
Tools for Sequence-Based miRNA Target Prediction: What to Choose?

Abstract
:1. Introduction
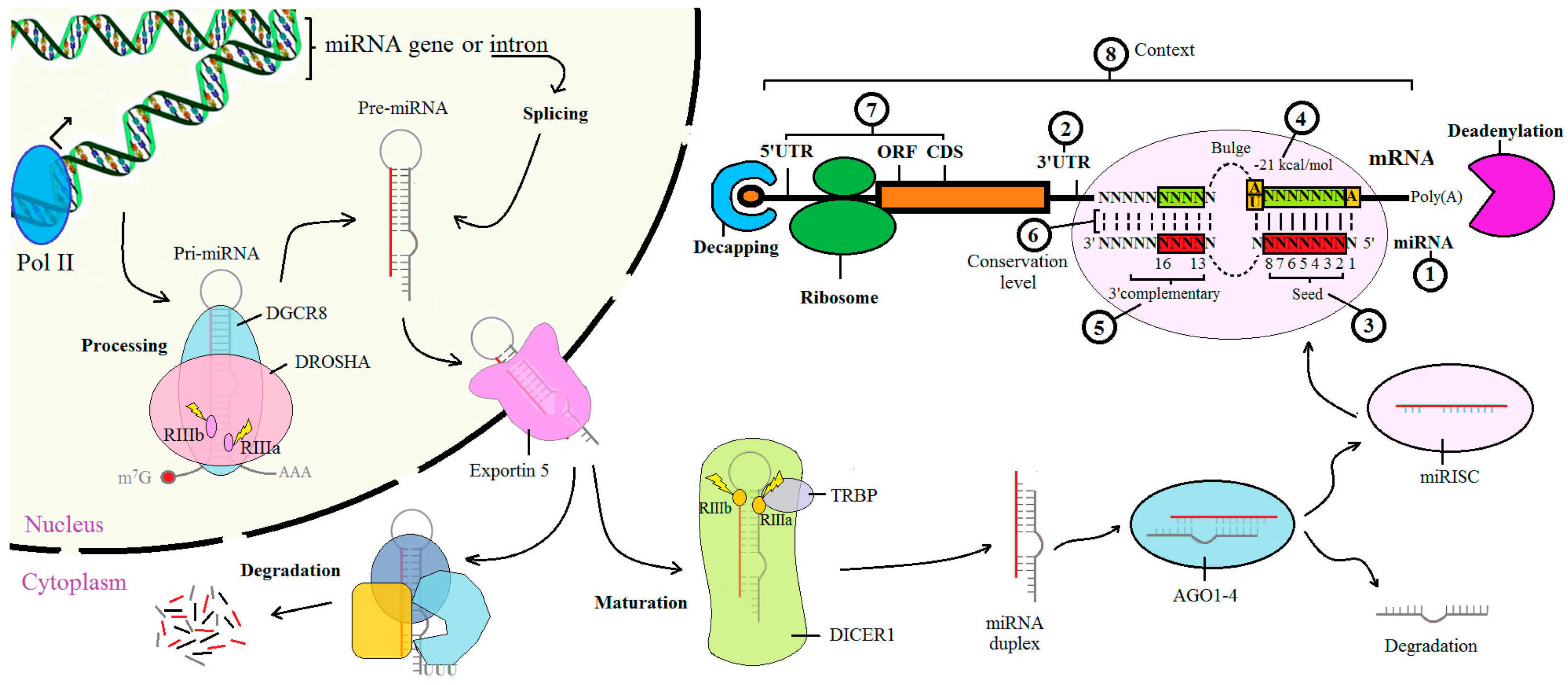
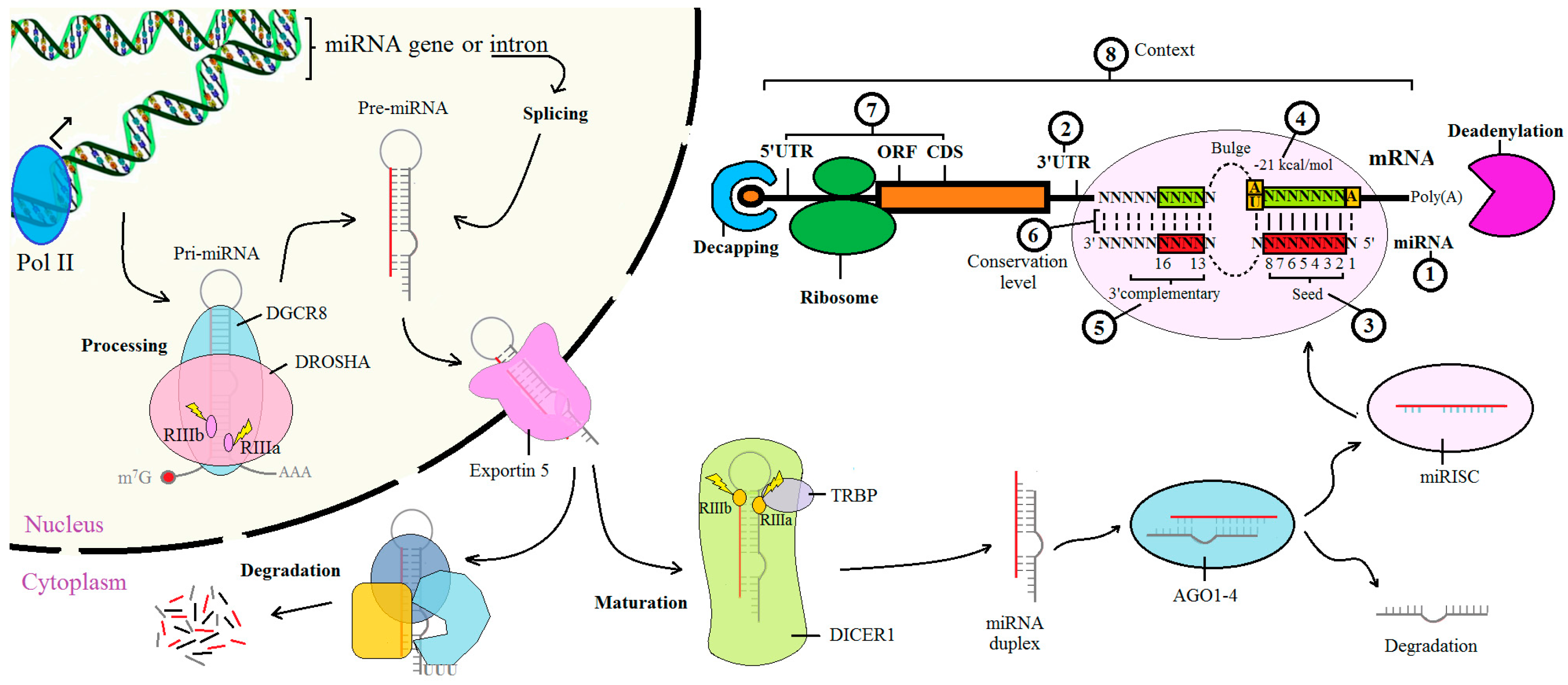
2. Biological Elements for Computational Analysis
2.1. The miRNA Sequences
2.2. The 3′ UTR of mRNA
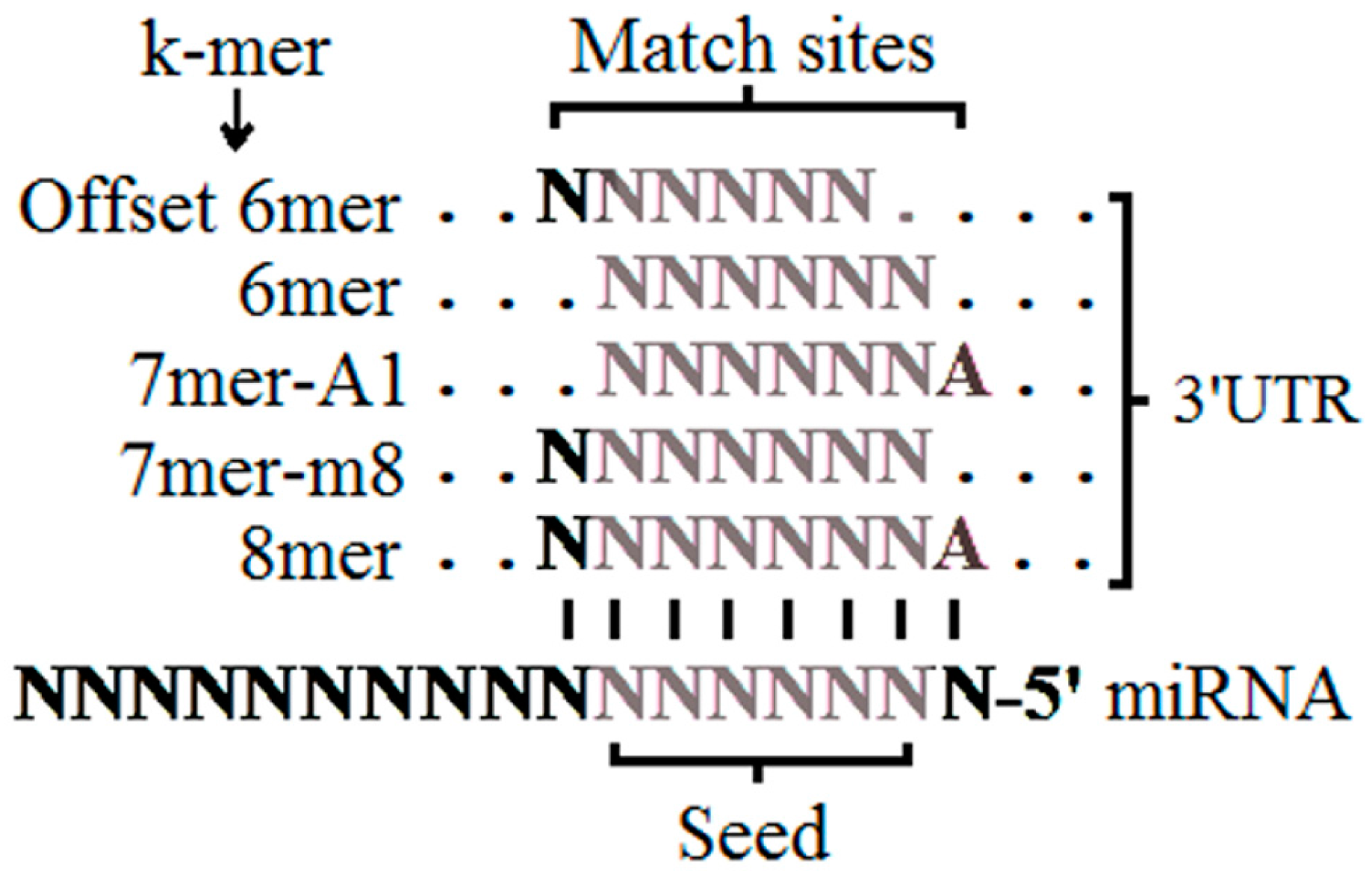
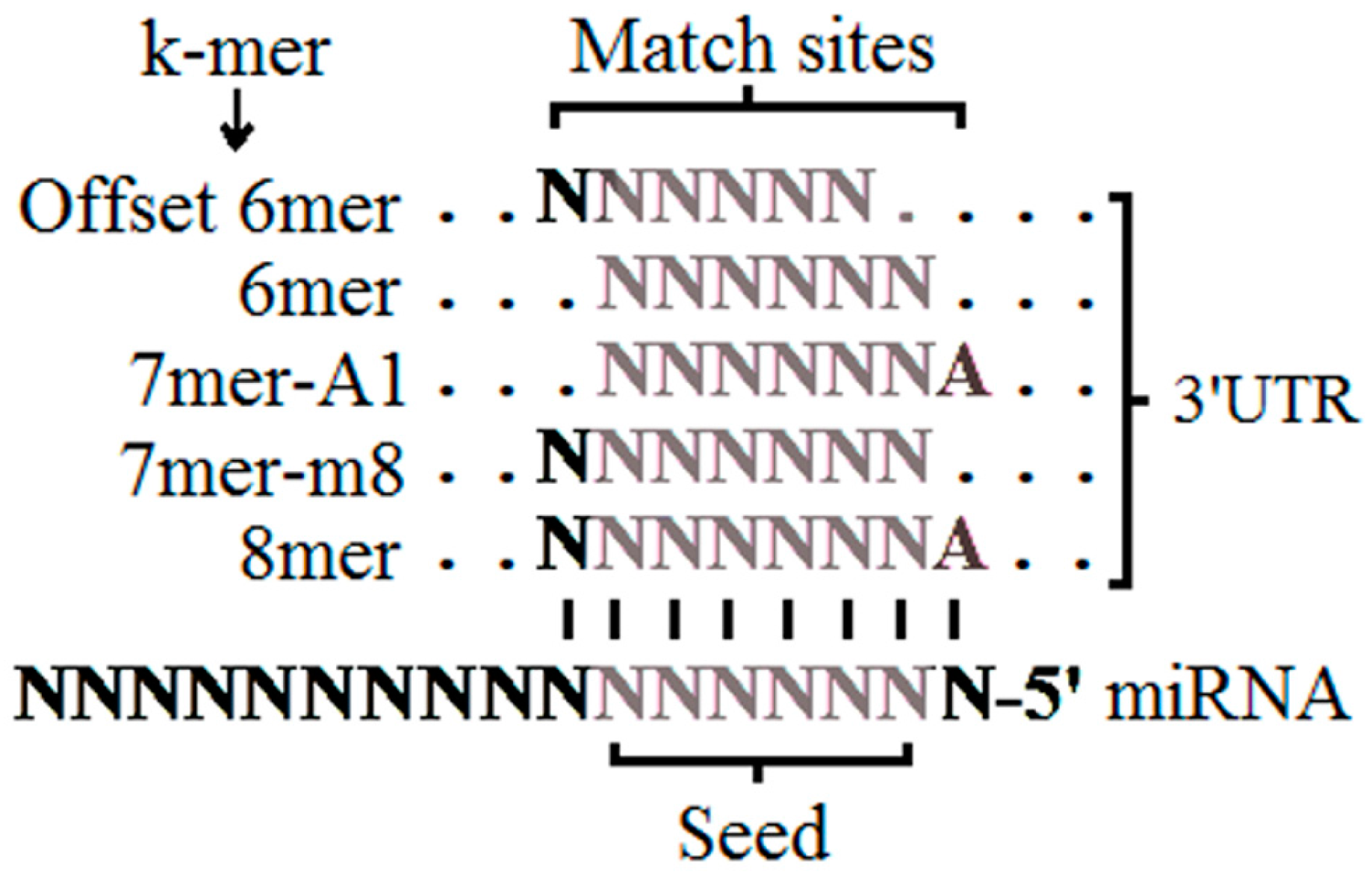
2.3. The Seed Region
2.4. Free Energy and Accessible Energy
2.5. miRNA 3′ Site
2.6. Conservation Status
2.7. Other Target Sites
2.8. The Contexts
3. Bioinformatic Tools for miRNA Target Prediction
3.1. TargetScan
3.2. miRanda
3.3. DIANA Tools
4. Discussion
Considerations for Using a miRNA Analysis Tool
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Ghildiyal, M.; Zamore, P.D. Small silencing RNAs: An expanding universe. Nat. Rev. Genet. 2009, 10, 94–108. [Google Scholar] [CrossRef] [PubMed]
- Krol, J.; Loedige, I.; Filipowicz, W. The widespread regulation of microRNA biogenesis, function and decay. Nat. Rev. Genet. 2010, 11, 597–610. [Google Scholar] [CrossRef] [PubMed]
- Jakub, O.; Westholm, E.C.L. Mirtrons: MicroRNA biogenesis via splicing. Biochimie 2011, 93, 1897–1904. [Google Scholar]
- Borchert, G.M.; Lanier, W.; Davidson, B.L. RNA polymerase III transcribes human microRNAs. Nat. Struct. Mol. Biol. 2006, 13, 1097–1101. [Google Scholar] [CrossRef] [PubMed]
- Paroo, Z.; Liu, Q.; Wang, X. Biochemical mechanisms of the RNA-induced silencing complex. Cell Res. 2007, 17, 187–194. [Google Scholar] [CrossRef] [PubMed]
- Wilson, R.C.; Tambe, A.; Kidwell, M.A.; Nolands, C.L.; Schneider, C.P.; Doudna, J.A. Dicer–TRBP complex formation ensures accurate mammalian microRNA biogenesis. Mol. Cell 2015, 57, 397–407. [Google Scholar] [CrossRef] [PubMed]
- Ha, M.; Kim, V.N. Regulation of microRNA biogenesis. Nat. Rev. Mol. Cell Biol. 2014, 15, 509–524. [Google Scholar] [CrossRef] [PubMed]
- Sekar, D.; Venugopal, B.; Sekar, P.; Ramalingam, K. Role of microRNA 21 in diabetes and associated/related diseases. Gene 2016, 582, 14–18. [Google Scholar] [CrossRef] [PubMed]
- Shi, C.; Huang, F.; Gu, X.; Zhang, M.; Wen, J.; You, L.; Cui, X.; Ji, C.; Guo, X. Adipogenic miRNA and Meta-signature miRNAs involved in human adipocyte differentiation and obesity. Oncotarget 2016, 7, 40830–40845. [Google Scholar] [CrossRef] [PubMed]
- Vicente, R.; Noël, D.; Pers, Y.-M.; Apparailly, F.; Jorgensen, C. Deregulation and therapeutic potential of microRNAs in arthritic diseases. Nat. Rev. Rheumatol. 2016, 12, 211–220. [Google Scholar] [CrossRef] [PubMed]
- Denby, L.; Baker, A.H. Targeting non-coding RNA for the therapy of renal disease. Curr. Opin. Pharmacol. 2016, 27, 70–77. [Google Scholar] [CrossRef] [PubMed]
- Hackfort, B.T.; Mishra, P.K. Emerging role of hydrogen sulfide-microRNA cross-talk in cardiovascular diseases. Am. J. Physiol. Heart Circ. Physiol. 2016, 10, H802–H812. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.; Gregory, R.I. MicroRNA biogenesis pathways in cancer. Nat. Rev. Cancer 2015, 15, 321–333. [Google Scholar] [CrossRef] [PubMed]
- Ling, H.; Fabbri, M.; Calin, G.A. MicroRNAs and other non-coding RNAs as targets for anticancer drug development. Nat. Rev. Drug Discov. 2013, 11, 847–865. [Google Scholar] [CrossRef] [PubMed]
