
Probing the Ion Binding Site in a DNA Holliday Junction Using Förster Resonance Energy Transfer (FRET)


Abstract
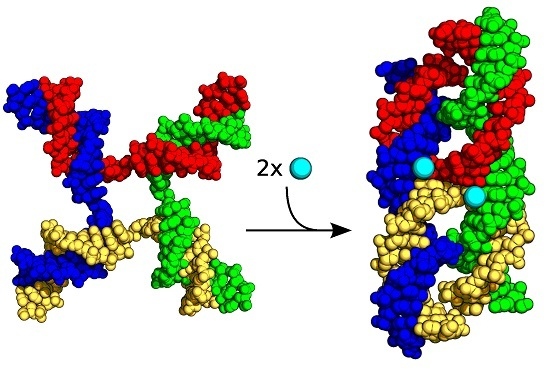
:
1. Introduction
2. Results and Discussion
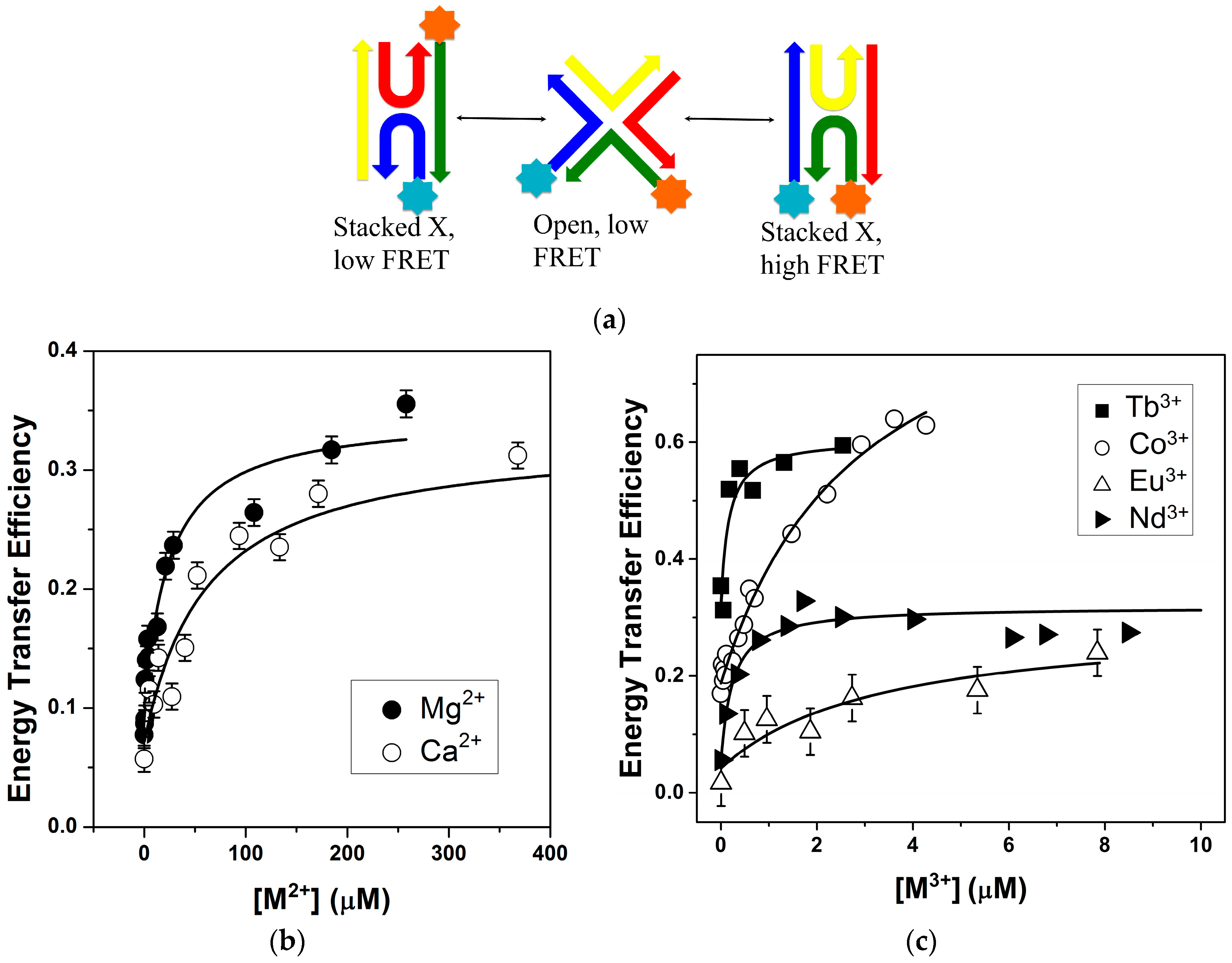
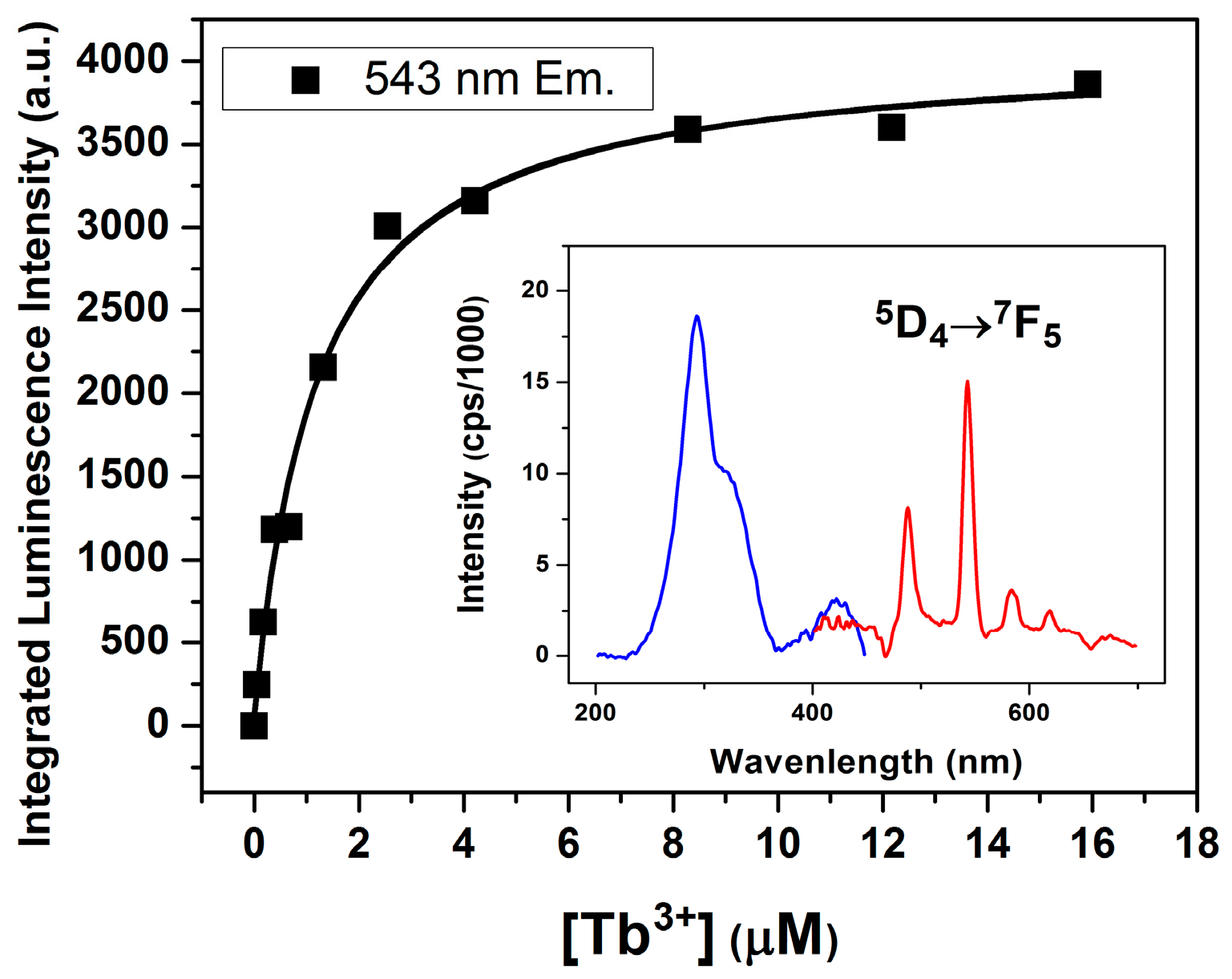
2.1. FRET Measurements of Ion Affinities and Junction Folding
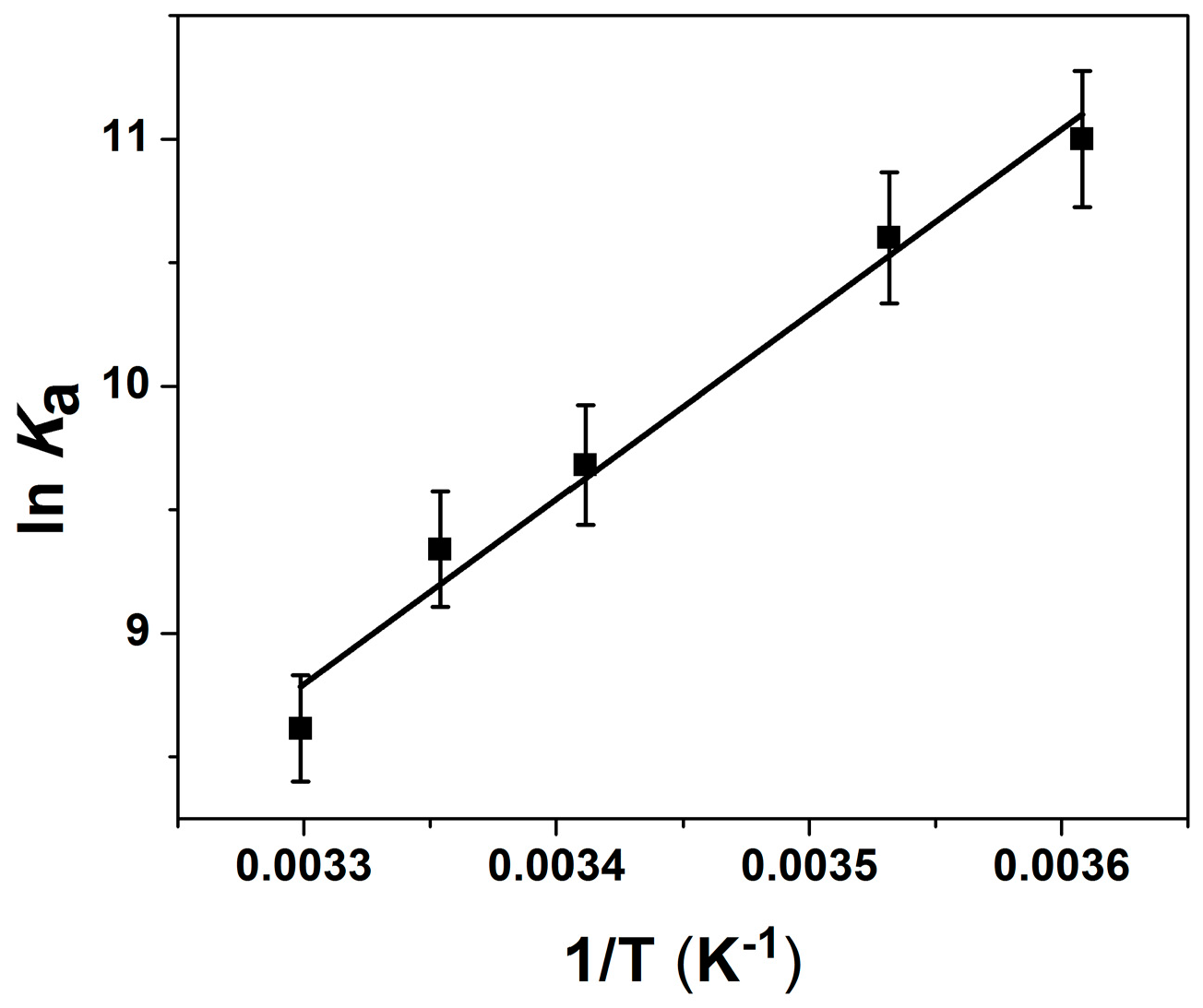
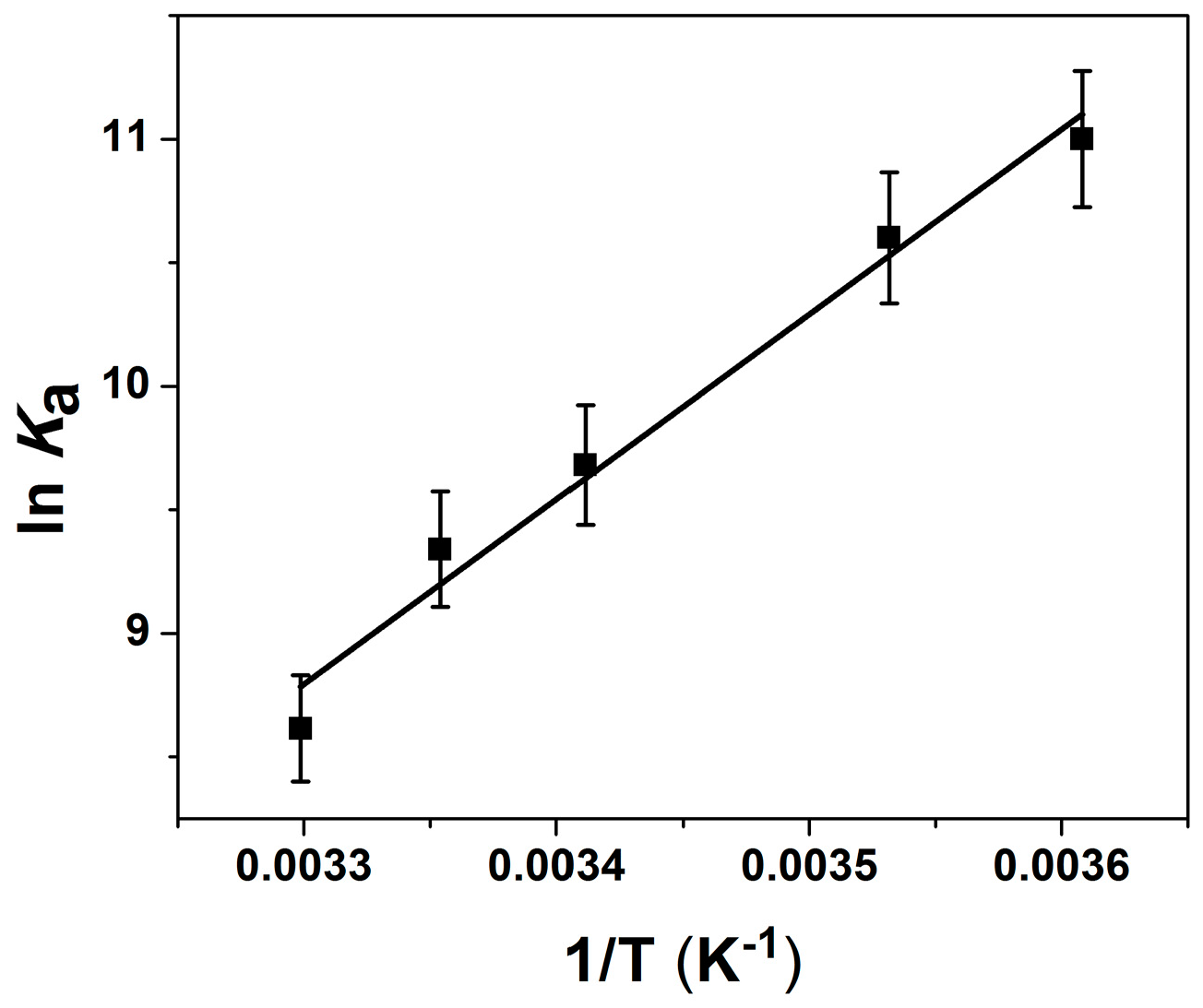
2.2. Thermodynamic Parameters of Ion Binding and Stoichiometry
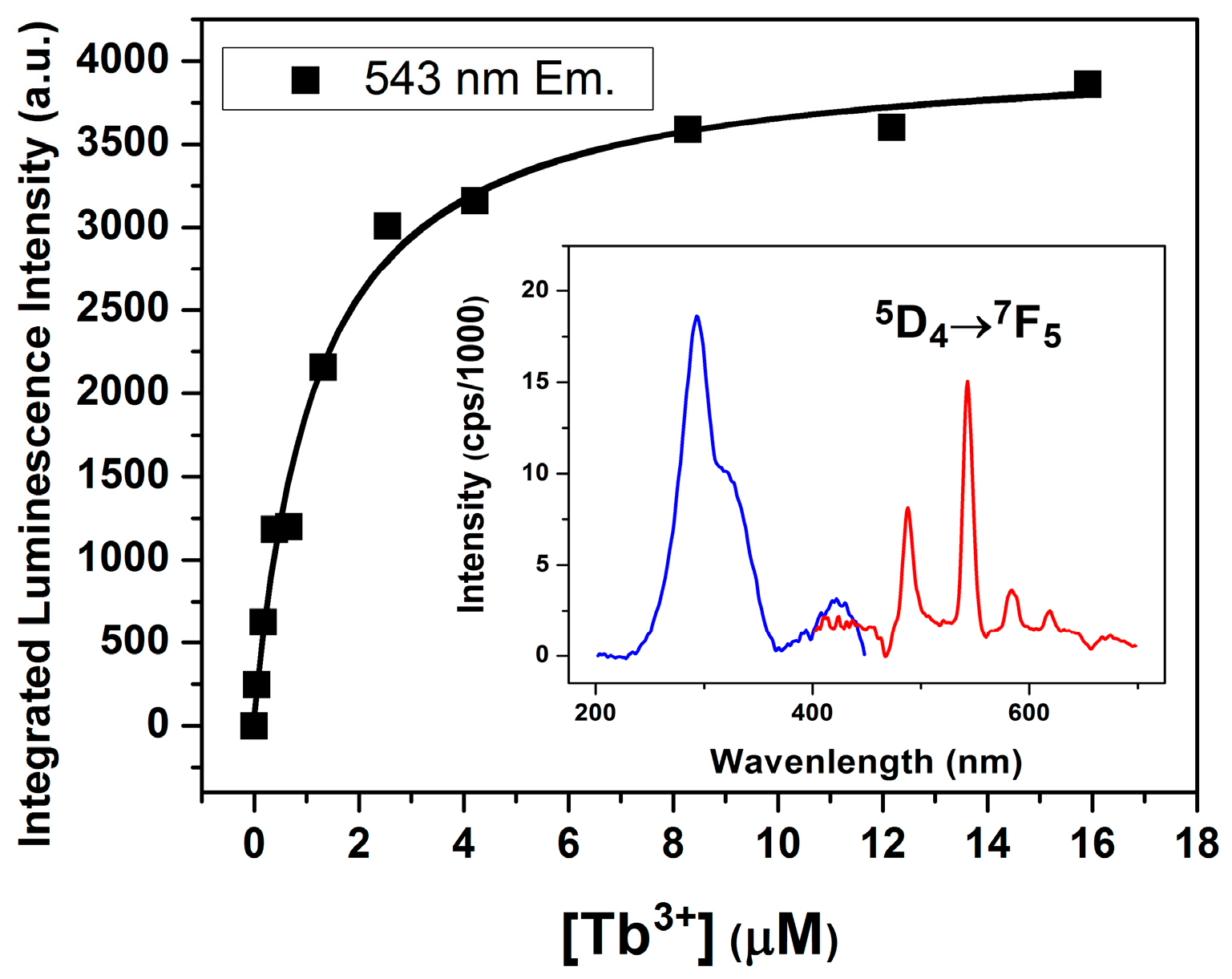
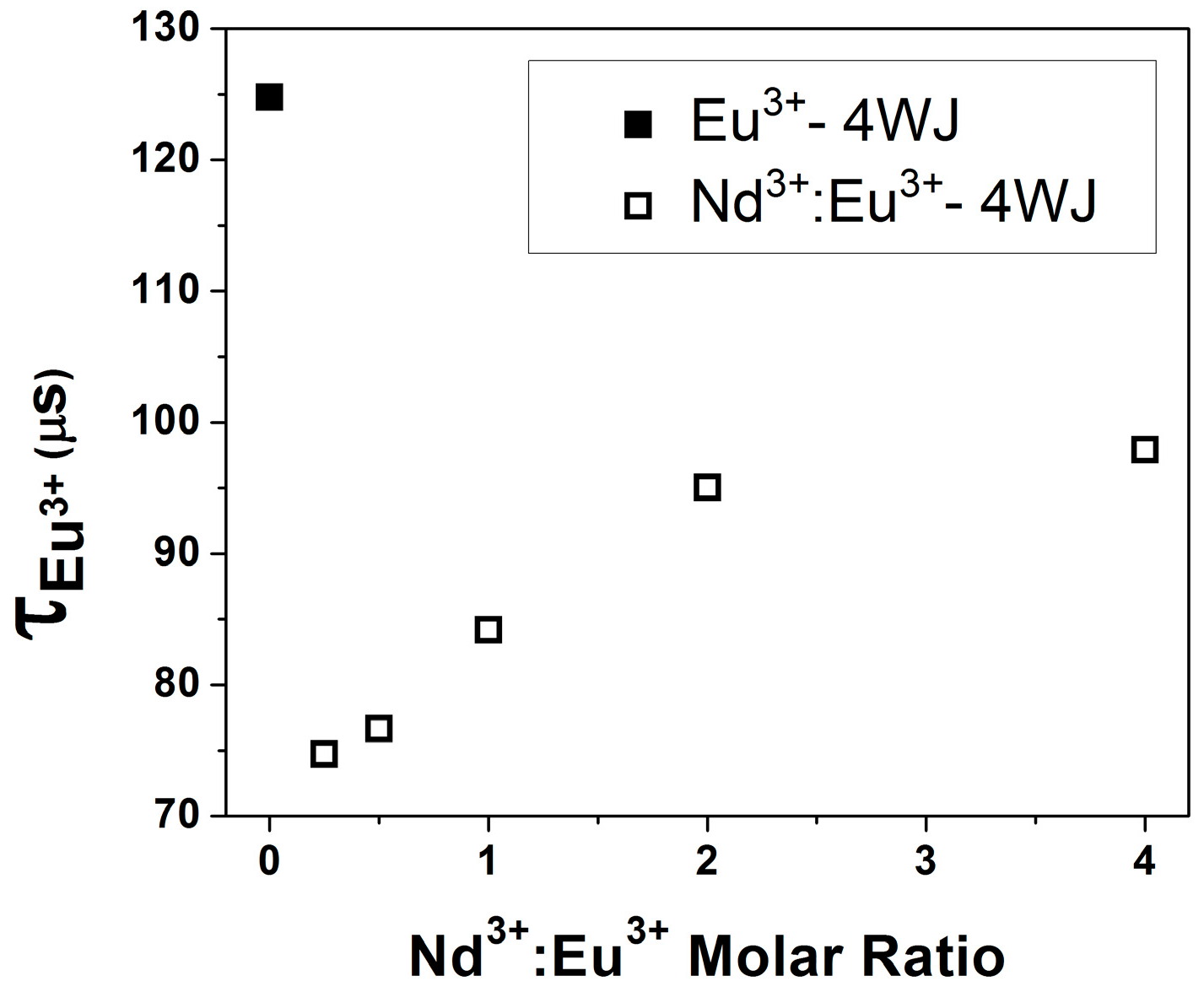
2.3. Lanthanide Luminescence and Multiplicity of Binding Sites
Coordination Geometry and Number of Ions Bound
3. Experimental Section
3.1. Oligonucleotide Preparation and Labeling
3.2. Junction Construction
3.3. FRET Measurements and Analysis
3.4. Isothermal Titration Calorimetry
3.5. Lanthanide Luminescence
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Brazda, V.; Laister, R.C.; Jagelska, E.B.; Arrowsmith, C. Cruciform structures are a common DNA feature important for regulating biological processes. BMC Mol. Biol. 2011, 12, 33. [Google Scholar] [CrossRef] [PubMed]
- Lu, S.; Wang, G.; Bacolla, A.; Zhao, J.; Spitser, S.; Vasquez, K.M. Short inverted repeats are hotspots for genetic instability: Relevance to cancer genomes. Cell Rep. 2015. [Google Scholar] [CrossRef] [PubMed]
- Khuu, P.A.; Voth, A.R.; Hays, F.A.; Ho, P.S. The stacked-x DNA Holliday junction and protein recognition. J. Mol. Recognit. 2006, 19, 234–242. [Google Scholar] [CrossRef] [PubMed]
