
Validation of FRET Assay for the Screening of Growth Inhibitors of Escherichia coli Reveals Elongasome Assembly Dynamics


Abstract
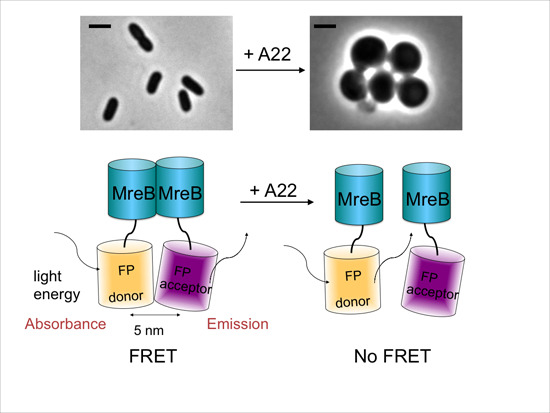
:
1. Introduction
2. Results and Discussion
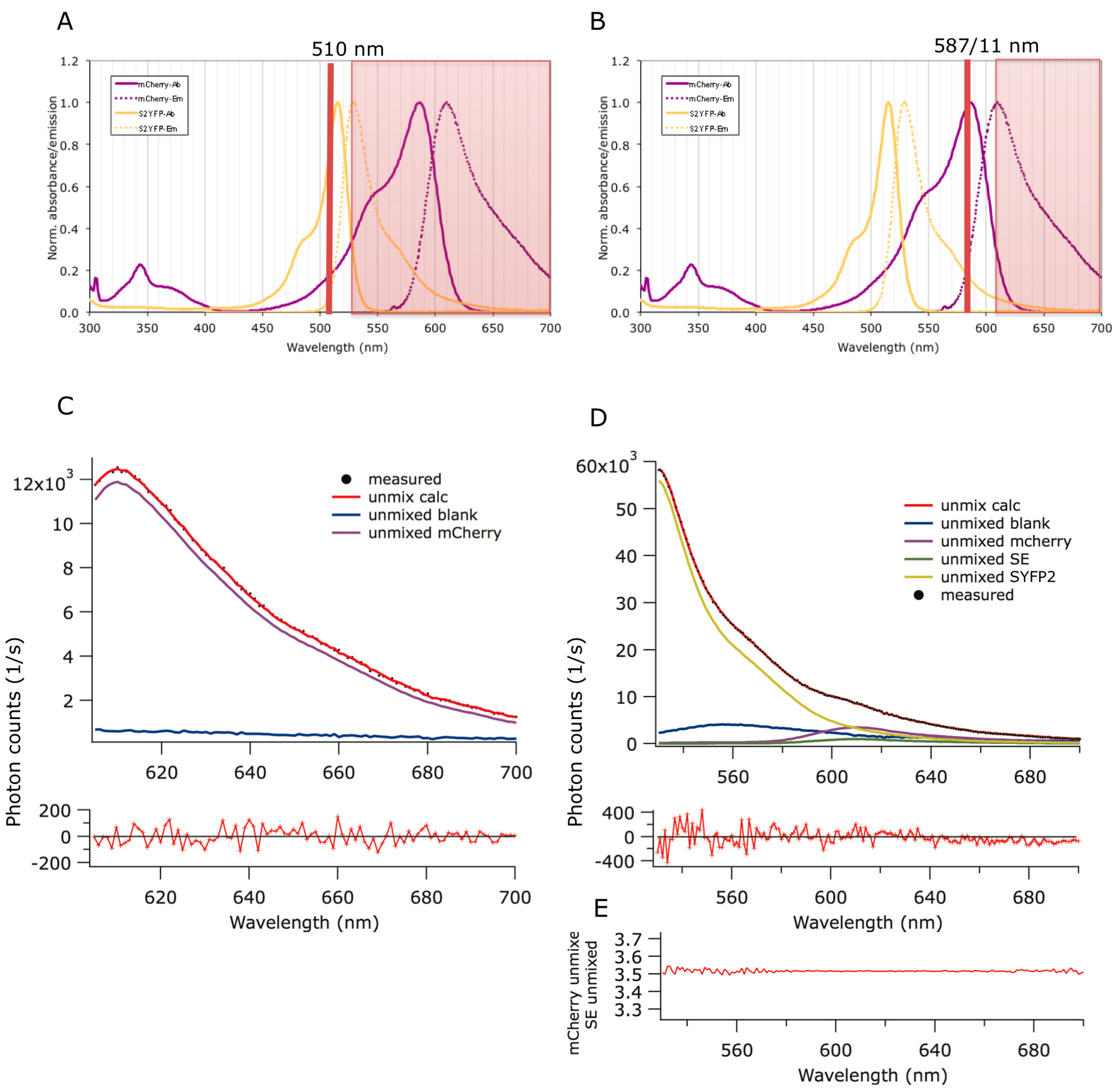
2.1. Förster Resonance Energy Transfer (FRET) Couple Super Yellow Fluorescent Protein 2 (SYFP2) and mCherry
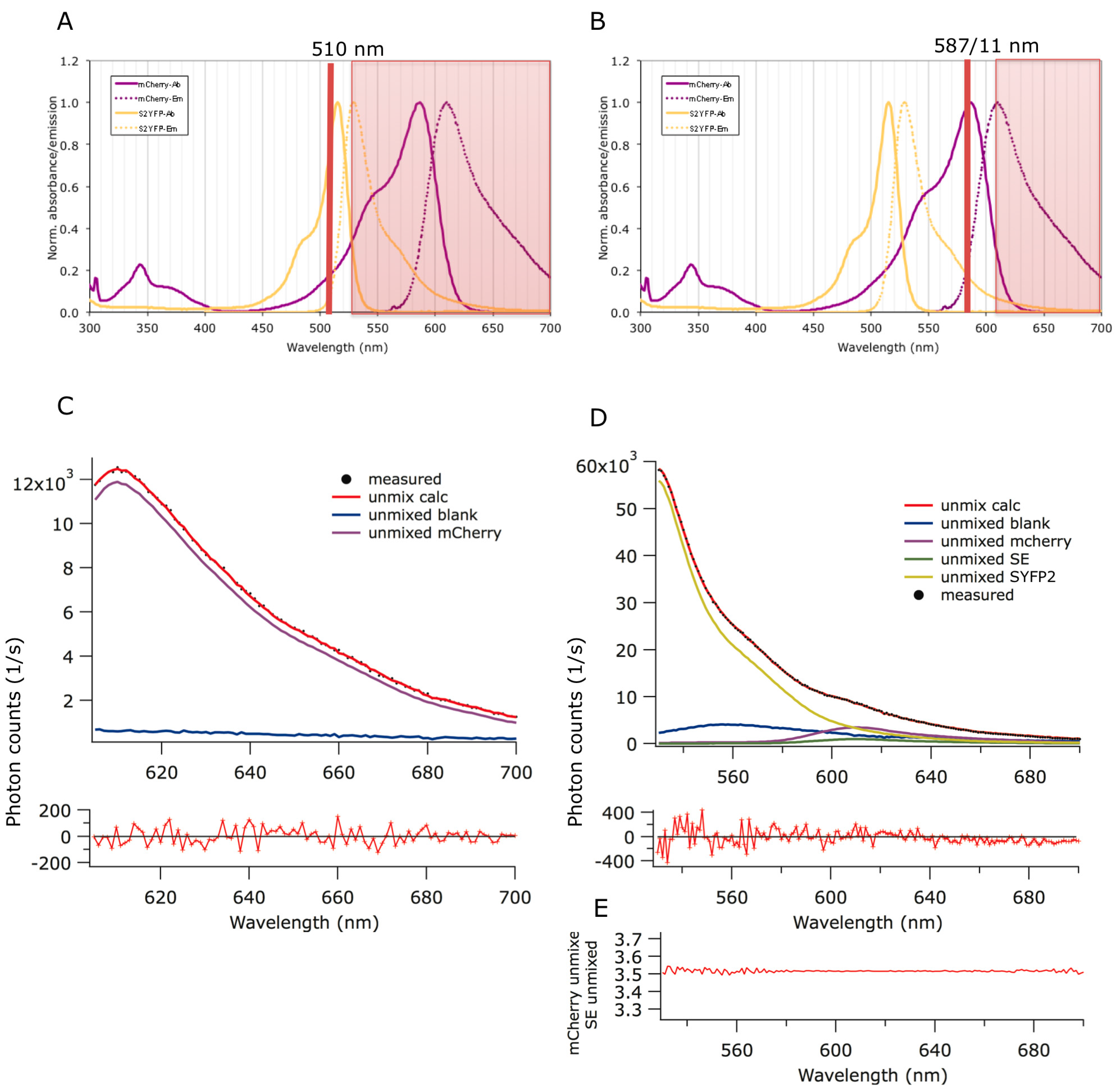
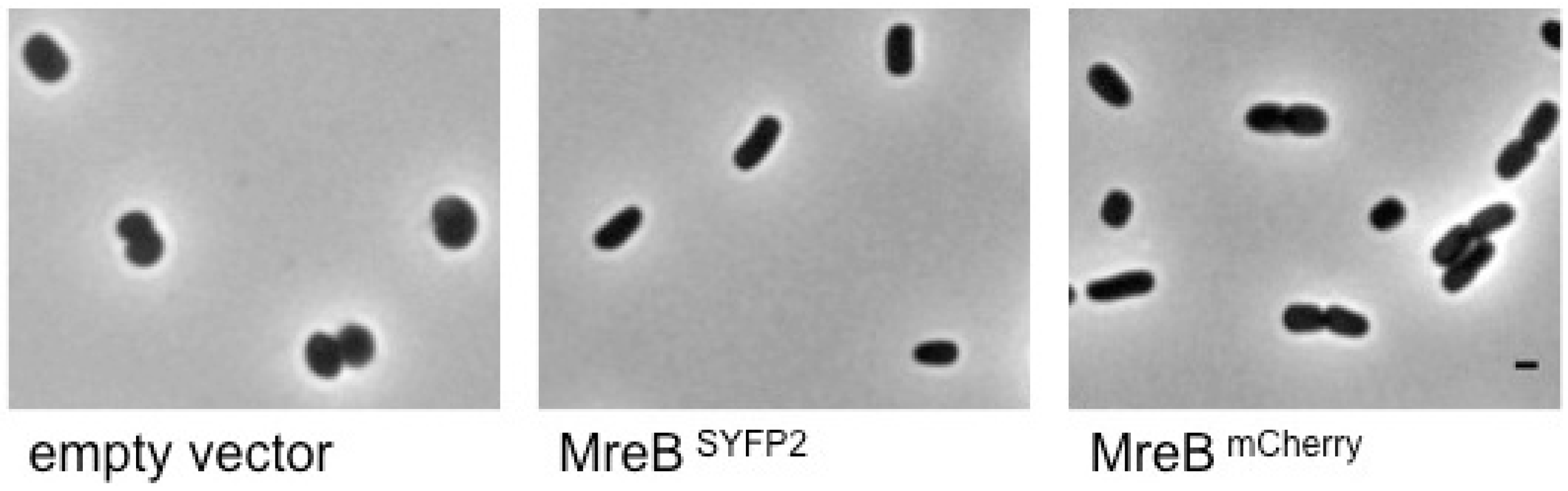
2.2. Spectral FRET Is Insensitive to Shape Changes
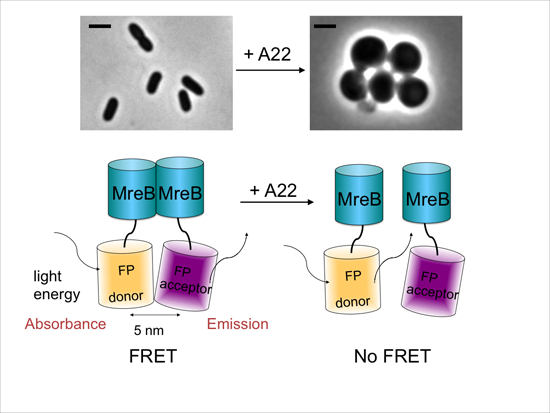
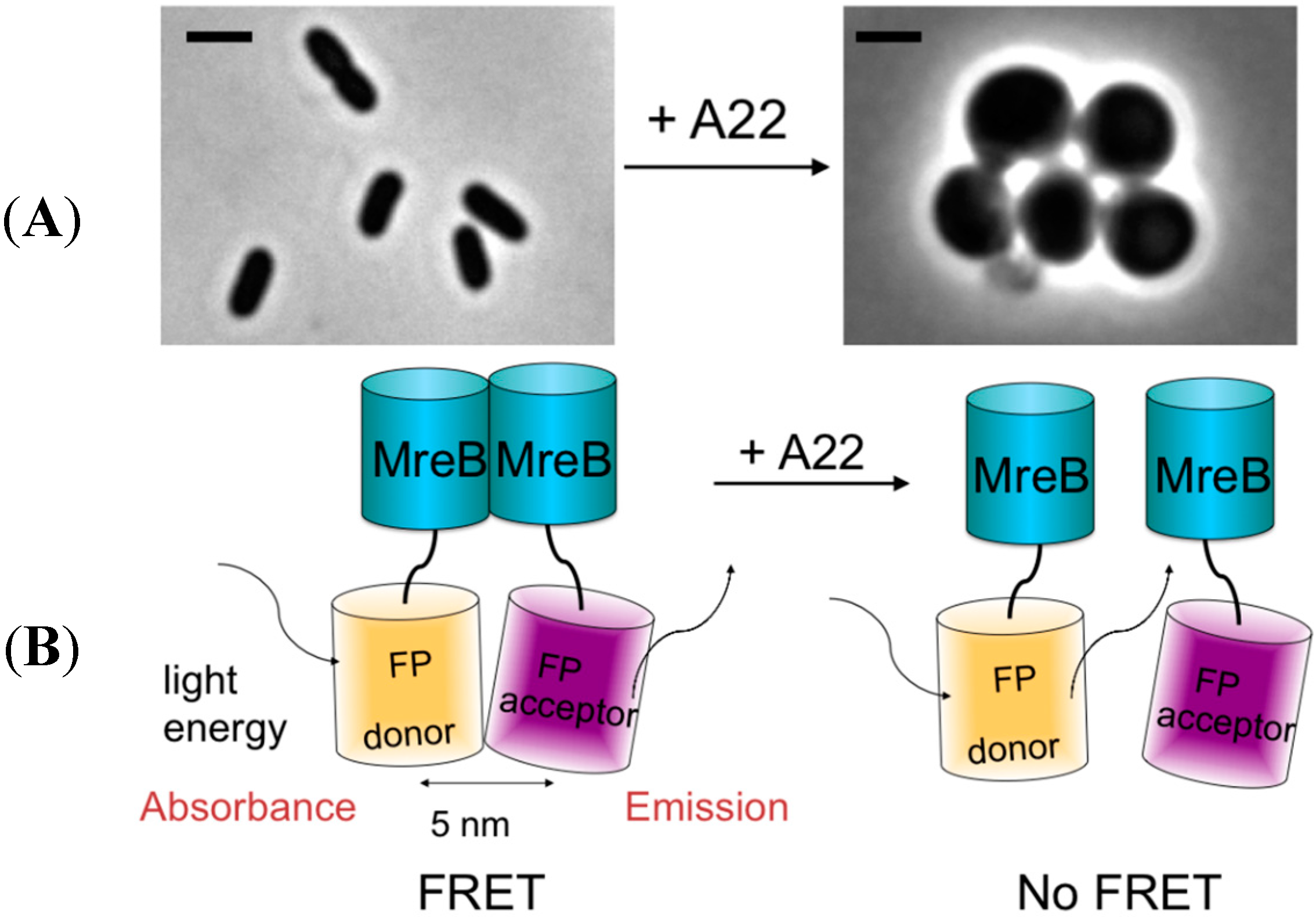
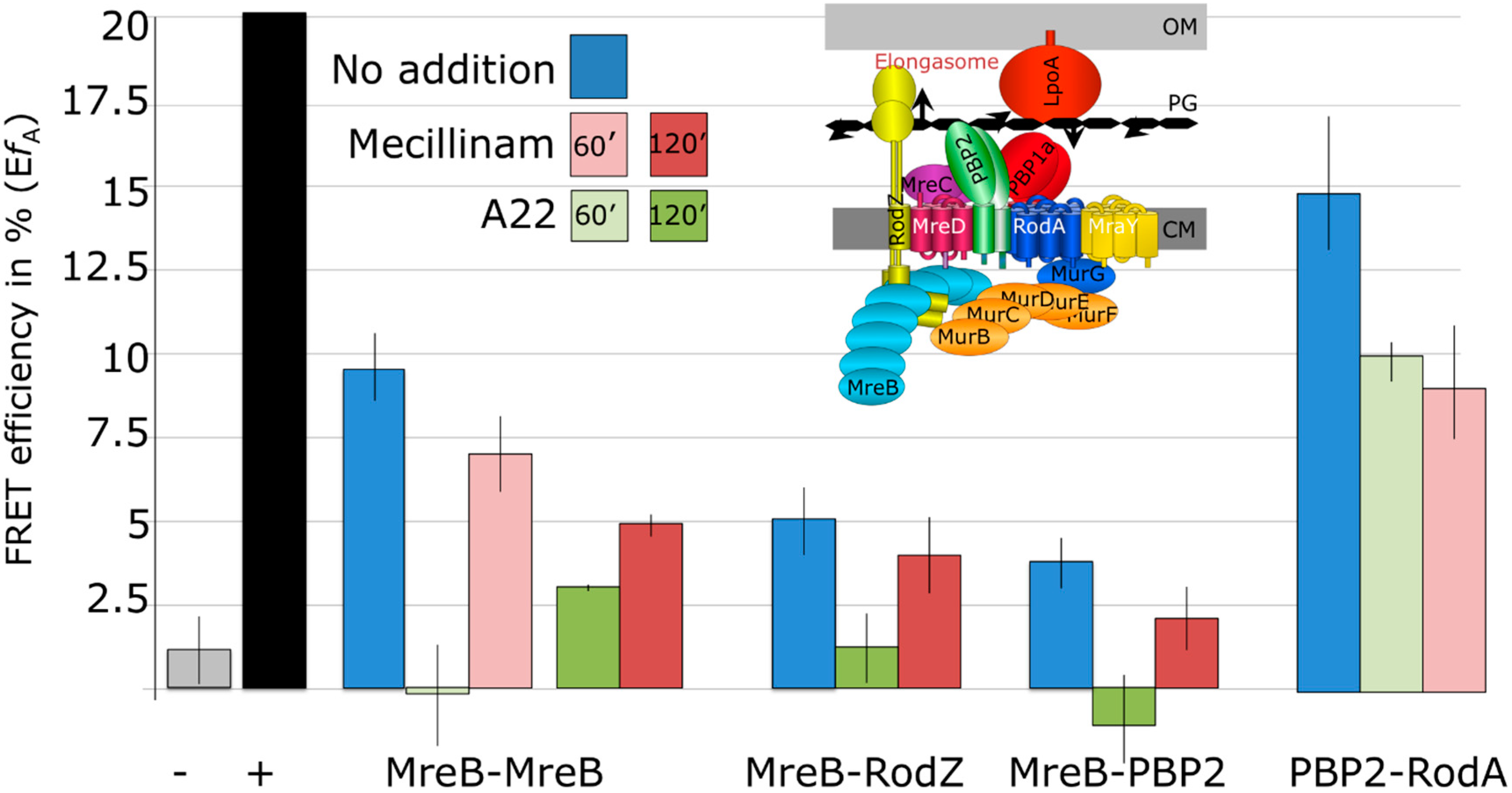
2.3. MreB Interactions Are Lost in the Presence of S-(3,4-Dichlorobenzyl) Isothiourea (A22)
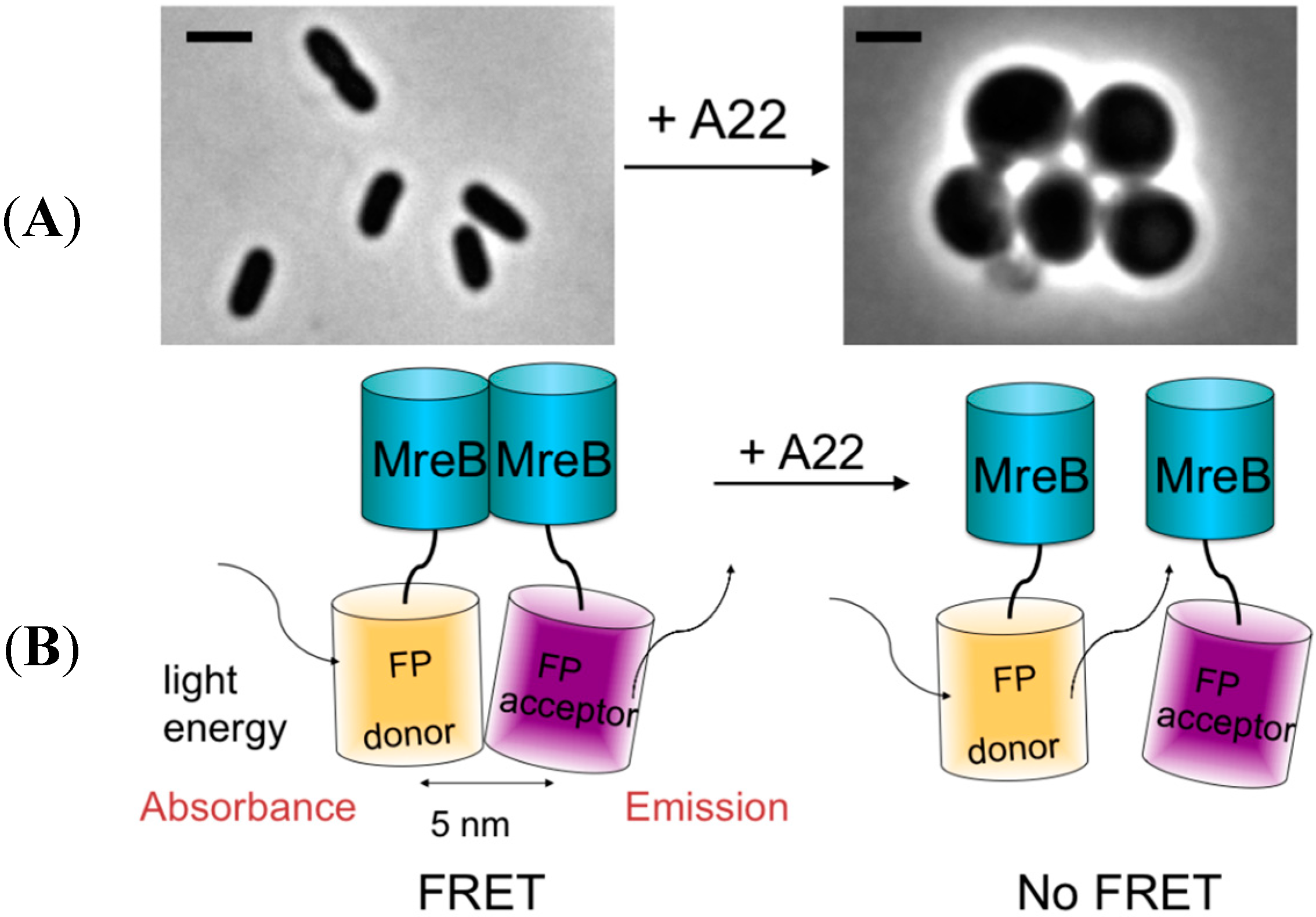

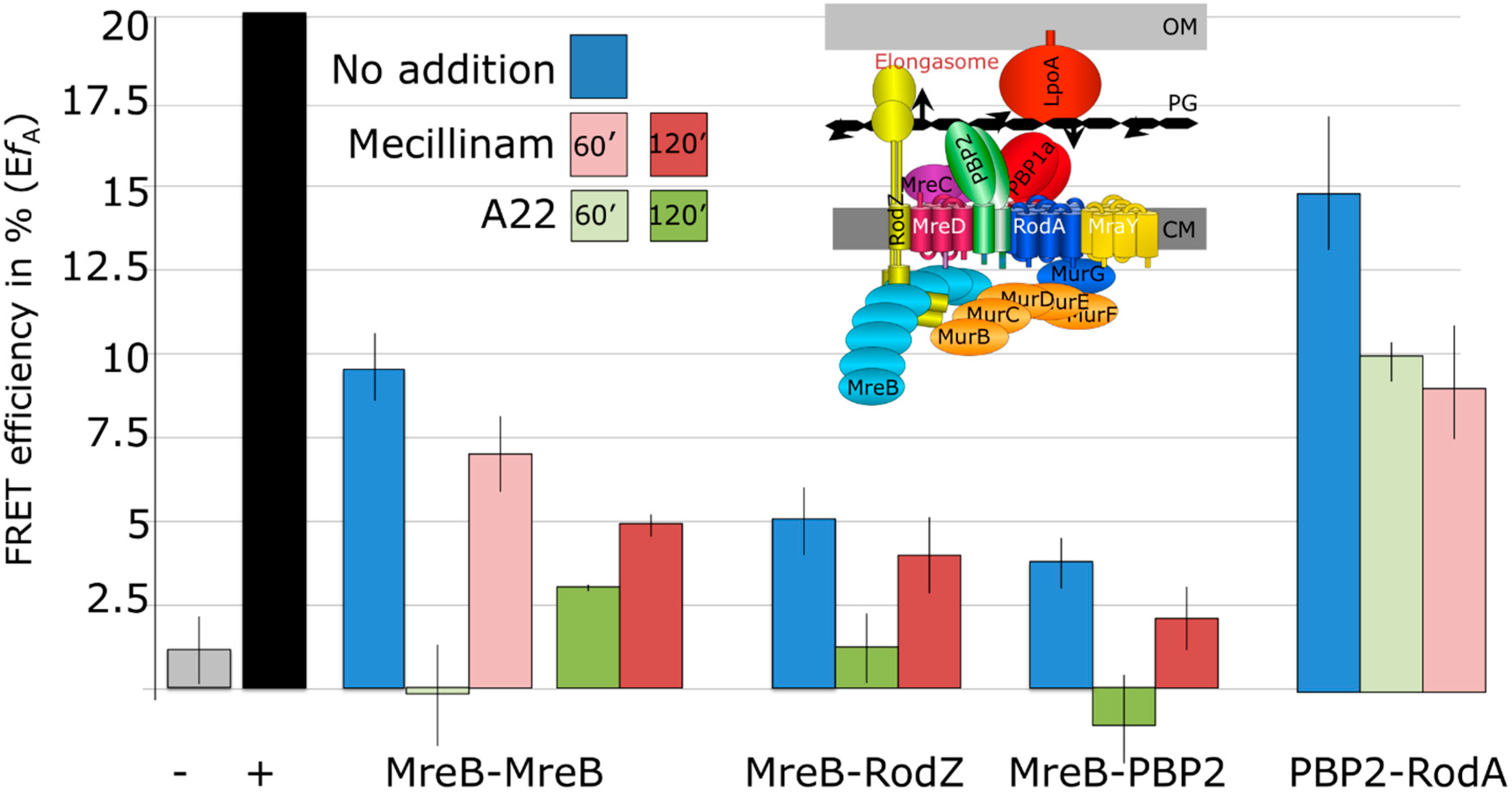
2.4. MreB Polymerization and PBP2 Activity Are Essential to Maintain Elongasome Protein Interactions

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Protein | Minimal a | Rich b | Reference |
|---|---|---|---|
| PBP1A | 135, 116 | 220, 554 | [44,45] |
| PBP2 | 58, 76 | 120, 324 | [25,45] |
| MreB | 9.000, 2.393 | 15.000, 11.304 | [8,45] |
| MurG | 168 | 518 | [45] |
| RodZ | 150, 576 | 650, 1309 | [25,45] |
3. Experimental Section
3.1. Bacterial Strains and Growth Conditions
3.2. Construction of the Plasmids
3.3. Microscopy and Image Analysis
3.4. FRET Experiment and Data Analysis
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Lu, P.-L.; Liu, Y.-C.; Toh, H.-S.; Lee, Y.-L.; Liu, Y.-M.; Ho, C.-M.; Huang, C.-C.; Liu, C.-E.; Ko, W.-C.; Wang, J.-H.; et al. Epidemiology and antimicrobial susceptibility profiles of Gram-negative bacteria causing urinary tract infections in the Asia-Pacific region: 2009–2010 results from the Study for Monitoring Antimicrobial Resistance Trends (SMART). Int. J. Antimicrob. Agents 2012, 40, S37–S43. [Google Scholar] [CrossRef]
- Silver, L.L. Challenges of antibacterial discovery. Clin. Microbiol. Rev. 2011, 24, 71–109. [Google Scholar] [CrossRef] [PubMed]
- Day, R.N.; Davidson, M.W. The fluorescent protein palette: Tools for cellular imaging. Chem. Soc. Rev. 2009, 38, 2887–2921. [Google Scholar] [CrossRef] [PubMed]
