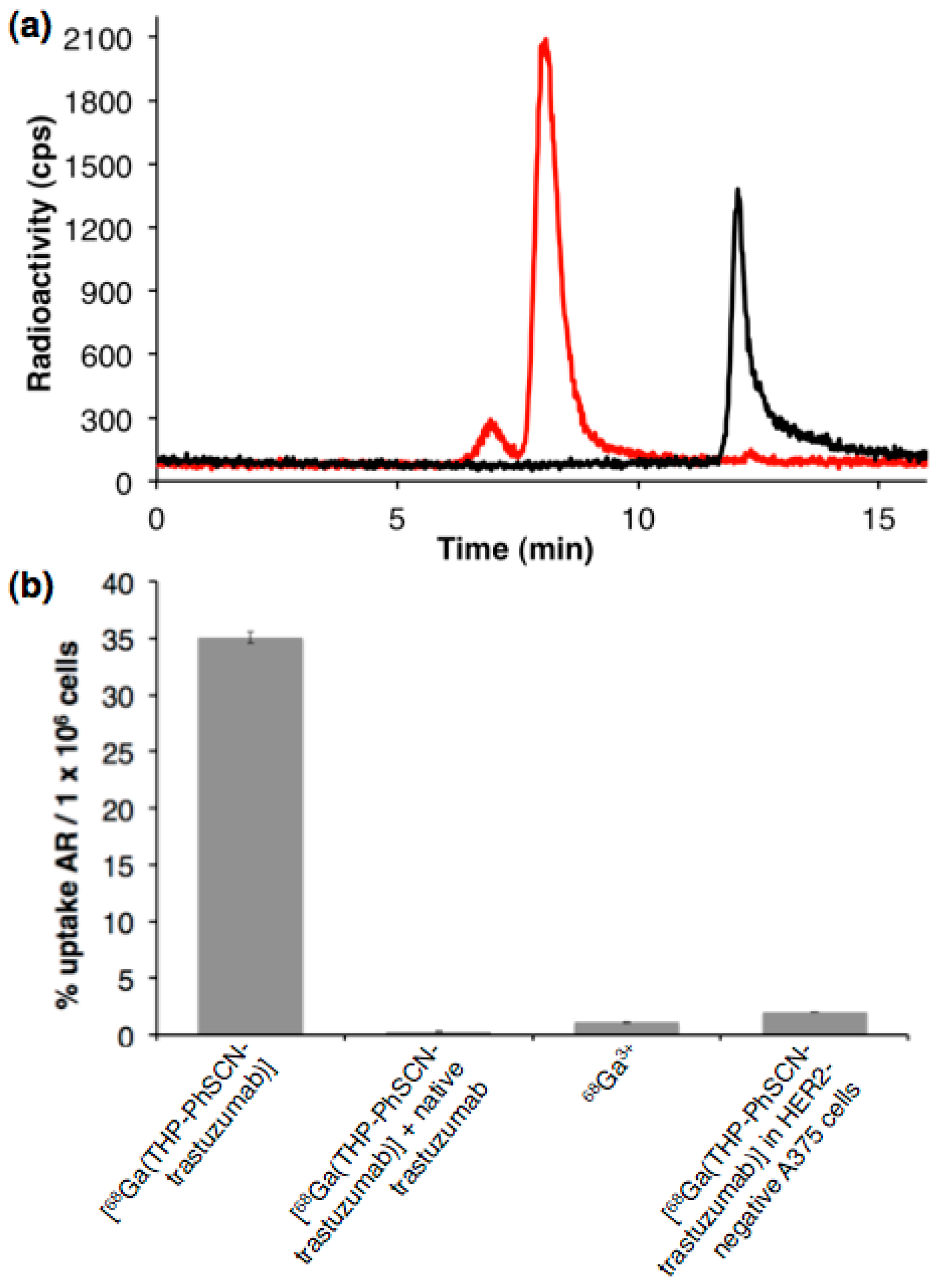
5.3. Preparation, Radiolabelling and In Vitro Uptake of a Trastuzumab Immunoconjugate
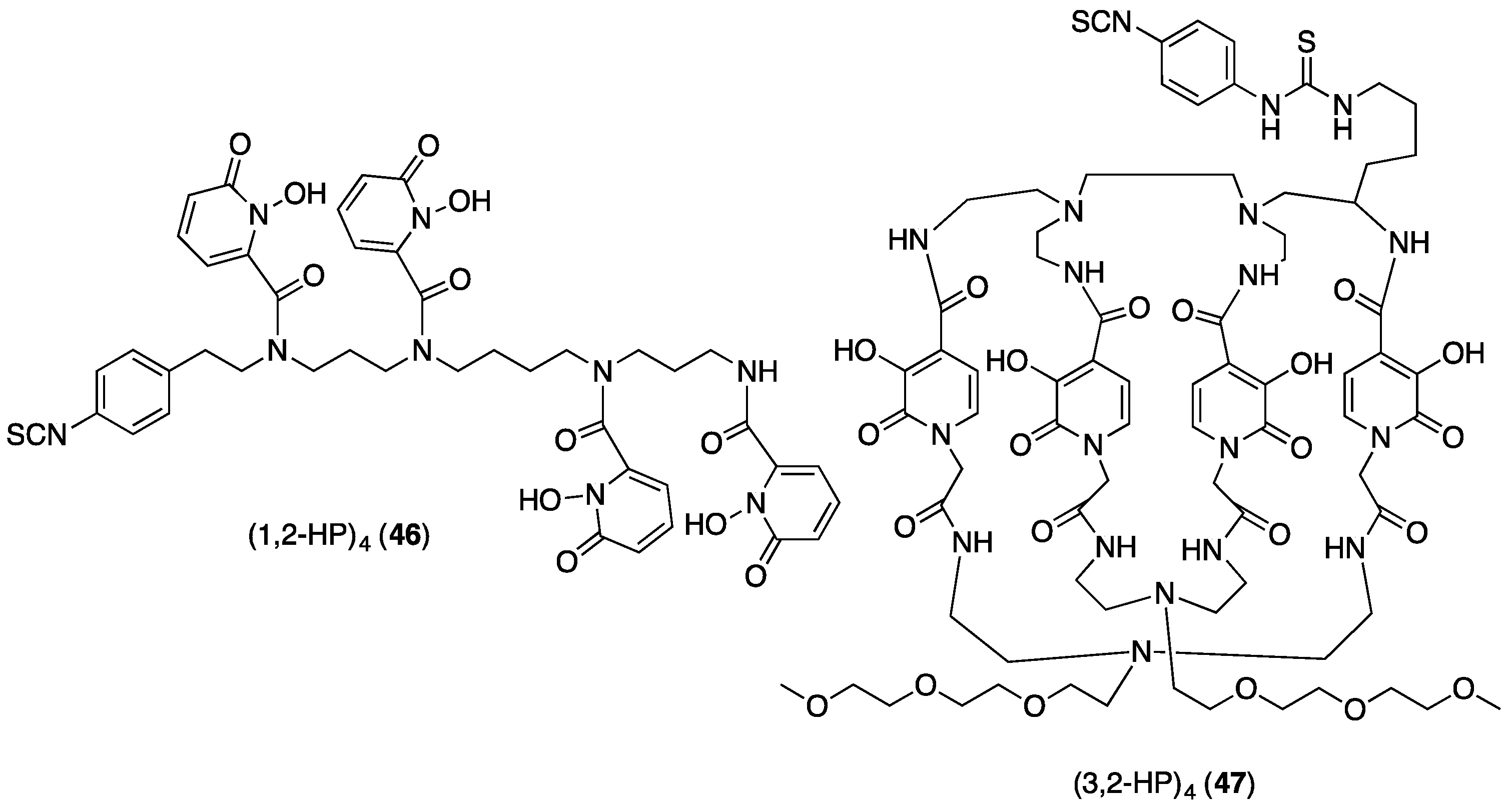
5.4. Other THP Derivatives
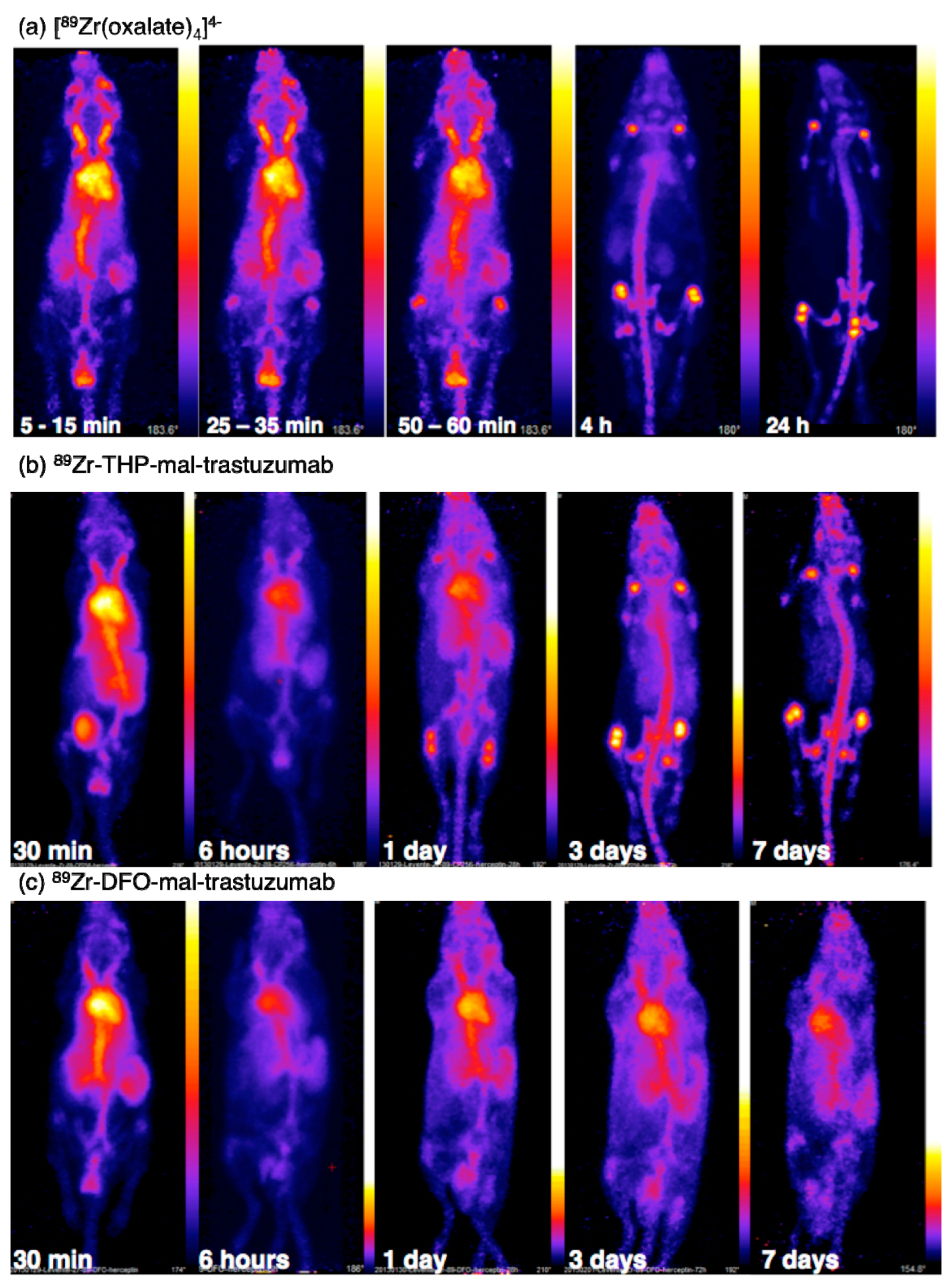
6. Hydroxypyridinones for Radiolabelling 89Zr4+
7. Concluding Remarks
Acknowledgments
Conflicts of Interest
Abbreviations
| Bn | Benzyl |
| DFO | Deferrioxamine B |
| EDTA | Ethylenediaminetetraacetate |
| GIT | Gastrointestinal tract |
| HER2 | Human epidermal growth factor receptor 2 |
| HP | Hydroxypyridinone |
| HPLC | High performance liquid chromatography |
| PET | Positron Emission Tomography |
| RGD | Cyclic RGDfK peptide |
| TATE | Octreotate |
| THP | Tris(hydroxypyridinone) |
| SSTR2 | Somatostatin 2 receptor |
References
- Blower, P.J. A nuclear chocolate box: The periodic table of nuclear medicine. Dalton Trans. 2015, 44, 4819–4844. [Google Scholar] [CrossRef] [PubMed]
- Hofman, M.S.; Kong, G.; Neels, O.C.; Eu, P.; Hong, E.; Hicks, R.J. High management impact of Ga-68 DOTATATE (Ga Tate) PET/CT for imaging neuroendocrine and other somatostatin expressing tumours. J. Med. Imaging Radiat. Oncol. 2012, 56, 40–47. [Google Scholar] [CrossRef] [PubMed]
- Afshar-Oromieh, A.; Zechmann, C.M.; Malcher, A.; Eder, M.; Eisenhut, M.; Linhart, H.G.; Holland-Letz, T.; Hadaschik, B.A.; Giesel, F.L.; Debus, J.; et al. Comparison of PET imaging with a 68Ga-labelled PSMA ligand and 18F-choline-based PET/CT for the diagnosis of recurrent prostate cancer. Eur. J. Nucl. Med. Mol. Imaging 2014, 41, 11–20. [Google Scholar] [CrossRef] [PubMed]
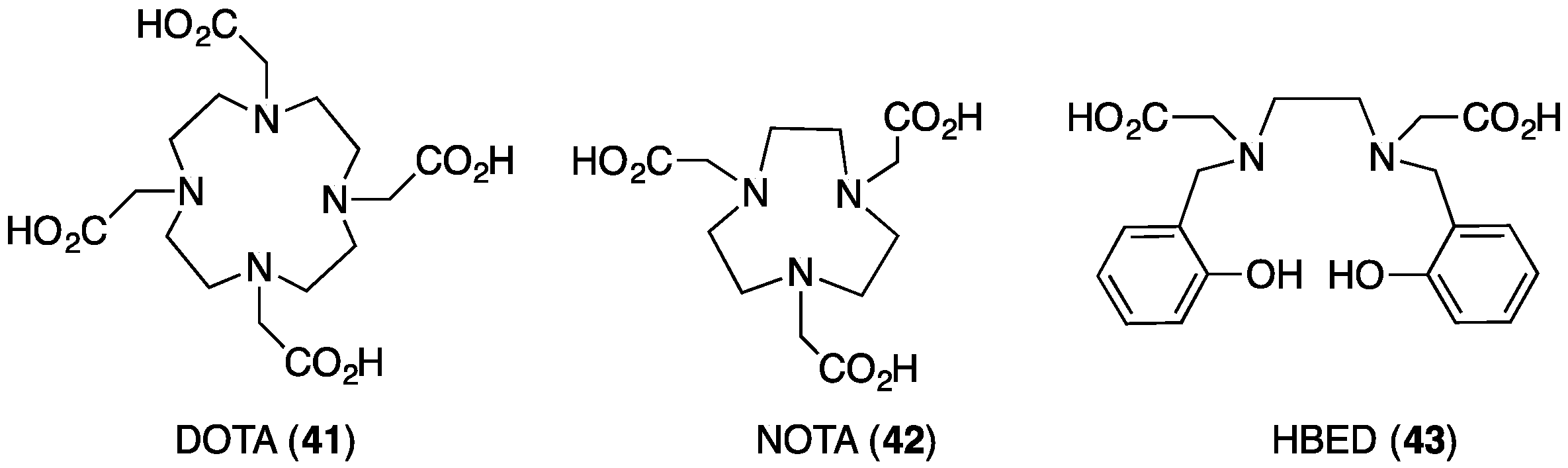
- Ma, M.T.; Blower, P.J. Chelators for diagnostic molecular imaging with radioisotopes of copper, gallium and zirconium. In Metal Chelation in Medicine; Crichton, R.R., Ward, R.J., Hider, R.C., Eds.; The Royal Society of Chemistry: Cambridge, UK, 2017; pp. 260–312. [Google Scholar]
- Price, E.W.; Orvig, C. Matching chelators to radiometals for radiopharmaceuticals. Chem. Soc. Rev. 2014, 43, 260–290. [Google Scholar] [CrossRef] [PubMed]
- Price, T.W.; Greenman, J.; Stasiuk, G.J. Current advances in ligand design for inorganic positron emission tomography tracers 68Ga, 64Cu, 89Zr and 44Sc. Dalton Trans. 2016, 45, 15702–15724. [Google Scholar] [CrossRef] [PubMed]
- Spang, P.; Herrmann, C.; Roesch, F. Bifunctional gallium-68 chelators: Past, present, and future. Semin. Nucl. Med. 2016, 46, 373–394. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.D.; Hider, R.C. Design of iron chelators with therapeutic application. Coord. Chem. Rev. 2002, 232, 151–171. [Google Scholar] [CrossRef]
- Liu, Z.D.; Hider, R.C. Design of clinically useful iron(III)-selective chelators. Med. Res. Rev. 2002, 22, 26–64. [Google Scholar] [CrossRef] [PubMed]