- Jones, M.R.; Seeman, N.C.; Mirkin, C.A. Nanomaterials. Programmable materials and the nature of the DNA bond. Science 2015, 347, 1260901. [Google Scholar] [CrossRef] [PubMed]
- Duckett, D.R.; Murchie, A.I.; Diekmann, S.; von Kitzing, E.; Kemper, B.; Lilley, D.M. The structure of the Holliday junction, and its resolution. Cell 1988, 55, 79–89. [Google Scholar] [CrossRef]
- Churchill, M.E.; Tullius, T.D.; Kallenbach, N.R.; Seeman, N.C. A Holliday recombination intermediate is twofold symmetric. Proc. Natl. Acad. Sci. USA 1988, 85, 4653–4656. [Google Scholar] [CrossRef] [PubMed]
- Cooper, J.P.; Hagerman, P.J. Geometry of a branched DNA structure in solution. Proc. Natl. Acad. Sci. USA 1989, 86, 7336–7340. [Google Scholar] [CrossRef] [PubMed]
- Miick, S.M.; Fee, R.S.; Millar, D.P.; Chazin, W.J. Crossover isomer bias is the primary sequence-dependent property of immobilized Holliday junctions. Proc. Natl. Acad. Sci. USA 1997, 94, 9080–9084. [Google Scholar] [CrossRef] [PubMed]
- Declais, A.C.; Lilley, D.M. New insight into the recognition of branched DNA structure by junction-resolving enzymes. Curr. Opin. Struct. Biol. 2008, 18, 86–95. [Google Scholar] [CrossRef] [PubMed]
- Hays, F.A.; Watson, J.; Ho, P.S. Caution! DNA crossing: Crystal structures of Holliday junctions. J. Biol. Chem. 2003, 278, 49663–49666. [Google Scholar] [CrossRef] [PubMed]
- Eichman, B.F.; Ortiz-Lombardia, M.; Aymami, J.; Coll, M.; Ho, P.S. The inherent properties of DNA four-way junctions: Comparing the crystal structures of Holliday junctions. J. Mol. Biol. 2002, 320, 1037–1051. [Google Scholar] [CrossRef]