- Pietraszewska-Bogiel, A.; Gadella, T.W.J. FRET microscopy: From principle to routine technology in cell biology. J. Microsc. 2010, 241, 111–118. [Google Scholar] [CrossRef] [PubMed]
- Typas, A.; Banzhaf, M.; Gross, C.A.; Vollmer, W. From the regulation of peptidoglycan synthesis to bacterial growth and morphology. Nat. Rev. Microbiol. 2011, 10, 123–136. [Google Scholar] [CrossRef] [PubMed]
- Den Blaauwen, T.; de Pedro, M.A.; Nguyen-Distèche, M.; Ayala, J.A. Morphogenesis of rod-shaped sacculi. FEMS Microbiol. Rev. 2008, 32, 321–344. [Google Scholar] [CrossRef] [PubMed]
- Den Blaauwen, T.; Aarsman, M.E.G.; Vischer, N.O.E.; Nanninga, N. Penicillin-binding protein PBP2 of Escherichia coli localizes preferentially in the lateral wall and at mid-cell in comparison with the old cell pole. Mol. Microbiol. 2003, 47, 539–547. [Google Scholar] [CrossRef] [PubMed]
- Karczmarek, A.; Martínez-Arteaga, R.; Baselga, R.M.-A.; Alexeeva, S.; Hansen, F.G.; Vicente, M.; Nanninga, N.; den Blaauwen, T. DNA and origin region segregation are not affected by the transition from rod to sphere after inhibition of Escherichia coli MreB by A22. Mol. Microbiol. 2007, 65, 51–63. [Google Scholar] [CrossRef] [PubMed]
- Bean, G.J.; Flickinger, S.T.; Westler, W.M.; Mccully, M.E.; Sept, D.; Weibel, D.B.; Amann, K.J. A22 disrupts the bacterial actin cytoskeleton by directly binding and inducing a low-affinity state in MreB. Biochemistry 2009, 48, 4852–4857. [Google Scholar] [CrossRef] [PubMed]
- Nurse, P.; Marians, K.J. Purification and characterization of Escherichia coli MreB protein. J. Biol. Chem. 2013, 288, 3469–3475. [Google Scholar] [CrossRef] [PubMed]
- Van den Ent, F.; Amos, L.A.; Löwe, J. Prokaryotic origin of the actin cytoskeleton. Nature 2001, 413, 39–44. [Google Scholar] [CrossRef] [PubMed]
- Salje, J.; van den Ent, F.; de Boer, P.; Löwe, J. Direct membrane binding by bacterial actin MreB. Mol. Cell 2011, 43, 478–487. [Google Scholar] [CrossRef] [PubMed]