- Zhou, T.; Ma, Y.; Kong, X.; Hider, R.C. Design of iron chelators with therapeutic application. Dalton Trans. 2012, 41, 6371–6389. [Google Scholar] [CrossRef] [PubMed]
- Hider, R.C.; Kontoghiorghes, G.; Silver, J. Pharmaceutically Active 3-Hydroxypyrid-2-and-4-ones. UK Patent Application GB 2118176A, 1982. [Google Scholar]
- Shannon, R.D. Revised effective ionic radii and systematic studies of interatomic distances in halides and chalcogenides. Acta Crystallogr. Sect. A 1976, A32, 751–767. [Google Scholar] [CrossRef]
- Berry, D.J.; Ma, Y.; Ballinger, J.R.; Tavare, R.; Koers, A.; Sunassee, K.; Zhou, T.; Nawaz, S.; Mullen, G.E.D.; Hider, R.C.; et al. Efficient bifunctional gallium-68 chelators for positron emission tomography: Tris(hydroxypyridinone) ligands. Chem. Commun. 2011, 47, 7068–7070. [Google Scholar] [CrossRef] [PubMed]
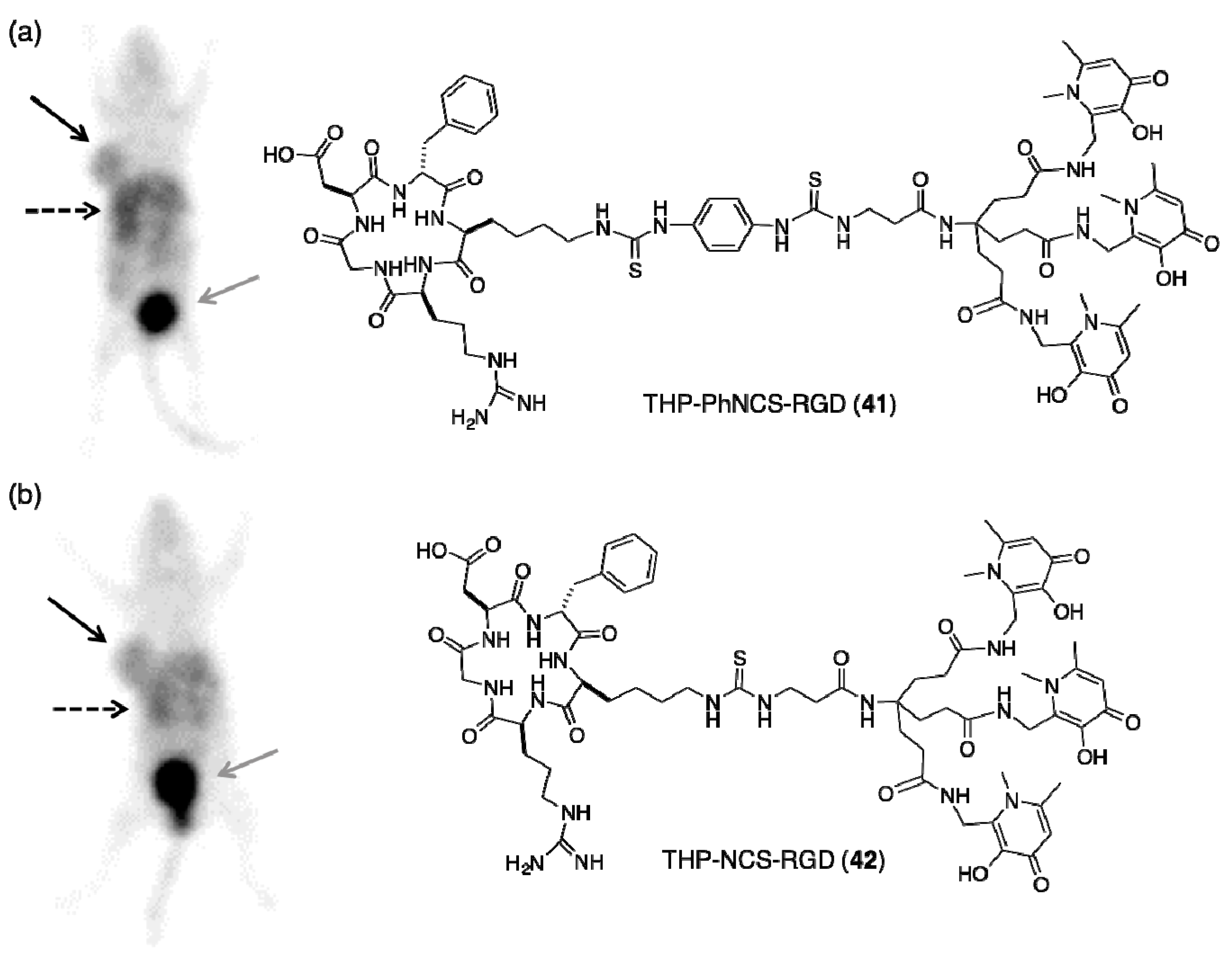
- Ma, M.T.; Cullinane, C.; Imberti, C.; Baguna Torres, J.; Terry, S.Y.A.; Roselt, P.; Hicks, R.J.; Blower, P.J. New tris(hydroxypyridinone) bifunctional chelators containing isothiocyanate groups provide a versatile platform for rapid one-step labeling and pet imaging with 68Ga3+. Bioconjug. Chem. 2016, 27, 309–318. [Google Scholar] [CrossRef] [PubMed]
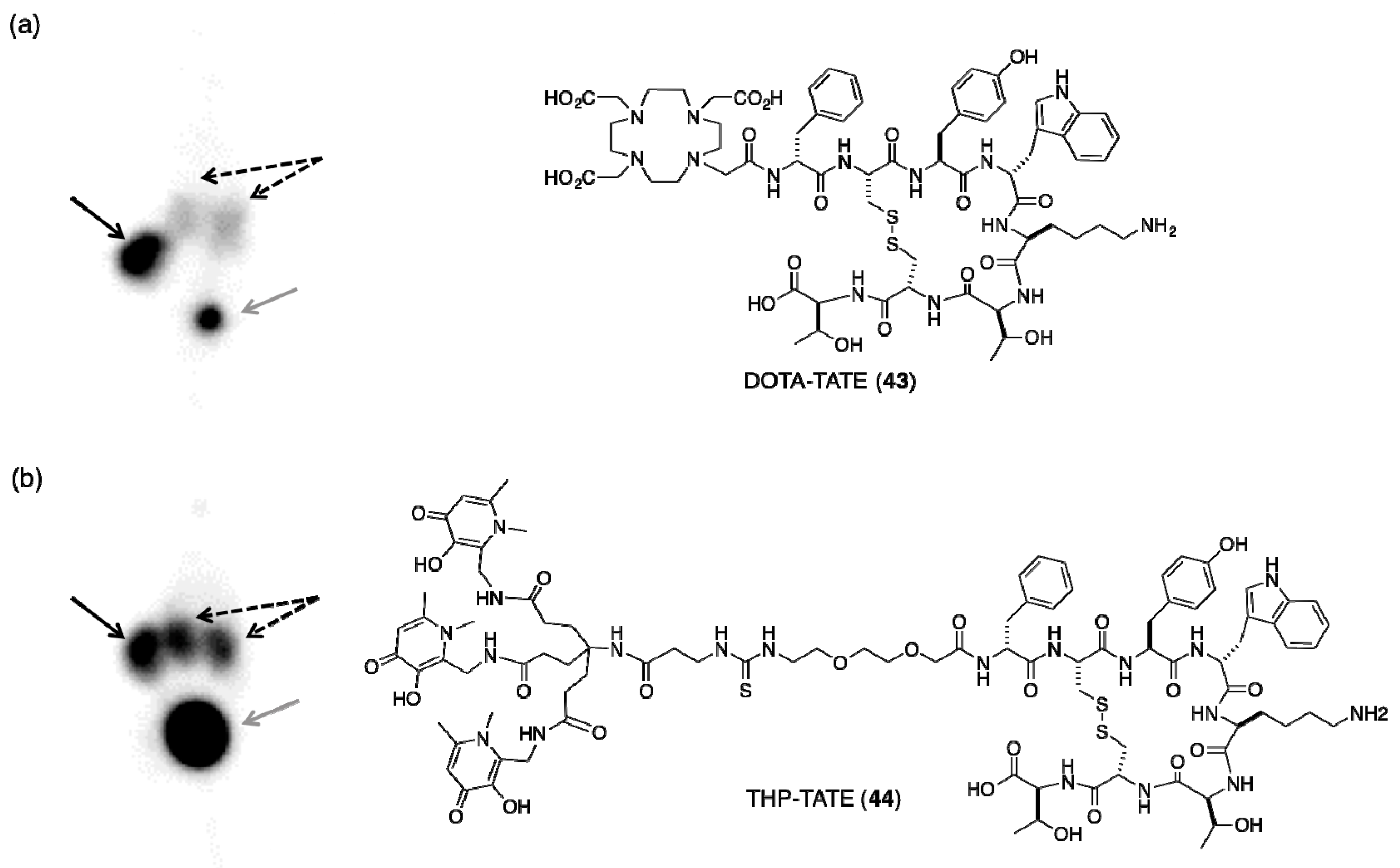
- Ma, M.T.; Cullinane, C.; Waldeck, K.; Roselt, P.; Hicks, R.J.; Blower, P.J. Rapid kit-based 68Ga-labelling and PET imaging with THP-Tyr3-octreotate: A preliminary comparison with DOTA-Tyr3-octreotate. EJNMMI Res. 2015, 5, 52. [Google Scholar] [CrossRef] [PubMed]
- Ma, M.T.; Meszaros, L.K.; Paterson, B.M.; Berry, D.J.; Cooper, M.S.; Ma, Y.; Hider, R.C.; Blower, P.J. Tripodal tris(hydroxypyridinone) ligands for immunoconjugate PET imaging with 89Zr4+: Comparison with desferrioxamine-B. Dalton Trans. 2015, 44, 4884–4900. [Google Scholar] [CrossRef] [PubMed]
- Gateau, C.; Mintz, E.; Delangle, P. Rational design of copper and iron chelators to treat Wilson’s disease and hemochromatosis. In Ligand Design in Medicinal Inorganic Chemistry; Storr, T., Ed.; John Wiley & Sons, Ltd.: Chichester, UK, 2014; pp. 287–319. [Google Scholar]
- Gumienna-Kontecka, E.; Pyrkosz-Bulska, M.; Szebesczyk, A.; Ostrowska, M. Iron chelating strategies in systemic metal overload, neurodegeneration and cancer. Curr. Med. Chem. 2014, 21, 3741–3767. [Google Scholar] [CrossRef] [PubMed]
- Price, E.W.; Orvig, C. The Chemistry of Inorganic Nuclides (86Y 68Ga 64Cu 89Zr 124I). In The Chemistry of Molecular Imaging; Long, N., Wong, W.-T., Eds.; John Wiley & Sons: Hoboken, NJ, USA, 2015; pp. 105–136. [Google Scholar]