- Samanta, S.; Balasubramanian, S.; Rajasingh, S.; Patel, U.; Dhanasekaran, A.; Dawn, B.; Rajasingh, J. MicroRNA: A new therapeutic strategy for cardiovascular diseases. Trends Cardiovasc. Med. 2016, 26, 207–419. [Google Scholar] [CrossRef] [PubMed]
- Riquelme, I.; Letelier, P.; Riffo-Campos, A.L.; Brebi, P.; Roa, J. Emerging role of miRNAs in the drug resistance of gastric cancer. Int. J. Mol. Sci. 2016, 17, 424. [Google Scholar] [CrossRef] [PubMed]
- Lee, R.C.; Feinbaum, R.L.; Ambros, V. The C. elegans heterochronic gene lin-4 encodes small RNAs with antisense complementarity to & II-14. Cell 1993, 75, 843–854. [Google Scholar] [PubMed]
- Wightman, B.; Ha, I.; Ruvkun, G. Posttranscriptional regulation of the heterochronic gene lin-14 by lin-4 mediates temporal pattern formation in C. elegans. Cell 1993, 75, 855–862. [Google Scholar] [CrossRef]
- Lau, N.C.; Lim, L.P.; Weinstein, E.G.; Bartel, D.P. An abundant class of tiny RNAs with probable regulatory roles in Caenorhabditis elegans. Science 2001, 294, 858–862. [Google Scholar] [CrossRef] [PubMed]
- Lander, E.S.; Linton, L.M.; Birren, B.; Nusbaum, C.; Zody, M.C.; Baldwin, J.; Devon, K.; Dewar, K.; Doyle, M.; FitzHugh, W.; et al. Initial sequencing and analysis of the human genome. Nature 2001, 409, 860–921. [Google Scholar] [CrossRef] [PubMed]
- Venter, J.C.; Adams, M.D.; Myers, E.W.; Li, P.W.; Mural, R.J.; Sutton, G.G.; Smith, H.O.; Yandell, M.; Evans, C.A.; Holt, R.A.; et al. The sequence of the human genome. Science 2001, 291, 1304–1351. [Google Scholar] [CrossRef] [PubMed]
- Argaman, L.; Hershberg, R.; Vogel, J.; Bejerano, G.; Wagner, G.H.; Margalit, H.; Altuvia, S. Novel small RNA-encoding genes in the intergenic regions of Escherichia coli. Curr. Biol. 2001, 11, 941–950. [Google Scholar] [CrossRef]
- Rivas, E.; Klein, R.J.; Jones, T.A.; Eddy, S.R. Computational identification of noncoding RNAs in E. coli by comparative genomics. Curr. Biol. 2001, 11, 1369–1373. [Google Scholar] [CrossRef]
- Wassarman, K.M.; Repoila, F.; Rosenow, C.; Storz, G.; Gottesman, S. Identification of novel small RNAs using comparative genomics and microarrays. Genes Dev. 2001, 15, 1637–1651. [Google Scholar] [CrossRef] [PubMed]
- Reinhart, B.; Weinstein, E.G.; Rhoades, M.W.; Barted, B.; Bartel, D.P. MicroRNAs in plants. Genes Dev. 2002, 16, 1616–1626. [Google Scholar] [CrossRef] [PubMed]
- Lee, R.C.; Ambros, V. An extensive class of small RNAs in Caenorhabditis elegans. Science 2001, 294, 862–864. [Google Scholar] [CrossRef] [PubMed]
- Lagos-quintana, M.; Rauhut, R.; Meyer, J.; Borkhardt, A.; Tuschl, T. New microRNAs from mouse and human new microRNAs from mouse and human. RNA 2003, 9, 175–179. [Google Scholar] [CrossRef] [PubMed]
- Lagos-Quintana, M.; Rauhut, R.; Yalcin, A.; Meyer, J.; Lendeckel, W.; Tuschl, T. Identification of tissue-specific MicroRNAs from mouse. Curr. Biol. 2002, 12, 735–739. [Google Scholar] [CrossRef]
- Lim, L.P.; Glasner, M.E.; Yekta, S.; Burge, C.B.; Bartel, D.P. Vertebrate microRNA genes. Science 2003, 299, 1540. [Google Scholar] [CrossRef] [PubMed]
- Griffiths-Jones, S.; Bateman, A.; Marshall, M.; Khanna, A.; Eddy, S.R. Rfam: An RNA family database. Nucleic Acids Res. 2003, 31, 439–441. [Google Scholar] [CrossRef] [PubMed]