- Reimold, C.; Soufo, H.J.D.; Dempwolff, F.; Graumann, P.L. Motion of variable-length MreB filaments at the bacterial cell membrane influences cell morphology. Mol. Biol. Cell 2013, 24, 2340–2349. [Google Scholar] [CrossRef] [PubMed]
- Divakaruni, A.V.; Baida, C.; White, C.L.; Gober, J.W. The cell shape proteins MreB and MreC control cell morphogenesis by positioning cell wall synthetic complexes. Mol. Microbiol. 2007, 66, 174–188. [Google Scholar] [CrossRef] [PubMed]
- Mohammadi, T.; van Dam, V.; Sijbrandi, R.; Vernet, T.; Zapun, A.; Bouhss, A.; Diepeveen-de Bruin, M.; Nguyen-DisteChe, M.; de Kruijff, B.; Breukink, E. Identification of FtsW as a transporter of lipid-linked cell wall precursors across the membrane. EMBO J. 2011, 30, 1425–1432. [Google Scholar] [CrossRef] [PubMed]
- Mohammadi, T.; Karczmarek, A.; Crouvoisier, M.; Bouhss, A.; Mengin-Lecreulx, D.; den Blaauwen, T. The essential peptidoglycan glycosyltransferase MurG forms a complex with proteins involved in lateral envelope growth as well as with proteins involved in cell division in Escherichia coli. Mol. Microbiol. 2007, 65, 1106–1121. [Google Scholar] [CrossRef] [PubMed]
- Banzhaf, M.; van den Berg van Saparoea, B.; Terrak, M.; Fraipont, C.; Egan, A.; Philippe, J.; Zapun, A.; Breukink, E.; Nguyen-Distèche, M.; den Blaauwen, T.; et al. Cooperativity of peptidoglycan synthases active in bacterial cell elongation. Mol. Microbiol. 2012, 85, 179–194. [Google Scholar] [CrossRef] [PubMed]
- Typas, A.; Banzhaf, M.; van den Berg van Saparoea, B.; Verheul, J.; Biboy, J.; Nichols, R.J.; Zietek, M.; Beilharz, K.; Kannenberg, K.; von Rechenberg, M.; et al. Regulation of peptidoglycan synthesis by outer-membrane proteins. Cell 2010, 143, 1097–1109. [Google Scholar] [CrossRef] [PubMed]
- Kruse, T.; Bork-Jensen, J.; Gerdes, K. The morphogenetic MreBCD proteins of Escherichia coli form an essential membrane-bound complex. Mol. Microbiol. 2005, 55, 78–89. [Google Scholar] [CrossRef] [PubMed]
- Shiomi, D.; Margolin, W. Compensation for the loss of the conserved membrane targeting sequence of FtsA provides new insights into its function. Mol. Microbiol. 2008, 67, 558–569. [Google Scholar] [CrossRef] [PubMed]