- Dobbin, P.S.; Hider, R.C.; Hall, A.D.; Taylor, P.D.; Sarpong, P.; Porter, J.B.; Xiao, G.; van der Helm, D. Synthesis, physicochemical properties, and biological evaluation of N-substituted 2-alkyl-3-hydroxy-4(1H)-pyridinones: Orally active iron chelators with clinical potential. J. Med. Chem. 1993, 36, 2448–2458. [Google Scholar] [CrossRef] [PubMed]
- Hider, R.C.; Hall, A.D. Clinically useful chelators of tripositive elements. Prog. Med. Chem. 1991, 28, 41–173. [Google Scholar] [PubMed]
- Li, Y.J.; Martell, A.E. Potentiometric and spectrophotometric determination of stabilities of the 1-hydroxy-2-pyridinone complexes of trivalent and divalent metal ions. Inorg. Chim. Acta 1993, 214, 103–111. [Google Scholar]
- Scarrow, R.C.; Riley, P.E.; Abu-Dari, K.; White, D.L.; Raymond, K.N. Ferric ion sequestering agents. 13. Synthesis, structures, and thermodynamics of complexation of cobalt(III) and iron(III) tris complexes of several chelating hydroxypyridinones. Inorg. Chem. 1985, 24, 954–967. [Google Scholar] [CrossRef]
- Clarke, E.T.; Martell, A.E. 1-Methyl-3-hydroxy-2-pyridinone and 1,4-dihydroxy-2-pyridinone complexes of the trivalent metal ions of iron(III), gallium(III), aluminum(III), indium(III) and gadolinium(III): Potentiometric and spectrophotometric determination of stabilities. Inorg. Chim. Acta 1992, 196, 185–194. [Google Scholar] [CrossRef]
- Clarke, E.T.; Martell, A.E. Stabilities of 1,2-dimethyl-3-hydroxy-4-pyridinone chelates of divalent and trivalent metal ions. Inorg. Chim. Acta 1992, 191, 56–63. [Google Scholar] [CrossRef]
- Amelia Santos, M. Hydroxypyridinone complexes with aluminum. In vitro/vivo studies and perspectives. Coord. Chem. Rev. 2002, 228, 187–203. [Google Scholar] [CrossRef]
- Hsieh, W.-Y.; Liu, S. Synthesis and characterization of Cr(III) complexes with 3-hydroxy-4-pyrones and 1,2-dimethyl-3-hydroxy-4-pyridinone (DMHP): X-ray crystal structures of Cr(DMHP)3·12H2O and Cr(ma)3. Synth. React. Inorg. Met.-Org. Nano-Met. Chem. 2005, 35, 61–70. [Google Scholar] [CrossRef]
- Hsieh, W.-Y.; Liu, S. Synthesis, characterization, and structures of Mn(DMHP)3·12H2O and Mn(DMHP)2Cl·0.5H2O. Inorg. Chem. 2005, 44, 2031–2038. [Google Scholar] [CrossRef] [PubMed]
- Nelson, W.O.; Karpishin, T.B.; Rettig, S.J.; Orvig, C. Aluminum and gallium compounds of 3-hydroxy-4-pyridinones: Synthesis, characterization, and crystallography of biologically active complexes with unusual hydrogen bonding. Inorg. Chem. 1988, 27, 1045–1051. [Google Scholar] [CrossRef]
- Matsuba, C.A.; Nelson, W.O.; Rettig, S.J.; Orvig, C. Neutral water-soluble indium complexes of 3-hydroxy-4-pyrones and 3-hydroxy-4-pyridinones. Inorg. Chem. 1988, 27, 3935–3939. [Google Scholar] [CrossRef]