- Ambros, V.; Bartel, B.; Bartel, D.P.; Burge, C.B.; Carrington, J.C.; Chen, X.; Dreyfuss, G.; Eddy, S.R.; Griffiths-Jones, S.; Marshall, M.; et al. A uniform system for microRNA annotation. RNA 2003, 9, 277–279. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.; Kim, M.; Han, J.J.; Yeom, K.H.; Lee, S.; Baek, S.H.; Kim, V.N. MicroRNA genes are transcribed by RNA polymerase II. EMBO J. 2004, 23, 4051–4060. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.; Ahn, C.; Han, J.; Choi, H.; Kim, J.; Yim, J.; Lee, J.; Provost, P.; Rådmark, O.; Kim, S.; et al. The nuclear RNase III Drosha initiates microRNA processing. Nature 2003, 425, 415–419. [Google Scholar] [CrossRef] [PubMed]
- Denli, A.M.; Tops, B.B.J.; Plasterk, R.H.A.; Ketting, R.F.; Hannon, G.J. Processing of primary microRNAs by the Microprocessor complex. Nature 2004, 432, 231–235. [Google Scholar] [CrossRef] [PubMed]
- Han, J.; Lee, Y.; Yeom, K.H.; Nam, J.W.; Heo, I.; Rhee, J.K.; Sohn, S.Y.; Cho, Y.; Zhang, B.T.; Kim, V.N. Molecular basis for the recognition of primary microRNAs by the Drosha-DGCR8 complex. Cell 2006, 125, 887–901. [Google Scholar] [CrossRef] [PubMed]
- Lund, E.; Guttinger, S.; Calado, A.; Dahlberg, J.; Kutay, U. Nuclear export of microRNA precursors. Science 2004, 303, 95–98. [Google Scholar] [CrossRef] [PubMed]
- Chendrimada, T.P.; Gregory, R.I.; Kumaraswamy, E.; Cooch, N.; Nishikura, K.; Shiekhattar, R. TRBP recruits the Dicer complex to Ago2 for microRNA processing and gene silencing. Nature 2005, 436, 740–744. [Google Scholar] [CrossRef] [PubMed]
- Yates, L.A.; Norbury, C.J.; Gilbert, R.J.C. The long and short of microRNA. Cell 2013, 153, 516–519. [Google Scholar] [CrossRef] [PubMed]
- Filipowicz, W.; Bhattacharyya, S.N.; Sonenberg, N. Mechanisms of post-transcriptional regulation by microRNAs: Are the answers in sight? Nat. Rev. Genet. 2008, 9, 102–114. [Google Scholar] [CrossRef] [PubMed]
- Nawrocki, E.P.; Burge, S.W.; Bateman, A.; Daub, J.; Eberhardt, R.Y.; Eddy, S.R.; Floden, E.W.; Gardner, P.P.; Jones, T.A.; Tate, J.; et al. Rfam 12.0: Updates to the RNA families database. Nucleic Acids Res. 2015, 43, D130–D137. [Google Scholar] [CrossRef] [PubMed]
- Kozomara, A.; Griffiths-Jones, S. MiRBase: Annotating high confidence microRNAs using deep sequencing data. Nucleic Acids Res. 2014, 42, 68–73. [Google Scholar] [CrossRef] [PubMed]
- Sethupathy, P.; Corda, B.; Hatzigeorgiou, A.G. TarBase: A comprehensive database of experimentally supported animal microRNA targets TarBase. RNA 2006, 12, 192–197. [Google Scholar] [CrossRef] [PubMed]
- Xiao, F.; Zuo, Z.; Cai, G.; Kang, S.; Gao, X.; Li, T. miRecords: An integrated resource for microRNA-target interactions. Nucleic Acids Res. 2009, 37, 105–110. [Google Scholar] [CrossRef] [PubMed]
- Lewis, B.P.; Shih, I.-H.; Jones-Rhoades, M.W.; Bartel, D.P. Prediction of mammalian MicroRNA targets. Cell 2003, 115, 787–798. [Google Scholar] [CrossRef]
- Consortium, T.E.P. An integrated encyclopedia of DNA elements in the human genome. Nature 2012, 489, 91–100. [Google Scholar]