- Vats, P.; Shih, Y.-L.; Rothfield, L. Assembly of the MreB-associated cytoskeletal ring of Escherichia coli. Mol. Microbiol. 2009, 72, 170–182. [Google Scholar] [CrossRef] [PubMed]
- El Ghachi, M.; Matteï, P.-J.; Ecobichon, C.; Martins, A.; Hoos, S.; Schmitt, C.; Colland, F.; Ebel, C.; Prévost, M.-C.; Gabel, F.; et al. Characterization of the elongasome core PBP2:MreC complex of Helicobacter pylori. Mol. Microbiol. 2011, 82, 68–86. [Google Scholar] [CrossRef] [PubMed]
- Shiomi, D.; Sakai, M.; Niki, H. Determination of bacterial rod shape by a novel cytoskeletal membrane protein. EMBO J. 2008, 27, 3081–3091. [Google Scholar] [CrossRef] [PubMed]
- Alyahya, S.A.; Alexander, R.; Costa, T.; Henriques, A.O.; Emonet, T.; Jacobs-Wagner, C. RodZ, a component of the bacterial core morphogenic apparatus. Proc. Natl. Acad. Sci. USA 2009, 106, 1239–1244. [Google Scholar] [CrossRef] [PubMed]
- Bendezú, F.O.; Hale, C.A.; Bernhardt, T.G.; de Boer, P.A.J. RodZ (YfgA) is required for proper assembly of the MreB actin cytoskeleton and cell shape in E. coli. EMBO J. 2009, 28, 193–204. [Google Scholar] [CrossRef] [PubMed]
- Van den Ent, F.; Johnson, C.M.; Persons, L.; de Boer, P.; Löwe, J. Bacterial actin MreB assembles in complex with cell shape protein RodZ. EMBO J. 2010, 29, 1081–1090. [Google Scholar] [CrossRef] [PubMed]
- Muchová, K.; Chromiková, Z.; Barák, I. Control of Bacillus subtilis cell shape by RodZ. Environ. Microbiol. 2013, 15, 3259–3271. [Google Scholar] [CrossRef] [PubMed]
- Shiomi, D.; Toyoda, A.; Aizu, T.; Ejima, F.; Fujiyama, A.; Shini, T.; Kohara, Y.; Niki, H. Mutations in cell elongation genes mreB, mrdA and mrdB suppress the shape defect of RodZ-deficient cells. Mol. Microbiol. 2013, 87, 1029–1044. [Google Scholar] [CrossRef] [PubMed]
- Iwai, N.; Nagai, K.; Wachi, M. Novel S-benzylisothiourea compound that induces spherical cells in Escherichia coli probably by acting on a rod-shape-determining protein(s) other than penicillin-binding protein 2. Biosci. Biotechnol. Biochem. 2002, 66, 2658–2662. [Google Scholar] [CrossRef] [PubMed]