- Ho, P.S.; Eichman, B.F. The crystal structures of DNA Holliday junctions. Curr. Opin. Struct. Biol. 2001, 11, 302–308. [Google Scholar] [CrossRef]
- Ortiz-Lombardia, M.; Gonzalez, A.; Eritja, R.; Aymami, J.; Azorin, F.; Coll, M. Crystal structure of a DNA Holliday junction. Nat. Struct. Biol. 1999, 6, 913–917. [Google Scholar] [PubMed]
- Clegg, R.M.; Murchie, A.I.; Zechel, A.; Carlberg, C.; Diekmann, S.; Lilley, D.M. Fluorescence resonance energy transfer analysis of the structure of the four-way DNA junction. Biochemistry 1992, 31, 4846–4856. [Google Scholar] [CrossRef] [PubMed]
- Murchie, A.I.; Clegg, R.M.; von Kitzing, E.; Duckett, D.R.; Diekmann, S.; Lilley, D.M. Fluorescence energy transfer shows that the four-way DNA junction is a right-handed cross of antiparallel molecules. Nature 1989, 341, 763–766. [Google Scholar] [CrossRef] [PubMed]
- Vitoc, C.I.; Mukerji, I. HU binding to a DNA four-way junction probed by Förster resonance energy transfer. Biochemistry 2011, 50, 1432–1441. [Google Scholar] [CrossRef] [PubMed]
- Joo, C.; McKinney, S.A.; Lilley, D.M.J.; Ha, T. Exploring rare conformational species and ionic effects in DNA Holliday junctions using single-molecule spectroscopy. J. Mol. Biol. 2004, 341, 739–751. [Google Scholar] [CrossRef] [PubMed]
- McKinney, S.A.; Declais, A.C.; Lilley, D.M.; Ha, T. Structural dynamics of individual Holliday junctions. Nat. Struct. Biol. 2003, 10, 93–97. [Google Scholar] [CrossRef] [PubMed]