- Charalambous, J.; Dodd, A.; McPartlin, M.; Matondo, S.O.C.; Pathirana, N.D.; Powell, H.R. Synthesis and X-ray crystal structure of tris(1,2-dimethyl-3-hydroxypyrid-4-onato)iron(III). Polyhedron 1988, 7, 2235–2237. [Google Scholar] [CrossRef]
- Evans, R.W.; Rafique, R.; Zarea, A.; Rapisarda, C.; Cammack, R.; Evans, P.J.; Porter, J.B.; Hider, R.C. Nature of non-transferrin-bound iron: Studies on iron citrate complexes and thalassemic sera. J. Biol. Inorg. Chem. 2008, 13, 57–74. [Google Scholar] [CrossRef] [PubMed]
- Fenton, H.J.H. LXXIII.—Oxidation of tartaric acid in presence of iron. J. Chem. Soc. Trans. 1894, 65, 899–910. [Google Scholar] [CrossRef]
- Liu, Z.D.; Khodr, H.H.; Liu, D.Y.; Lu, S.L.; Hider, R.C. Synthesis, physicochemical characterization, and biological evaluation of 2-(1′-hydroxyalkyl)-3-hydroxypyridin-4-ones: Novel iron chelators with enhanced pFe3+ values. J. Med. Chem. 1999, 42, 4814–4823. [Google Scholar] [CrossRef] [PubMed]
- Motekaitis, R.J.; Martell, A.E. Stabilities of the iron(III) chelates of 1,2-dimethyl-3-hydroxy-4-pyridinone and related ligands. Inorg. Chim. Acta 1991, 183, 71–80. [Google Scholar] [CrossRef]
- Xie, Y.-Y.; Lu, Z.; Kong, X.-L.; Zhou, T.; Bansal, S.; Hider, R. Systematic comparison of the mono-, dimethyl- and trimethyl 3-hydroxy-4(1H)-pyridones—Attempted optimization of the orally active iron chelator, deferiprone. Eur. J. Med. Chem. 2016, 115, 132–140. [Google Scholar] [CrossRef] [PubMed]
- Clevette, D.J.; Lyster, D.M.; Nelson, W.O.; Rihela, T.; Webb, G.A.; Orvig, C. Solution chemistry of gallium and indium 3-hydroxy-4-pyridinone complexes in vitro and in vivo. Inorg. Chem. 1990, 29, 667–672. [Google Scholar] [CrossRef]
- Piyamongkol, S.; Ma, Y.M.; Kong, X.L.; Liu, Z.D.; Aytemir, M.D.; van der Helm, D.; Hider, R.C. Amido-3-hydroxypyridin-4-ones as iron(III) ligands. Chem. Eur. J. 2010, 16, 6374–6381. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Lu, Z.; Kong, X.; Ma, Y.; Zhang, X.; Bansal, S.S.; Abbate, V.; Hider, R.C. Design and synthesis of novel pegylated iron chelators with decreased metabolic rate. Future Med. Chem. 2015, 7, 2439–2449. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Luo, W.; Quinn, P.J.; Liu, Z.; Hider, R.C. Design, synthesis, physicochemical properties, and evaluation of novel iron chelators with fluorescent sensors. J. Med. Chem. 2004, 47, 6349–6362. [Google Scholar] [CrossRef] [PubMed]
- Abbate, V.; Reelfs, O.; Kong, X.; Pourzand, C.; Hider, R.C. Dual selective iron chelating probes with a potential to monitor mitochondrial labile iron pools. Chem. Commun. 2016, 52, 784–787. [Google Scholar] [CrossRef] [PubMed]
- Galanello, R. Deferiprone in the treatment of transfusion-dependent thalassemia: A review and perspective. Ther. Clin. Risk Manag. 2007, 3, 795–805. [Google Scholar] [PubMed]