- Dye, N.A.; Pincus, Z.; Fisher, I.C.; Shapiro, L.; Theriot, J.A. Mutations in the nucleotide binding pocket of MreB can alter cell curvature and polar morphology in Caulobacter. Mol. Microbiol. 2011, 81, 368–394. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Furchtgott, L.; Huang, K.C.; Shaevitz, J.W. Helical insertion of peptidoglycan produces chiral ordering of the bacterial cell wall. Proc. Natl. Acad. Sci. USA 2012, 109, E595–E604. [Google Scholar] [CrossRef] [PubMed]
- Van Teeffelen, S.; Wang, S.; Furchtgott, L.; Huang, K.C.; Wingreen, N.S.; Shaevitz, J.W.; Gitai, Z. The bacterial actin MreB rotates, and rotation depends on cell-wall assembly. Proc. Natl. Acad. Sci. USA 2011, 108, 15822–15827. [Google Scholar] [CrossRef] [PubMed]
- Clegg, R.M. FRET and FLIM Techniques, 1st ed.; Gadella, T.W.J., Ed.; Elsevier: Amsterdam, The Netherland, 2009; Volume 33, pp. 1–48. [Google Scholar]
- Goedhart, J.; von Stetten, D.; Noirclerc-Savoye, M.; Lelimousin, M.; Joosen, L.; Hink, M.A.; van Weeren, L.; Gadella, T.W.J., Jr.; Royant, A. Structure-guided evolution of cyan fluorescent proteins towards a quantum yield of 93%. Nat. Commun. 2012, 3, 751. [Google Scholar] [CrossRef] [PubMed]
- Kremers, G.-J.; Goedhart, J.; van Munster, E.B.; Gadella, T.W.J., Jr. Cyan and yellow super fluorescent proteins with improved brightness, protein folding, and FRET förster radius. Biochemistry 2006, 45, 6570–6580. [Google Scholar] [CrossRef] [PubMed]
- Shaner, N.C.; Campbell, R.E.; Steinbach, P.A.; Giepmans, B.N.G.; Palmer, A.E.; Tsien, R.Y. Improved monomeric red, orange and yellow fluorescent proteins derived from Discosoma sp. red fluorescent protein. Nat. Biotechnol. 2004, 22, 1567–1572. [Google Scholar] [CrossRef] [PubMed]
- Alexeeva, S.; Gadella, T.W.J., Jr.; Verheul, J.; Verhoeven, G.S.; den Blaauwen, T. Direct interactions of early and late assembling division proteins in Escherichia coli cells resolved by FRET. Mol. Microbiol. 2010, 77, 384–398. [Google Scholar] [CrossRef] [PubMed]
- Spratt, B.G. Distinct penicillin binding proteins involved in the division, elongation, and shape of Escherichia coli K12. Proc. Natl. Acad. Sci. USA 1975, 72, 2999–3003. [Google Scholar] [CrossRef] [PubMed]