- Duckett, D.R.; Murchie, A.I.; Lilley, D.M. The role of metal ions in the conformation of the four-way DNA junction. EMBO J. 1990, 9, 583–590. [Google Scholar] [PubMed]
- Mollegaard, N.E.; Murchie, A.I.; Lilley, D.M.; Nielsen, P.E. Uranyl photoprobing of a four-way DNA junction: Evidence for specific metal ion binding. EMBO J. 1994, 13, 1508–1513. [Google Scholar] [PubMed]
- Thorpe, J.H.; Gale, B.C.; Teixeira, S.C.; Cardin, C.J. Conformational and hydration effects of site-selective sodium, calcium and strontium ion binding to the DNA Holliday junction structure d(TCGGTACCGA)(4). J. Mol. Biol. 2003, 327, 97–109. [Google Scholar] [CrossRef]
- Hyeon, C.; Lee, J.; Yoon, J.; Hohng, S.; Thirumalai, D. Hidden complexity in the isomerization dynamics of Holliday junctions. Nat. Chem. 2012, 4, 907–914. [Google Scholar] [CrossRef] [PubMed]
- Wheatley, E.G.; Pieniazek, S.N.; Mukerji, I.; Beveridge, D.L. Molecular dynamics of a DNA Holliday junction: The inverted repeat sequence d(CCGGTACCGG)(4). Biophys. J. 2012, 102, 552–560. [Google Scholar] [CrossRef] [PubMed]
- Watson, J.; Hays, F.A.; Ho, P.S. Definitions and analysis of DNA Holliday junction geometry. Nucleic Acids Res. 2004, 32, 3017–3027. [Google Scholar] [CrossRef] [PubMed]
- Auclair, S.M.; Oliver, D.B.; Mukerji, I. Defining the solution state dimer structure of Escherichia coli SecA using Förster resonance energy transfer. Biochemistry 2013, 52, 2388–2401. [Google Scholar] [CrossRef] [PubMed]