- Vlachos, I.S.; Paraskevopoulou, M.D.; Karagkouni, D.; Georgakilas, G.; Vergoulis, T.; Kanellos, I.; Anastasopoulos, I.L.; Maniou, S.; Karathanou, K.; Kalfakakou, D.; et al. DIANA-TarBase v7.0: Indexing more than half a million experimentally supported miRNA:mRNA interactions. Nucleic Acids Res. 2015, 43, D153–D159. [Google Scholar] [CrossRef] [PubMed]
- Schnall-Levin, M.; Rissland, O.S.; Johnston, W.K.; Perrimon, N.; Bartel, D.P.; Berger, B. Unusually effective microRNA targeting within repeat-rich coding regions of mammalian mRNAs. Genome Res. 2011, 21, 1395–1403. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cunningham, F.; Amode, M.R.; Barrell, D.; Beal, K.; Billis, K.; Brent, S.; Carvalho-Silva, D.; Clapham, P.; Coates, G.; Fitzgerald, S.; et al. Ensembl 2015. Nucleic Acids Res. 2015, 43, D662–D669. [Google Scholar] [CrossRef] [PubMed]
- O’Leary, N.A.; Wright, M.W.; Brister, J.R.; Ciufo, S.; Haddad, D.; McVeigh, R.; Rajput, B.; Robbertse, B.; Smith-White, B.; Ako-Adjei, D.; et al. Reference sequence (RefSeq) database at NCBI: Current status, taxonomic expansion, and functional annotation. Nucleic Acids Res. 2016, 44, D733–D745. [Google Scholar] [CrossRef] [PubMed]
- Karolchik, D.; Baertsch, R.; Diekhans, M.; Furey, T.S.; Hinrichs, A.; Lu, Y.T.; Roskin, K.M.; Schwartz, M.; Sugnet, C.W.; Thomas, D.J.; et al. The UCSC genome browser database. Nucleic Acids Res. 2003, 31, 51–54. [Google Scholar] [CrossRef] [PubMed]
- Karolchik, D.; Hinrichs, A.S.; Kent, W.J. The UCSC genome browser. Curr. Protoc. Bioinform. 2009, 4, 1–35. [Google Scholar]
- Lewis, B.P.; Burge, C.B.; Bartel, D.P. Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets. Cell 2005, 120, 15–20. [Google Scholar] [CrossRef] [PubMed]
- Friedman, R.C.; Farh, K.K.-H.; Burge, C.B.; Bartel, D.P. Most mammalian mRNAs are conserved targets of microRNAs. Genome Res. 2009, 19, 92–105. [Google Scholar] [CrossRef]
- Krek, A.; Grün, D.; Poy, M.N.; Wolf, R.; Rosenberg, L.; Epstein, E.J.; MacMenamin, P.; da Piedade, I.; Gunsalus, K.C.; Stoffel, M.; et al. Combinatorial microRNA target predictions. Nat. Genet. 2005, 37, 495–500. [Google Scholar] [CrossRef] [PubMed]
- Kertesz, M.; Iovino, N.; Unnerstall, U.; Gaul, U.; Segal, E. The role of site accessibility in microRNA target recognition. Nat. Genet. 2007, 39, 1278–1284. [Google Scholar] [CrossRef] [PubMed]
- Krüger, J.; Rehmsmeier, M. RNAhybrid: MicroRNA target prediction easy, fast and flexible. Nucleic Acids Res. 2006, 34, 451–454. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y. miRU: An automated plant miRNA target prediction server. Nucleic Acids Res. 2005, 33, 701–704. [Google Scholar] [CrossRef] [PubMed]
- Doench, J.G.; Sharp, P.A. Specificity of microRNA target selection in translational repression. Genes Dev. 2004, 6, 504–511. [Google Scholar] [CrossRef] [PubMed]
- Mathews, D.H.; Sabina, J.; Zuker, M.; Turner, D.H. Expanded sequence dependence of thermodynamic parameters improves prediction of RNA secondary structure. J. Mol. Biol. 1999, 288, 911–940. [Google Scholar] [CrossRef] [PubMed]
- Hofacker, I.L.; Fontana, W.; Stadler, P.F.; Bonhoeffer, L.S.; Tacker, M.; Schuster, P. Fast folding and comparison of RNA secondary structures. Monatshefte Chem. 1994, 125, 167–188. [Google Scholar] [CrossRef]