- Van der Ploeg, R.; Verheul, J.; Vischer, N.O.E.; Alexeeva, S.; Hoogendoorn, E.; Postma, M.; Banzhaf, M.; Vollmer, W.; den Blaauwen, T. Colocalization and interaction between elongasome and divisome during a preparative cell division phase in Escherichia coli. Mol. Microbiol. 2013, 87, 1074–1087. [Google Scholar] [CrossRef] [PubMed]
- Park, J.T.; Burman, L. FL-1060: A new penicillin with a unique mode of action. Biochem. Biophys. Res. Commun. 1973, 51, 863–868. [Google Scholar] [CrossRef]
- Sykes, R.B.; Bonner, D.P.; Bush, K.; Georgopapadakou, N.H. Azthreonam (SQ 26,776), a synthetic monobactam specifically active against aerobic gram-negative bacteria. Antimicrob. Agents Chemother. 1982, 21, 85–92. [Google Scholar] [CrossRef] [PubMed]
- Pogliano, J.; Pogliano, K.; Weiss, D.S.; Losick, R.; Beckwith, J. Inactivation of FtsI inhibits constriction of the FtsZ cytokinetic ring and delays the assembly of FtsZ rings at potential division sites. Proc. Natl. Acad. Sci. USA 1997, 94, 559–564. [Google Scholar] [CrossRef] [PubMed]
- Buckstein, M.H.; He, J.; Rubin, H. Characterization of nucleotide pools as a function of physiological state in Escherichia coli. J. Bacteriol. 2008, 190, 718–726. [Google Scholar] [CrossRef] [PubMed]
- Dougherty, T.J.; Kennedy, K.; Kessler, R.E.; Pucci, M.J. Direct quantitation of the number of individual penicillin-binding proteins per cell in Escherichia coli. J. Bacteriol. 1996, 178, 6110–6115. [Google Scholar] [PubMed]
- Li, G.-W.; Burkhardt, D.; Gross, C.; Weissman, J.S. Quantifying absolute protein synthesis rates reveals principles underlying allocation of cellular resources. Cell 2014, 157, 624–635. [Google Scholar] [CrossRef] [PubMed]
- De Pedro, M.A.; Donachie, W.D.; Höltje, J.V.; Schwarz, H. Constitutive septal murein synthesis in Escherichia coli with impaired activity of the morphogenetic proteins RodA and penicillin-binding protein 2. J. Bacteriol. 2001, 183, 4115–4126. [Google Scholar] [CrossRef] [PubMed]