- Ohtaki, H.; Radnai, T. Structure and dynamics of hydrated ions. Chem. Rev. 1993, 93, 1157–1204. [Google Scholar] [CrossRef]
- Lipfert, J.; Doniach, S.; Das, R.; Herschlag, D. Understanding nucleic acid-ion interactions. Annu. Rev. Biochem. 2014, 83, 813–841. [Google Scholar] [CrossRef] [PubMed]
- Jen-Jacobson, L.; Engler, L.E.; Jacobson, L.A. Structural and thermodynamic strategies for site-specific DNA binding proteins. Structure 2000, 8, 1015–1023. [Google Scholar] [CrossRef]
- Manning, G.S. The molecular theory of polyelectrolyte solutions with applications to the electrostatic properties of polynucleotides. Q. Rev. Biophys. 1978, 11, 179–246. [Google Scholar] [CrossRef] [PubMed]
- Matulis, D.; Rouzina, I.; Bloomfield, V.A. Thermodynamics of DNA binding and condensation: Isothermal titration calorimetry and electrostatic mechanism. J. Mol. Biol. 2000, 296, 1053–1063. [Google Scholar] [CrossRef] [PubMed]
- Morrow, J.R.; Andolina, C.M. Spectroscopic investigations of lanthanide ion binding to nucleic acids. In Interplay between Metal Ions and Nucleic Acids; Sigel, A., Sigel, H., Sigel, R.K.O., Eds.; Springer: Dordrecht, The Netherlands, 2012; pp. 171–199. [Google Scholar]
- Horrocks, W.D. Luminescence spectroscopy. In Metallobiochemistry Part c: Spectroscopic and Physical Methods for Probing Metal Ion Environments in Metalloenzymes and Metalloproteins; Riordan, J.F., Vallee, B.L., Eds.; Elsevier: Philadelphia, PA, USA, 1993; Volume 226, pp. 495–538. [Google Scholar]