- Lorenz, R.; Bernhart, S.; Siederdissen, C.; Tefer, H.; Flamm, C.; Stadler, P.F.; Hofacker, I.L. ViennaRNA package 2.0. Algorithms Mol. Biol. 2011, 6, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Brennecke, J.; Stark, A.; Russell, R.B.; Cohen, S.M. Principles of microRNA-target recognition. PLoS Biol. 2005, 3, 0404–0418. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grimson, A.; Farh, K.K.-H.; Johnston, W.K.; Garrett-Engele, P.; Lim, L.P.; Bartel, D.P. MicroRNA targeting specificity in mammals: Determinants beyond seed pairing. Mol. Cell 2007, 27, 91–105. [Google Scholar] [CrossRef] [PubMed]
- Betel, D.; Koppal, A.; Agius, P.; Sander, C.; Leslie, C. Comprehensive modeling of microRNA targets predicts functional non-conserved and non-canonical sites. Genome Biol. 2010, 11, R90. [Google Scholar] [CrossRef] [PubMed]
- Nam, J.-W.; Rissland, O.S.; Koppstein, D.; Abreu-Goodger, C.; Jan, C.H.; Agarwal, V.; Yildirim, M.A.; Rodriguez, A.; Bartel, D.P. Global analyses of the effect of different cellular contexts on MicroRNA targeting. Mol. Cell 2014, 53, 1031–1043. [Google Scholar] [CrossRef] [PubMed]
- Reczko, M.; Maragkakis, M.; Alexiou, P.; Grosse, I.; Hatzigeorgiou, A.G. Functional microRNA targets in protein coding sequences. Bioinformatics 2012, 28, 771–776. [Google Scholar] [CrossRef] [PubMed]
- Gaidatzis, D.; van Nimwegen, E.; Hausser, J.; Zavolan, M. Inference of miRNA targets using evolutionary conservation and pathway analysis. BMC Bioinform. 2007, 8, 69. [Google Scholar] [CrossRef] [PubMed]
- Miranda, K.C.; Huynh, T.; Tay, Y.; Ang, Y.S.; Tam, W.L.; Thomson, A.M.; Lim, B.; Rigoutsos, I. A pattern-based method for the identification of microRNA binding sites and their corresponding heteroduplexes. Cell 2006, 126, 1203–1217. [Google Scholar] [CrossRef] [PubMed]
- Ørom, U.A.; Nielsen, F.C.; Lund, A.H. MicroRNA-10a binds the 5′ UTR of ribosomal protein mRNAs and enhances their translation. Mol. Cell 2008, 30, 460–471. [Google Scholar] [CrossRef] [PubMed]
- Gu, W.; Xu, Y.; Xie, X.; Wang, T.; Ko, J.; Zhou, T. microRNA-mediated gene regulation The role of RNA structure at 5′ untranslated region in microRNA-mediated gene regulation. RNA 2014, 20, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Mandke, P.; Wyatt, N.; Fraser, J.; Bates, B.; Berberich, S.J.; Markey, M.P. MicroRNA-34a modulates MDM4 expression via a target site in the open reading frame. PLoS ONE 2012, 7, e42034. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Xiao, Y.F.; Zhang, J.W.; Xie, R.; Hu, C.J.; Tang, B.; Wang, S.M.; Wu, Y.Y.; Hao, N.B.; Yang, S.M. MiR-1182 attenuates gastric cancer proliferation and metastasis by targeting the open reading frame of hTERT. Cancer Lett. 2015, 360, 151–159. [Google Scholar] [CrossRef] [PubMed]
- Guo, Z.-W.; Xie, C.; Yang, J.-R.; Li, J.-H.; Yang, J.-H.; Zheng, L. MtiBase: A database for decoding microRNA target sites located within CDS and 5′UTR regions from CLIP-Seq and expression profile datasets. Database 2015, 2015, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Hsu, J.B.-K.; Chiu, C.-M.; Hsu, S.-D.; Huang, W.-Y.; Chien, C.-H.; Lee, T.-Y.; Huang, H.D. miRTar: An integrated system for identifying miRNA-target interactions in human. BMC Bioinform. 2011, 12, 300. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.; San Lucas, A.; Wang, Z.; Liu, Y. Identifying microRNA targets in different gene regions. BMC Bioinform. 2014, 15 (Suppl. 7), S4. [Google Scholar] [CrossRef] [PubMed]