- Heinz, U.; Hegetschweiler, K.; Acklin, P.; Faller, B.; Lattmann, R.; Schnebli, H.P. 4-[3,5-bis(2-hydroxyphenyl)-1,2,4-triazol-1-yl]-benzoic acid: A novel efficient and eelective iron(III) complexing agent. Angew. Chem. Int. Ed. 1999, 38, 2568–2570. [Google Scholar] [CrossRef]
- Xia, S.; Zhang, W.; Huang, L.; Jiang, H. Comparative efficacy and safety of deferoxamine, deferiprone and deferasirox on severe thalassemia: A meta-analysis of 16 randomized controlled trials. PLoS ONE 2013, 8, e82662. [Google Scholar] [CrossRef] [PubMed]
- Cermak, J.; Jonasova, A.; Vondrakova, J.; Cervinek, L.; Belohlavkova, P.; Neuwirtova, R. A comparative study of deferasirox and deferiprone in the treatment of iron overload in patients with myelodysplastic syndromes. Leuk. Res. 2013, 37, 1612–1615. [Google Scholar] [CrossRef] [PubMed]
- Zachariah, M.; Tony, S.; Bashir, W.; Al Rawas, A.; Wali, Y.; Pathare, A. Comparative assessment of deferiprone and deferasirox in thalassemia major patients in the first two decades-single centre experience. J. Pediatr. Hematol. Oncol. 2013, 30, 104–112. [Google Scholar] [CrossRef] [PubMed]
- Pepe, A.; Meloni, A.; Capra, M.; Cianciulli, P.; Prossomariti, L.; Malaventura, C.; Putti, M.C.; Lippi, A.; Romeo, M.A.; Bisconte, M.G.; et al. Deferasirox, deferiprone and desferrioxamine treatment in thalassemia major patients: Cardiac iron and function comparison determined by quantitative magnetic resonance imaging. Haematologica 2010, 96, 41. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Z.D.; Piyamongkol, S.; Liu, D.Y.; Khodr, H.H.; Lu, S.L.; Hider, R.C. Synthesis of 2-amido-3-hydroxypyridin-4(1H)-ones: Novel iron chelators with enhanced pFe3+ values. Bioorg. Med. Chem. 2001, 9, 563–573. [Google Scholar] [CrossRef]
- Liu, Z.D.; Kayyali, R.; Hider, R.C.; Porter, J.B.; Theobald, A.E. Design, synthesis, and evaluation of novel 2-substituted 3-hydroxypyridin-4-ones: Structure-activity investigation of metalloenzyme inhibition by iron chelators. J. Med. Chem. 2002, 45, 631–639. [Google Scholar] [CrossRef] [PubMed]
- Piyamongkol, S.; Zhou, T.; Liu, Z.D.; Khodr, H.H.; Hider, R.C. Design and characterization of novel hexadentate 3-hydroxypyridin-4-one ligands. Tetrahedron Lett. 2005, 46, 1333–1336. [Google Scholar] [CrossRef]
- Zhou, T.; Neubert, H.; Liu, D.Y.; Liu, Z.D.; Ma, Y.M.; Kong, X.L.; Luo, W.; Mark, S.; Hider, R.C. Iron binding dendrimers: A novel approach for the treatment of hemochromatosis. J. Med. Chem. 2006, 49, 4171–4182. [Google Scholar] [CrossRef] [PubMed]
- Xie, Y.-Y.; Liu, M.-S.; Hu, P.-P.; Kong, X.-L.; Qiu, D.-H.; Xu, J.-L.; Hider, R.C.; Zhou, T. Synthesis, physico-chemical properties, and antimicrobial evaluation of a new series of iron(III) hexadentate chelators. Med. Chem. Res. 2013, 22, 2351–2359. [Google Scholar] [CrossRef]