- Pechlaner, M.; Sigel, R.K.O. Characterization of metal ion-nucleic acid interactions in solution. In Interplay between Metal Ions and Nucleic Acids; Sigel, A., Sigel, H., Sigel, R.K.O., Eds.; Springer: Dordrecht, The Netherlands, 2012; pp. 1–42. [Google Scholar]
- Feig, A.L.; Panek, M.; Horrocks, W.D., Jr.; Uhlenbeck, O.C. Probing the binding of Tb(III) and Eu(III) to the hammerhead ribozyme using luminescence spectroscopy. Chem. Biol. 1999, 6, 801–810. [Google Scholar] [CrossRef]
- Feig, A.L.; Scott, W.G.; Uhlenbeck, O.C. Inhibition of the hammerhead ribozyme cleavage reaction by site-specific binding of Tb. Science 1998, 279, 81–84. [Google Scholar] [CrossRef] [PubMed]
- Escudier, J.M.; Dupouy, C.; Fountain, M.A.; del Mundo, I.M.; Jacklin, E.M.; Morrow, J.R. Synthesis and luminescence properties of a trinucleotide-europium(III) complex conjugate. Org. Biomol. Chem. 2009, 7, 3251–3257. [Google Scholar] [CrossRef] [PubMed]
- Mathews, R.A.; Rossiter, C.S.; Morrow, J.R.; Richard, J.P. A minimalist approach to understanding the efficiency of mononuclear Zn(II) complexes as catalysts of cleavage of an RNA analog. Dalton Trans. 2007, 34, 3804–3811. [Google Scholar] [CrossRef] [PubMed]
- Kimura, T.; Nagaishi, R.; Kato, Y.; Yoshida, Z. Luminescence study on preferential solvation of Europium(III) in water/non-aqueous solvent mixtures. J. Alloy. Compd. 2001, 323–324, 164–168. [Google Scholar] [CrossRef]
- Andolina, C.M.; Holthoff, W.G.; Page, P.M.; Mathews, R.A.; Morrow, J.R.; Bright, F.V. Spectroscopic system for direct lanthanide photoluminescence spectroscopy with nanomolar detection limits. Appl. Spectrosc. 2009, 63, 483–493. [Google Scholar] [CrossRef] [PubMed]