- Hafner, M.; Landthaler, M.; Burger, L.; Khorshid, M.; Berninger, P.; Rothballer, A.; Ascano, M., Jr.; Jungkamp, A.C.; Munschauer, M.; Ulrich, A.; et al. Transcritpome wide identification of RNA binding protein and microRNA target sites by PAR-CLIP. Cell 2010, 141, 129–141. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Li, Y.; Toth, J.I.; Petroski, M.D.; Zhang, Z.; Zhao, J.C. N6-methyladenosine modification destabilizes developmental regulators in embryonic stem cells. Nat. Cell Biol. 2014, 16, 191–198. [Google Scholar] [CrossRef] [PubMed]
- Salmena, L.; Poliseno, L.; Tay, Y.; Kats, L.; Pandolfi, P.P. A ceRNA hypothesis: The rosetta stone of a hidden RNA language? Cell 2011, 146, 353–358. [Google Scholar] [CrossRef] [PubMed]
- Jeggari, A.; Marks, D.S.; Larsson, E. miRcode: A map of putative microRNA target sites in the long non-coding transcriptome. Bioinformatics 2012, 28, 2062–2063. [Google Scholar] [CrossRef] [PubMed]
- Garcia, D.M.; Baek, D.; Shin, C.; Bell, G.W.; Grimson, A.; Bartel, D.P. Weak seed-pairing stability and high target-site abundance decreases the proficiency of Isy-6 and other miRNA’s. Nat. Struct. Mol. Biol. 2011, 18, 1139–1146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Enright, A.J.; John, B.; Gaul, U.; Tuschl, T.; Sander, C.; Marks, D.S. MicroRNA targets in Drosophila. Genome Biol. 2003, 5, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.C.; Morris, Q.D.; Frey, B.J. Bayesian inference of MicroRNA targets from sequence and expression data. J. Comput. Biol. 2007, 14, 550–563. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.C.; Frey, B.J.; Morris, Q.D. Comparing sequence and expression for predicting microRNA targets using GenMiR3. Pac. Symp. Biocomput. 2008, 63, 52–63. [Google Scholar]
- Joung, J.G.; Hwang, K.B.; Nam, J.W.; Kim, S.J.; Zhang, B.T. Discovery of microRNA-mRNA modules via population-based probabilistic learning. Bioinformatics 2007, 23, 1141–1147. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Li, Q.; Liu, J.; Zhou, X.J. A novel computational framework for simultaneous integration of multiple types of genomic data to identify microrna-gene regulatory modules. Bioinformatics 2011, 27, 401–409. [Google Scholar] [CrossRef] [PubMed]
- Yue, D.; Liu, H.; Huang, Y. Survey of computational algorithms for MicroRNA target prediction. Curr. Genom. 2009, 10, 478–492. [Google Scholar] [CrossRef] [PubMed]
- Witkos, T.M.; Koscianska, E.; Krzyzosiak, W.J. Practical aspects of microRNA target prediction. Curr. Mol. Med. 2011, 11, 93–109. [Google Scholar] [CrossRef] [PubMed]
- Peterson, S.M.; Thompson, J.A.; Ufkin, M.L.; Sathyanarayana, P.; Liaw, L.; Congdon, C.B. Common features of microRNA target prediction tools. Front. Genet. 2014, 5, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, V.; Bell, G.W.; Nam, J.W.; Bartel, D.P. Predicting effective microRNA target sites in mammalian mRNAs. eLife 2015, 4, 1–38. [Google Scholar] [CrossRef] [PubMed]