- Zhou, Y.-J.; Liu, M.-S.; Osamah, A.R.; Kong, X.-L.; Alsam, S.; Battah, S.; Xie, Y.-Y.; Hider, R.C.; Zhou, T. Hexadentate 3-hydroxypyridin-4-ones with high iron(III) affinity: Design, synthesis and inhibition on methicillin resistant staphylococcus aureus and pseudomonas strains. Eur. J. Med. Chem. 2015, 94, 8–21. [Google Scholar] [CrossRef] [PubMed]
- Nunes, A.; Podinovskaia, M.; Leite, A.; Gameiro, P.; Zhou, T.; Ma, Y.; Kong, X.; Schaible, U.E.; Hider, R.C.; Rangel, M. Fluorescent 3-hydroxy-4-pyridinone hexadentate iron chelators: Intracellular distribution and the relevance to antimycobacterial properties. J. Biol. Inorg. Chem. 2010, 15, 861–877. [Google Scholar] [CrossRef] [PubMed]
- Zhou, T.; Hider, R.C.; Kong, X. Mode of iron(III) chelation by hexadentate hydroxypyridinones. Chem. Commun. 2015, 51, 5614–5617. [Google Scholar] [CrossRef] [PubMed]
- Zhou, T.; Liu, Z.D.; Neubert, H.; Kong, X.L.; Ma, Y.M.; Hider, R.C. High affinity iron(III) scavenging by a novel hexadentate 3-hydroxypyridin-4-one-based dendrimer: Synthesis and characterization. Bioorg. Med. Chem. Lett. 2005, 15, 5007–5011. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.-J.; Kong, X.-L.; Li, J.-P.; Ma, Y.-M.; Hider, R.C.; Zhou, T. Novel 3-hydroxypyridin-4-one hexadentate ligand-based polymeric iron chelator: Synthesis, characterization and antimicrobial evaluation. Med. Chem. Commun. 2015, 6, 1620–1625. [Google Scholar] [CrossRef]
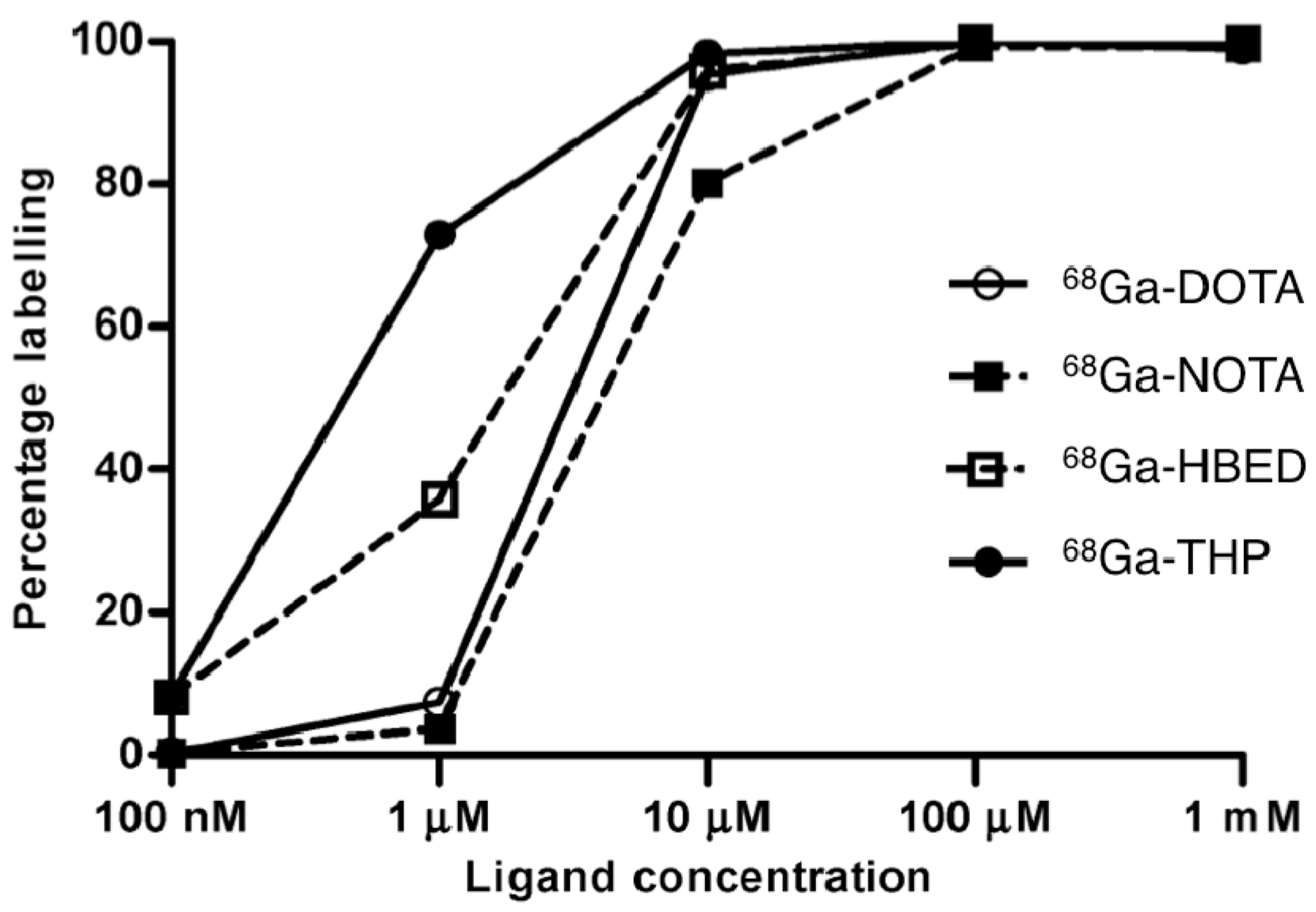
- Velikyan, I. Prospective of 68Ga-radiopharmaceutical development. Theranostics 2014, 4, 47–80. [Google Scholar] [CrossRef] [PubMed]
- Jackson, G.E.; Byrne, M.J. Metal ion speciation in blood plasma: Gallium-67-citrate and MRI contrast agents. J. Nucl. Med. 1996, 37, 379–386. [Google Scholar] [PubMed]
- Moerlein, S.M.; Welch, M.J. The chemistry of gallium and indium as related to radiopharmaceutical production. Int. J. Nucl. Med. Biol. 1981, 8, 277–287. [Google Scholar] [CrossRef]
- Harris, W.R.; Pecoraro, V.L. Thermodynamic binding constants for gallium transferrin. Biochemistry 1983, 22, 292–299. [Google Scholar] [CrossRef] [PubMed]
- Santos, M.A.; Gil, M.; Gano, L.; Chaves, S. Bifunctional 3-hydroxy-4-pyridinone derivatives as potential pharmaceuticals: Synthesis, complexation with Fe(III), Al(III) and Ga(III) and in vivo evaluation with 67Ga. J. Biol. Inorg. Chem. 2005, 10, 564–580. [Google Scholar] [CrossRef] [PubMed]
- Eder, M.; Krivoshein, A.V.; Backer, M.; Backer, J.M.; Haberkorn, U.; Eisenhut, M. ScVEGF-PEG-HBED-CC and scVEGF-PEG-NOTA conjugates: Comparison of easy-to-label recombinant proteins for [68Ga] PET imaging of VEGF receptors in angiogenic vasculature. Nucl. Med. Biol. 2010, 37, 405–412. [Google Scholar] [CrossRef] [PubMed]