- Ishino, F.; Park, W.; Tomioka, S.; Tamaki, S.; Takase, I.; Kunugita, K.; Matsuzawa, H.; Asoh, S.; Ohta, T.; Spratt, B.G. Peptidoglycan synthetic activities in membranes of Escherichia coli caused by overproduction of penicillin-binding protein 2 and rodA protein. J. Biol. Chem. 1986, 261, 7024–7031. [Google Scholar] [PubMed]
- Schirner, K.; Eun, Y.-J.; Dion, M.; Luo, Y.; Helmann, J.D.; Garner, E.C.; Walker, S. Lipid-linked cell wall precursors regulate membrane association of bacterial actin MreB. Nat. Chem. Biol. 2015, 11, 38–45. [Google Scholar] [CrossRef] [PubMed]
- Lages, M.C.A.; Beilharz, K.; Morales Angeles, D.; Veening, J.-W.; Scheffers, D.-J. The localization of key Bacillus subtilis penicillin binding proteins during cell growth is determined by substrate availability. Environ. Microbiol. 2013, 15, 3272–3281. [Google Scholar] [CrossRef] [PubMed]
- Potluri, L.; Karczmarek, A.; Verheul, J.; Piette, A.; Wilkin, J.-M.; Werth, N.; Banzhaf, M.; Vollmer, W.; Young, K.D.; Nguyen-Distèche, M.; et al. Septal and lateral wall localization of PBP5, the major d,d-carboxypeptidase of Escherichia coli, requires substrate recognition and membrane attachment. Mol. Microbiol. 2010, 77, 300–323. [Google Scholar] [CrossRef] [PubMed]
- Den Blaauwen, T.; Buddelmeijer, N.; Aarsman, M.E.; Hameete, C.M.; Nanninga, N. Timing of FtsZ assembly in Escherichia coli. J. Bacteriol. 1999, 181, 5167–5175. [Google Scholar] [PubMed]
- Shimozono, S.; Hosoi, H.; Mizuno, H.; Fukano, T.; Tahara, T.; Miyawaki, A. Concatenation of cyan and yellow fluorescent proteins for efficient resonance energy transfer. Biochemistry 2006, 45, 6267–6271. [Google Scholar] [CrossRef] [PubMed]
- Koppelman, C.-M.; Aarsman, M.E.G.; Postmus, J.; Pas, E.; Muijsers, A.O.; Scheffers, D.-J.; Nanninga, N.; den Blaauwen, T. R174 of Escherichia coli FtsZ is involved in membrane interaction and protofilament bundling, and is essential for cell division. Mol. Microbiol. 2004, 51, 645–657. [Google Scholar] [CrossRef] [PubMed]
- Karasawa, S.; Araki, T.; Nagai, T.; Mizuno, H.; Miyawaki, A. Cyan-emitting and orange-emitting fluorescent proteins as a donor/acceptor pair for fluorescence resonance energy transfer. Biochem. J. 2004, 381, 307–312. [Google Scholar] [PubMed]