- Andolina, C.M.; Morrow, J.R. Luminescence resonance energy transfer in heterodinuclear LnIII complexes for sensing biologically relevant anions. Eur. J. Inorg. Chem. 2011, 2011, 154–164. [Google Scholar] [CrossRef]
- Thermo Fisher Scientific. Amine Reactive Probe Labeling Protocol. Available online: https://www.thermofisher.com/us/en/home/references/protocols/cell-and-tissue-analysis/labeling-chemistry-protocols/amine-reactive-probe-labeling-protocol.html (accessed on 6 November 2015).
- Lakowicz, J.R. Principles of Fluorescence Spectroscopy; Springer: New York, NY, USA, 2006; p. XXVI, 954. [Google Scholar]
- Clegg, R.M.; Murchie, A.I.; Lilley, D.M. The solution structure of the four-way DNA junction at low-salt conditions: A fluorescence resonance energy transfer analysis. Biophys. J. 1994, 66, 99–109. [Google Scholar] [CrossRef]
- Freire, E.; Mayorga, O.L.; Straume, M. Isothermal titration. Anal. Chem. 1990, 62, 950–959. [Google Scholar] [CrossRef]
- Marky, L.A.; Breslauer, K.J. Calculating thermodynamic data for transitions of any molecularity from equilibrium melting curves. Biopolymers 1987, 26, 1601–1620. [Google Scholar] [CrossRef] [PubMed]
- Lyle, S.J.; Rahman, M.M. Complexometric titration of yttrium and the lanthanons. I. A comparison of direct methods. Talanta 1963, 10, 1177–1182. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Two Class Model a | Class 1 | Class 2 |
|---|---|---|