- John, B.; Enright, A.J.; Aravin, A.; Tuschl, T.; Sander, C.; Marks, D.S. Human microRNA targets. PLoS Biol. 2004, 2, 1862–1879. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Betel, D.; Wilson, M.; Gabow, A.; Marks, D.S.; Sander, C. The microRNA.org resource: Targets and expression. Nucleic Acids Res. 2008, 36, 149–153. [Google Scholar] [CrossRef] [PubMed]
- Smith, T.; Waterman, M. Identification of common molecular subsequences. J. Mol. Biol. 1981, 147, 195–197. [Google Scholar] [CrossRef]
- Siepel, A.; Bejerano, G.; Pedersen, J.S.; Hinrichs, A.S.; Hou, M.; Rosenbloom, K.; Clawson, H.; Spieth, J.; Hillier, L.W.; Richards, S.; et al. Evolutionarily conserved elements in vertebrate, insect, worm, and yeast genomes. Genome Res. 2005, 15, 1034–1050. [Google Scholar] [CrossRef] [PubMed]
- Kiriakidou, M.; Nelson, P.T.; Kouranov, A.; Fitziev, P.; Bouyioukos, C.; Mourelatos, Z.; Hatzigeorgiou, A. A combined computational-experimental approach predicts human microRNA targets. Genes Dev. 2004, 18, 1165–1178. [Google Scholar] [CrossRef] [PubMed]
- Pruitt, K.D.; Tatusova, T.; Maglott, D.R. NCBI Reference Sequence project: Update and current status. Nucleic Acids Res. 2003, 31, 34–37. [Google Scholar] [CrossRef] [PubMed]
- Maragkakis, M.; Reczko, M.; Simossis, V.A.; Alexiou, P.; Papadopoulos, G.L.; Dalamagas, T.; Giannopoulos, G.; Goumas, G.; Koukis, E.; Kourtis, K.; et al. DIANA-microT web server: Elucidating microRNA functions through target prediction. Nucleic Acids Res. 2009, 37, 273–276. [Google Scholar] [CrossRef] [PubMed]
- Maragkakis, M.; Vergoulis, T.; Alexiou, P.; Reczko, M.; Plomaritou, K.; Gousis, M.; Kourtis, K.; Koziris, N.; Dalamagas, T.; Hatzigeorgiou, A.G. DIANA-microT Web server upgrade supports Fly and Worm miRNA target prediction and bibliographic miRNA to disease association. Nucleic Acids Res. 2011, 39, 145–148. [Google Scholar] [CrossRef] [PubMed]
- Paraskevopoulou, M.D.; Georgakilas, G.; Kostoulas, N.; Vlachos, I.S.; Vergoulis, T.; Reczko, M.; Filippidis, C.; Dalamagas, T.; Hatzigeorgiou, A.G.; et al. DIANA-microT web server v5.0: Service integration into miRNA functional analysis workflows. Nucleic Acids Res. 2013, 41, 169–173. [Google Scholar] [CrossRef] [PubMed]
- Eden, E.; Navon, R.; Steinfeld, I.; Lipson, D.; Yakhini, Z. GOrilla: A tool for discovery and visualization of enriched GO terms in ranked gene lists. BMC Bioinform. 2009, 10, 48. [Google Scholar] [CrossRef] [PubMed]
- Steinfeld, I.; Navon, R.; Ach, R.; Yakhini, Z. MiRNA target enrichment analysis reveals directly active miRNAs in health and disease. Nucleic Acids Res. 2013, 41, e45. [Google Scholar] [CrossRef] [PubMed]
- Shirdel, E.A.; Xie, W.; Mak, T.W.; Jurisica, I. NAViGaTing the micronome—Using multiple microRNA prediction databases to identify signalling pathway-associated microRNAs. PLoS ONE 2011, 6, e17429. [Google Scholar] [CrossRef] [PubMed]
- Mullany, L.E.; Wolff, R.K.; Slattery, M.L. Effectiveness and usability of bioinformatics tools to analyze pathways associated with miRNA expression. Cancer Inform. 2015, 14, 121–130. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
| Tools | Parameter | Value Range | Meaning |
|---|---|---|---|
| TargetScan | Site type | 8mer > 7mer > 7mer-A1 > 6mer | The matching sites in the seed region (nucleotides 2 to 8 from 5′ of miRNA that have perfect WC pairing with the 3′ UTR), from the strictest to the least strict. |