- Clegg, R.M.; Murchie, A.I.; Zechel, A.; Carlberg, C.; Diekmann, S.; Lilley, D.M. Fluorescence resonance energy transfer analysis of the structure of the four-way DNA junction. Biochemistry 1992, 31, 4846–4856. [Google Scholar] [CrossRef] [PubMed]
- Clegg, R.M. Fluorescence resonance energy transfer and nucleic acids. Methods Enzymol. 1992, 211, 353–388. [Google Scholar] [PubMed]
- Hanahan, D. Studies on transformation of Escherichia coli with plasmids. J. Mol. Biol. 1983, 166, 557–580. [Google Scholar] [CrossRef]
- Taschner, P.E.; Huls, P.G.; Pas, E.; Woldringh, C.L. Division behavior and shape changes in isogenic ftsZ, ftsQ, ftsA, pbpB, and ftsE cell division mutants of Escherichia coli during temperature shift experiments. J. Bacteriol. 1988, 170, 1533–1540. [Google Scholar] [PubMed]
- Wachi, M.; Doi, M.; Tamaki, S.; Park, W.; Nakajima-Iijima, S.; Matsuhashi, M. Mutant isolation and molecular cloning of mre genes, which determine cell shape, sensitivity to mecillinam, and amount of penicillin-binding proteins in Escherichia coli. J. Bacteriol. 1987, 169, 4935–4940. [Google Scholar] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Van der Ploeg, R.; Goudelis, S.T.; Den Blaauwen, T. Validation of FRET Assay for the Screening of Growth Inhibitors of Escherichia coli Reveals Elongasome Assembly Dynamics. Int. J. Mol. Sci. 2015, 16, 17637-17654. https://doi.org/10.3390/ijms160817637
Van der Ploeg R, Goudelis ST, Den Blaauwen T. Validation of FRET Assay for the Screening of Growth Inhibitors of Escherichia coli Reveals Elongasome Assembly Dynamics. International Journal of Molecular Sciences. 2015; 16(8):17637-17654. https://doi.org/10.3390/ijms160817637
Chicago/Turabian StyleVan der Ploeg, René, Spyridon Theodoros Goudelis, and Tanneke Den Blaauwen. 2015. "Validation of FRET Assay for the Screening of Growth Inhibitors of Escherichia coli Reveals Elongasome Assembly Dynamics" International Journal of Molecular Sciences 16, no. 8: 17637-17654. https://doi.org/10.3390/ijms160817637