| Stoichiometry | 2.1 ± 0.4 | 15.9 ± 2.0 |
| Ka (M−1) | 40,180 ± 13,710 b | 25,000 ± 3000 |
| ∆H (kcal/mol) | −14.9 ± 0.6 b | 1.9 ± 0.5 |
| ∆S (cal/(°·mol)) | −31.5 ± 0.2 b | 26.8 ± 1.2 |
| Excitation Wavelength (nm) | Eu3+ (aq) | Eu3+–4WJ | ||
|---|---|---|---|---|
| (μs) | q | (μs) | q | |
| 464.1 | 105.9 | 9.9 | 121.4 | 8.6 |
| 464.5 | 117.8 | 8.9 | 124.4 | 8.4 |
| 465.0 | 140.0 | 7.5 | 128.7 | 8.2 |
| Average | 111.9 ± 8.4 a | 9.4 ± 1.2 | 124.8 ± 3.7 | 8.4 ± 0.2 |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Litke, J.L.; Li, Y.; Nocka, L.M.; Mukerji, I. Probing the Ion Binding Site in a DNA Holliday Junction Using Förster Resonance Energy Transfer (FRET). Int. J. Mol. Sci. 2016, 17, 366. https://doi.org/10.3390/ijms17030366
Litke JL, Li Y, Nocka LM, Mukerji I. Probing the Ion Binding Site in a DNA Holliday Junction Using Förster Resonance Energy Transfer (FRET). International Journal of Molecular Sciences. 2016; 17(3):366. https://doi.org/10.3390/ijms17030366
Chicago/Turabian StyleLitke, Jacob L., Yan Li, Laura M. Nocka, and Ishita Mukerji. 2016. "Probing the Ion Binding Site in a DNA Holliday Junction Using Förster Resonance Energy Transfer (FRET)" International Journal of Molecular Sciences 17, no. 3: 366. https://doi.org/10.3390/ijms17030366
APA StyleLitke, J. L., Li, Y., Nocka, L. M., & Mukerji, I. (2016). Probing the Ion Binding Site in a DNA Holliday Junction Using Förster Resonance Energy Transfer (FRET). International Journal of Molecular Sciences, 17(3), 366. https://doi.org/10.3390/ijms17030366