| Context++ score | From 1 to −1 | The sum of the contribution of 14 features for each of the four site types, the more negative the score, the greater the repression. | |
| Context++ score percentile | From i to 100 − i; | Percentage of sites for the miRNA with a less favorable context++score. | |
| Weighted context++ score | From 1 to −1 | The scores with a lower negative value indicate a greater prediction of repression. | |
| Cumulative weighted context++ score | C(i–1) + (1 − 2CSi)(AIRi-C(i–1)) | This score estimates the total repression expected from multiple sites of the same miRNA, for each mRNA target predicted. | |
| Branch-length score | 8mer: 1.8; 7mer-m8: 2.8; 7mer-A1: 3.6; 6mer: NA | This score is the sum of phylogenetic branch lengths between species that contain a matching site. | |
| PCT score | Between 0 and 1 | The higher the score, the greater the conservation and the greater mRNA destabilization expected. | |
| Aggregate PCT | Value = 1 − ((1 – PCT) site1 × (1 − PCT) site2 | For each miRNA, this parameter includes the conserved 3′ UTR targets with multiple sites that were missed in the human 3′ UTR annotation, but were present in the mouse annotations. | |
| Conserved sites | ≥0 | Number of conserved sites identified. | |
| miRanda | mirSVR score | <0 | This score is an estimate of the miRNA effect on the mRNA expression level. The more negative the score, the greater effect. |
| PhastCons score | From 0 to 1 | This measures the conservation of nucleotide positions across multiple vertebrates. | |
| DIANA Tools | miTG score | From 0 to 1 | This is a general score for the predicted interaction, the closer to 1, the greater the confidence. |
| Also Predicted | red, blue and green | This compares with other tools; miRanda in red, TargetScan in blue and TarBase in green. | |
| Region | UTR3, CDS | Region of the mRNA where the interaction occurs. | |
| Binding Type | 6mer; 7mer; 8mer; 9mer; miRNA bugle | The matching sites between the miRNA and the mRNA. | |
| Score | From 0 to 1 | It is the site contribution score in the miTG score. | |
| Conservation | ≥0 | Number of species in which the predicted interaction is conserved. | |
| Signal-to-noise ratio (SNR) | >0 | This score is a measure of the “signal to noise” ratio, which enables the identification of the miTG score of each interaction without background noise. | |
| Precision | From 0 to 1 | This score is an indicator of the false-positive rate in a miTG interaction. |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Riffo-Campos, Á.L.; Riquelme, I.; Brebi-Mieville, P. Tools for Sequence-Based miRNA Target Prediction: What to Choose? Int. J. Mol. Sci. 2016, 17, 1987. https://doi.org/10.3390/ijms17121987
Riffo-Campos ÁL, Riquelme I, Brebi-Mieville P. Tools for Sequence-Based miRNA Target Prediction: What to Choose? International Journal of Molecular Sciences. 2016; 17(12):1987. https://doi.org/10.3390/ijms17121987
Chicago/Turabian StyleRiffo-Campos, Ángela L., Ismael Riquelme, and Priscilla Brebi-Mieville. 2016. "Tools for Sequence-Based miRNA Target Prediction: What to Choose?" International Journal of Molecular Sciences 17, no. 12: 1987. https://doi.org/10.3390/ijms17121987