- Velikyan, I.; Maecke, H.; Langstrom, B. Convenient preparation of 68Ga-based PET-radiopharmaceuticals at room temperature. Bioconjug. Chem. 2008, 19, 569–573. [Google Scholar] [CrossRef] [PubMed]
- Govindan, S.V.; Michel, R.B.; Griffiths, G.L.; Goldenberg, D.M.; Mattes, M.J. Deferoxamine as a chelator for 67Ga in the preparation of antibody conjugates. Nucl. Med. Biol. 2005, 32, 513–519. [Google Scholar] [CrossRef] [PubMed]
- Cooper, M.S.; Ma, M.T.; Sunassee, K.; Shaw, K.P.; Williams, J.D.; Paul, R.L.; Donnelly, P.S.; Blower, P.J. Comparison of 64Cu-complexing bifunctional chelators for radioimmunoconjugation: Labeling efficiency, specific activity, and in vitro/in vivo stability. Bioconjug. Chem. 2012, 23, 1029–1039. [Google Scholar] [CrossRef] [PubMed]
- Cooper, M.S.; Sabbah, E.; Mather, S.J. Conjugation of chelating agents to proteins and radiolabeling with trivalent metallic isotopes. Nat. Protoc. 2006, 1, 314–317. [Google Scholar] [CrossRef] [PubMed]
- Chaves, S.; Marques, S.M.; Matos, A.M.F.; Nunes, A.; Gano, L.; Tuccinardi, T.; Martinelli, A.; Santos, M.A. New tris(hydroxypyridinones) as iron and aluminium sequestering agents: Synthesis, complexation and in vivo studies. Chem. Eur. J. 2010, 16, 10535–10545. [Google Scholar] [CrossRef] [PubMed]
- Chaves, S.; Mendonca, A.C.; Marques, S.M.; Prata, M.I.; Santos, A.C.; Martins, A.F.; Geraldes, C.F.G.C.; Santos, M.A. A gallium complex with a new tripodal tris-hydroxypyridinone for potential nuclear diagnostic imaging: Solution and in vivo studies of 67Ga-labeled species. J. Inorg. Biochem. 2011, 105, 31–38. [Google Scholar] [CrossRef] [PubMed]
- Deri, M.A.; Ponnala, S.; Kozlowski, P.; Burton-Pye, B.P.; Cicek, H.T.; Hu, C.; Lewis, J.S.; Francesconi, L.C. p-SCN-Bn-HOPO: A superior bifunctional chelator for 89Zr immunoPET. Bioconjug. Chem. 2015, 26, 2579–2591. [Google Scholar] [CrossRef] [PubMed]
- Tinianow, J.N.; Pandya, D.N.; Pailloux, S.L.; Ogasawara, A.; Vanderbilt, A.N.; Gill, H.S.; Williams, S.-P.; Wadas, T.J.; Magda, D.; Marik, J. Evaluation of a 3-hydroxypyridin-2-one (2,3-HOPO) based macrocyclic chelator for 89Zr4+ and its use for immunopet imaging of HER2 positive model of ovarian carcinoma in mice. Theranostics 2016, 6, 511–521. [Google Scholar] [CrossRef] [PubMed]



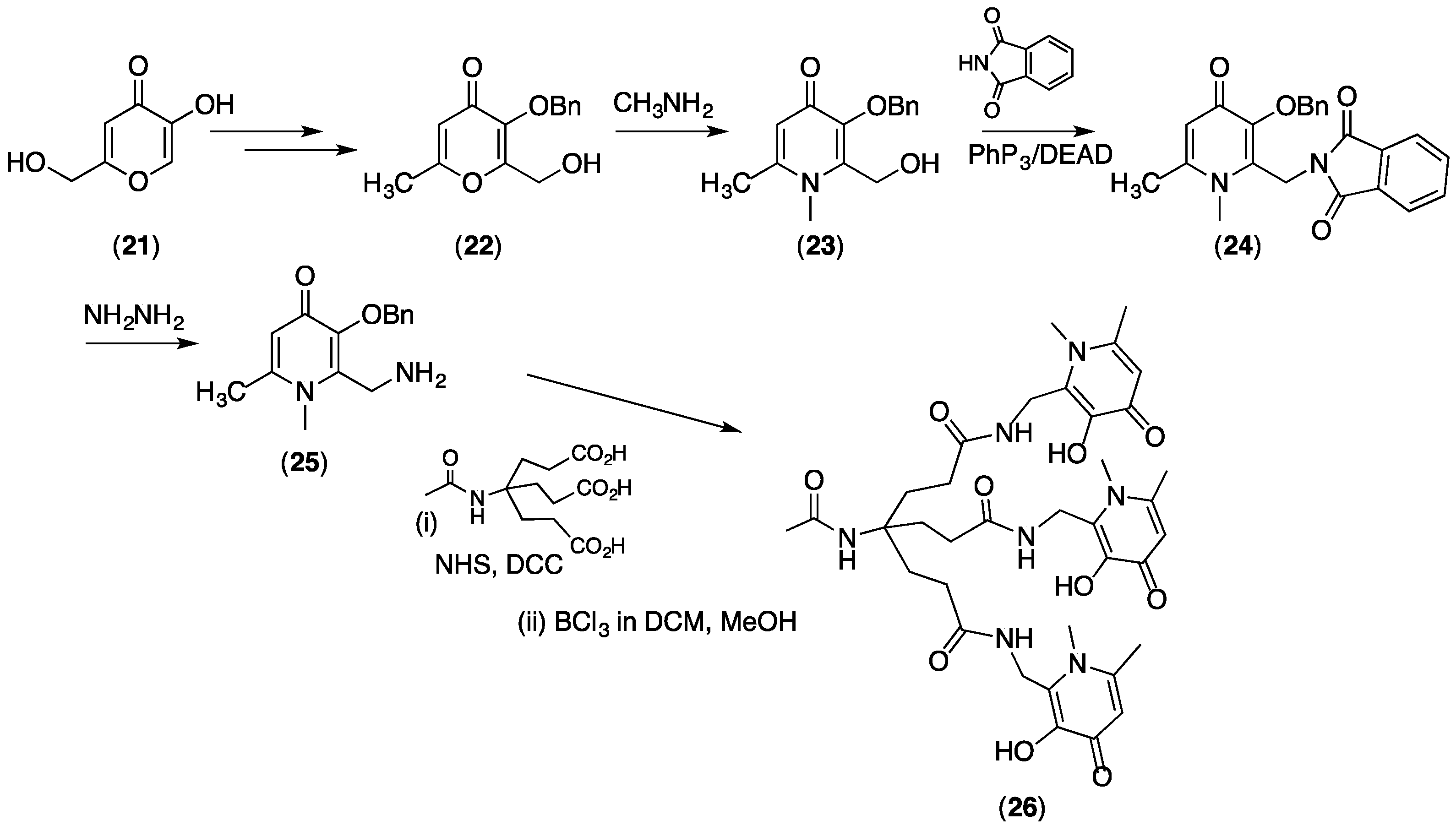
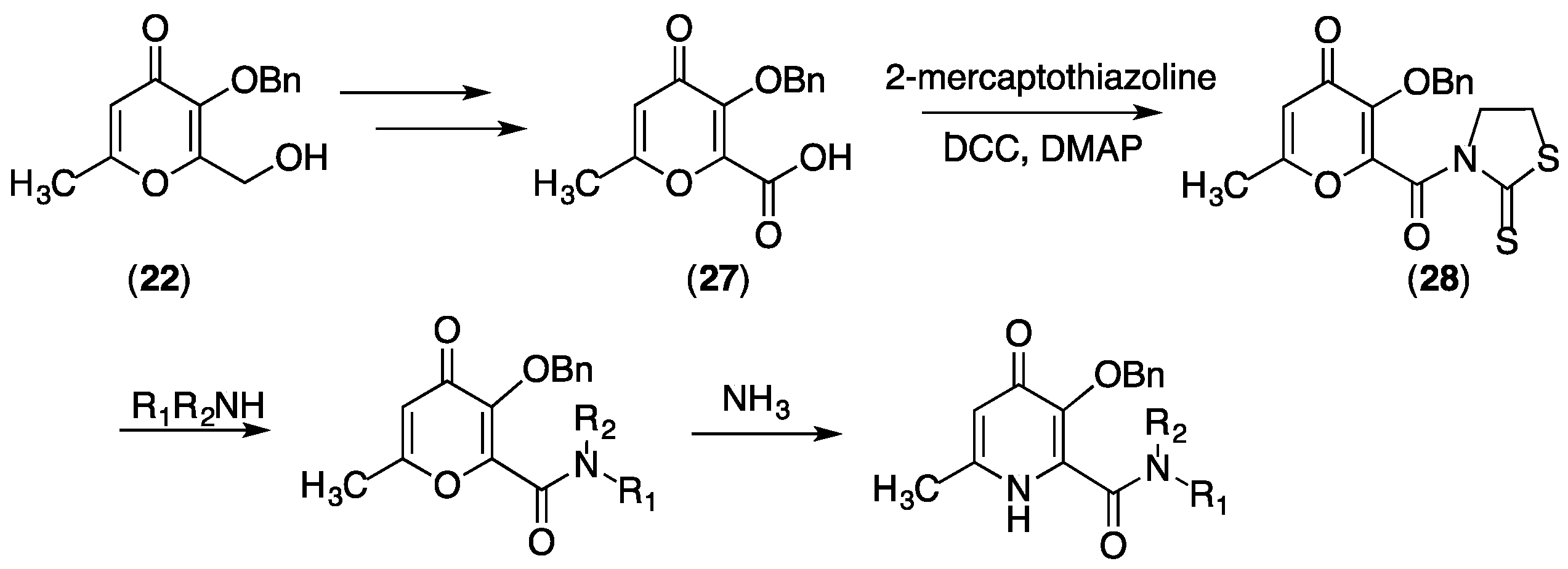
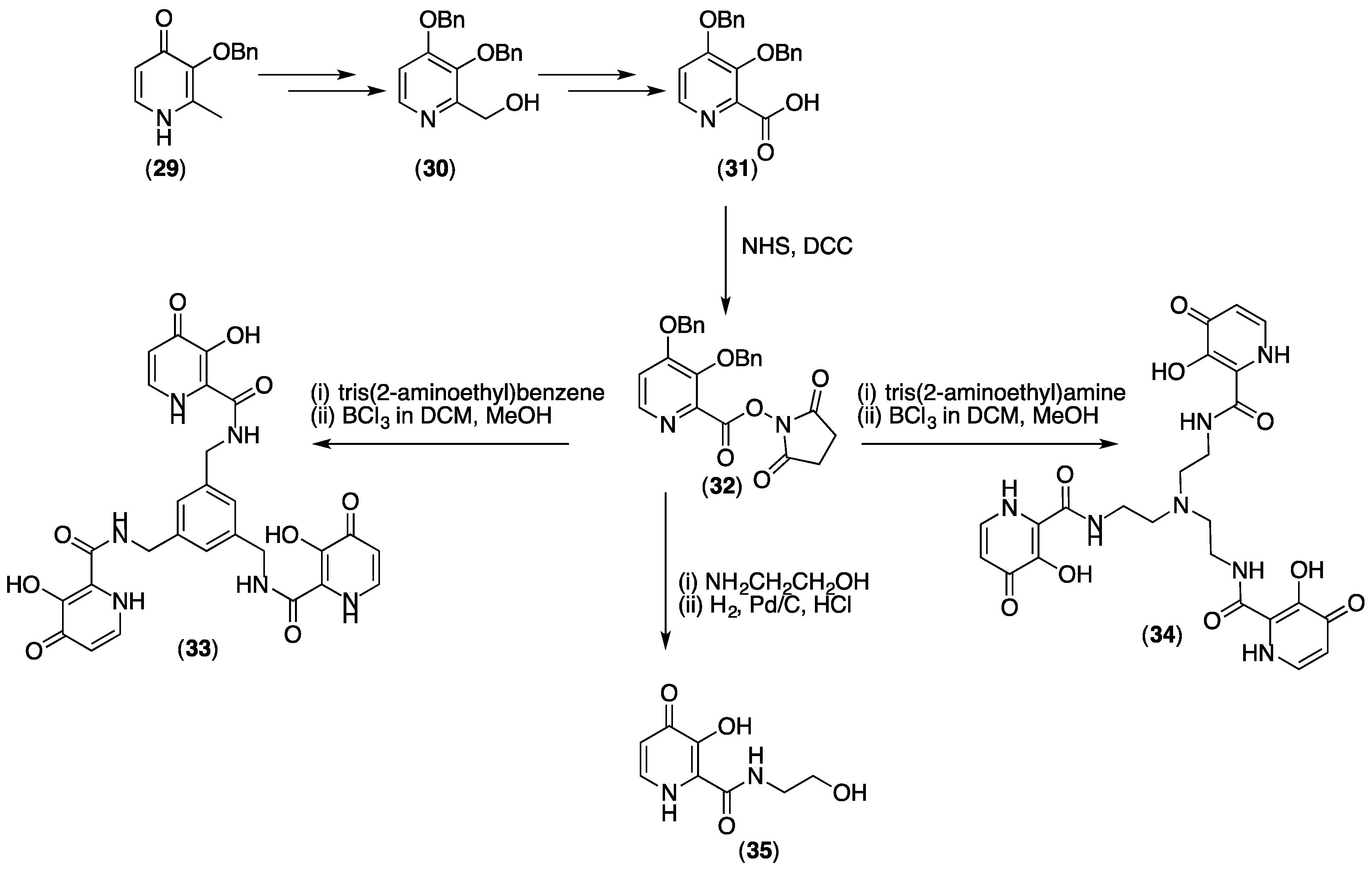

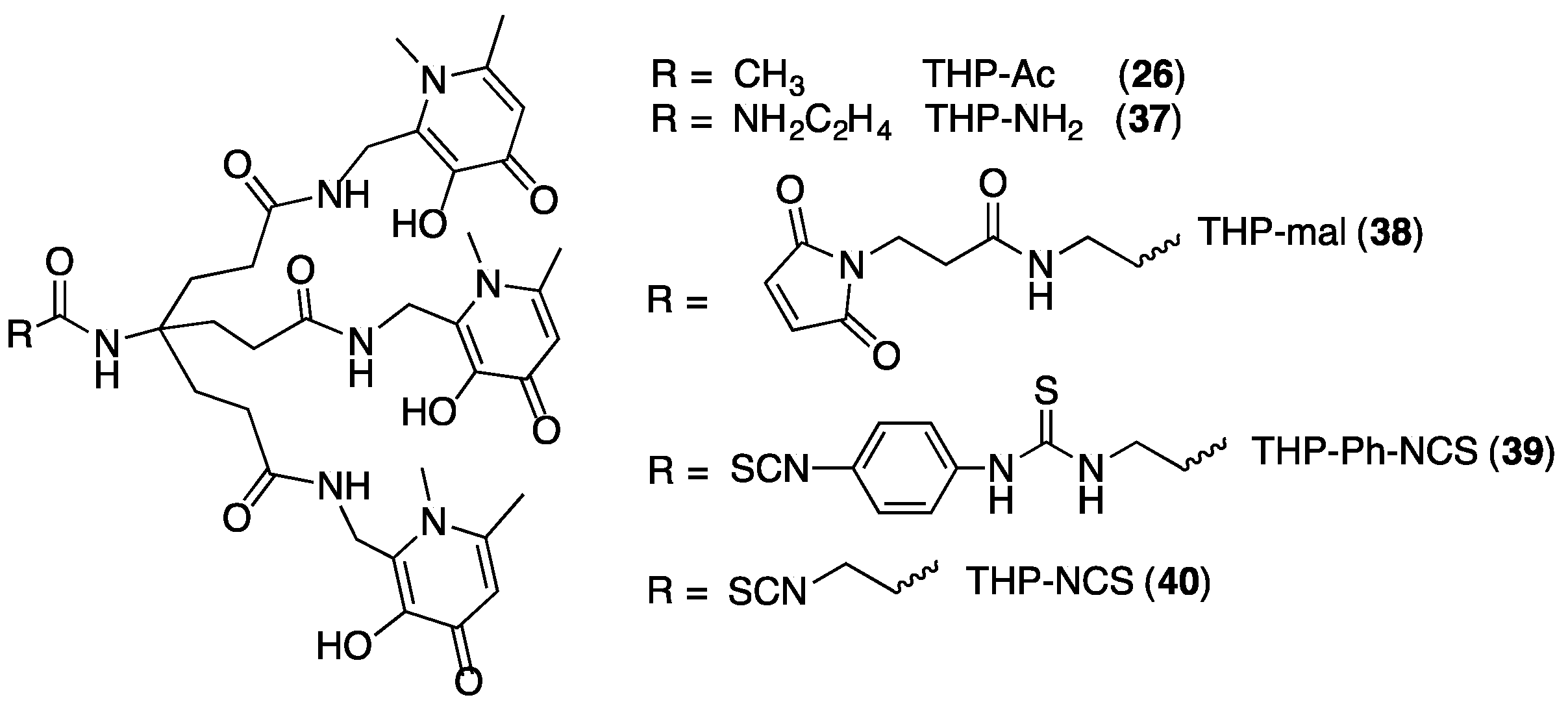



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | pKa1 | pKa2 | log K1 | log K2 | log K3 | log β3 Fe3+ | Ref. |
|---|---|---|---|---|---|---|---|
| 1,2-HP (2) | 1.2 | 5.86 | 10.6 | 9.5 | 7.1 | 27.2 | [22] |
| −0.9 | 5.78 | 10.3 | 9.0 | 7.6 | 26.9 | [23] | |
| 3,2-HP (3) | 0.1 | 8.66 | 11.7 | 9.8 | 8.1 | 29.6 | [23] |
| 3,4-HP (4) | 3.34 | 9.01 | 14.2 | 11.6 | 9.3 | 35.1 | [23] |
| 3.60 | 9.60 | – | – | – | 36.9 | [20] |
| Metal Ion | pKa1 | pKa2 | log K1 | log K2 | log K3 | log β3 | pM3+ | Ref. |
|---|---|---|---|---|---|---|---|---|
| Fe3+ | 3.56 | 9.64 | 14.92 | 12.23 | 9.79 | 37.2 | – | [20] |
| 3.68 | 9.77 | 14.56 | 12.19 | 9.69 | 36.4 | 19.4 | [34] | |
| 3.62 | 9.76 | 15.14 | 11.54 | 9.24 | 35.92 | 18.3 | [35] | |
| – | – | 15.10 | 11.51 | 9.27 | 35.88 | – | [25] | |
| 3.61 | 9.78 | 15.03 | 27.42 | – | 37.35 | 20.74 | [36] | |
| Ga3+ | – | – | 13.17 | 12.26 | 10.33 | 35.76 | – | [25] |
| 3.70 | 9.86 | 17.07 | 12.19 | 9.16 | 38.42 | – | [37] |
 | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Compound | R1 | R2 | R5 | R6 | pKa1 | pKa2 | log β3 (Fe3+) | pFe3+ | Ref. |
| 1 | CH3 | CH3 | H | H | 3.68 | 9.77 | 36.4 | 19.4 | [34] |
| 3.61 | 9.78 | 37.35 | 20.74 | [36] | |||||
| 4 | H | H | H | H | 3.34 | 9.01 | 35.1 | – | [20,23] |
| 5 | H | CH3 | H | H | 3.70 | 9.76 | 37.2 | – | [20] |
| 3.64 | 9.73 | 36.63 | 20.17 | [36] | |||||
| 6 | CH3 | CH2OH | H | H | 2.92 | 9.11 | 35.3 | 20.9 | [34] |
| 7 | CH2CH3 | CH2CH3 | H | H | 3.81 | 9.93 | 36.8 | 19.7 | [34] |
| 7 | CH2CH3 | CH2OH | H | H | 2.80 | 9.27 | 35.3 | 21.0 | [34] |
| 9 | CH2CH3 | CH(OH)CH3 | H | H | 3.03 | 8.77 | 35.1 | 21.4 | [34] |
| 10 | H | CON(CH3)2 | H | CH3 | 2.53 | 8.20 | 33.2 | 20.4 | [38] |
| 11 | H | CONHCH3 | H | CH3 | 6.66 | 2.32 | 32.5 | 22.8 | [38] |
| 12 | CH2CH3 | CH3 | H | H | 3.65 | 9.88 | 37.7 | – | [20] |
| 13 | CH3 | CH3 | CH3 | H | 3.37 | 10.32 | 37.93 | 19.71 | [36] |
| 14 | H | CH3 | CH3 | H | 3.43 | 10.27 | 37.28 | 19.22 | [36] |
© 2017 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cusnir, R.; Imberti, C.; Hider, R.C.; Blower, P.J.; Ma, M.T. Hydroxypyridinone Chelators: From Iron Scavenging to Radiopharmaceuticals for PET Imaging with Gallium-68. Int. J. Mol. Sci. 2017, 18, 116. https://doi.org/10.3390/ijms18010116
Cusnir R, Imberti C, Hider RC, Blower PJ, Ma MT. Hydroxypyridinone Chelators: From Iron Scavenging to Radiopharmaceuticals for PET Imaging with Gallium-68. International Journal of Molecular Sciences. 2017; 18(1):116. https://doi.org/10.3390/ijms18010116
Chicago/Turabian StyleCusnir, Ruslan, Cinzia Imberti, Robert C. Hider, Philip J. Blower, and Michelle T. Ma. 2017. "Hydroxypyridinone Chelators: From Iron Scavenging to Radiopharmaceuticals for PET Imaging with Gallium-68" International Journal of Molecular Sciences 18, no. 1: 116. https://doi.org/10.3390/ijms18010116