
Prediction of Protein Structural Class Based on Gapped-Dipeptides and a Recursive Feature Selection Approach


Abstract
:1. Introduction
2. Results and Discussion
2.1. Parameter Selection

{kind=link}
| Dataset | Accuracy (%) | Matthews Correlation Coefficient (MCC) | |||||||
|---|---|---|---|---|---|---|---|---|---|
| All-α | All-β | α/β | α + β | Overall | All-α | All-β | α/β | α + β | |
| Z277 | 97.1 | 98.4 | 97.5 | 96.9 | 97.5 | 0.96 | 0.98 | 0.97 | 0.96 |
| Z498 | 98.1 | 100 | 98.5 | 97.7 | 98.6 | 0.96 | 1 | 0.98 | 0.98 |
| 1189 | 94.2 | 93.2 | 92.5 | 83.0 | 90.9 | 0.89 | 0.91 | 0.89 | 0.82 |
| 25PDB | 94.8 | 92.3 | 87.0 | 86.4 | 90.3 | 0.88 | 0.89 | 0.87 | 0.84 |
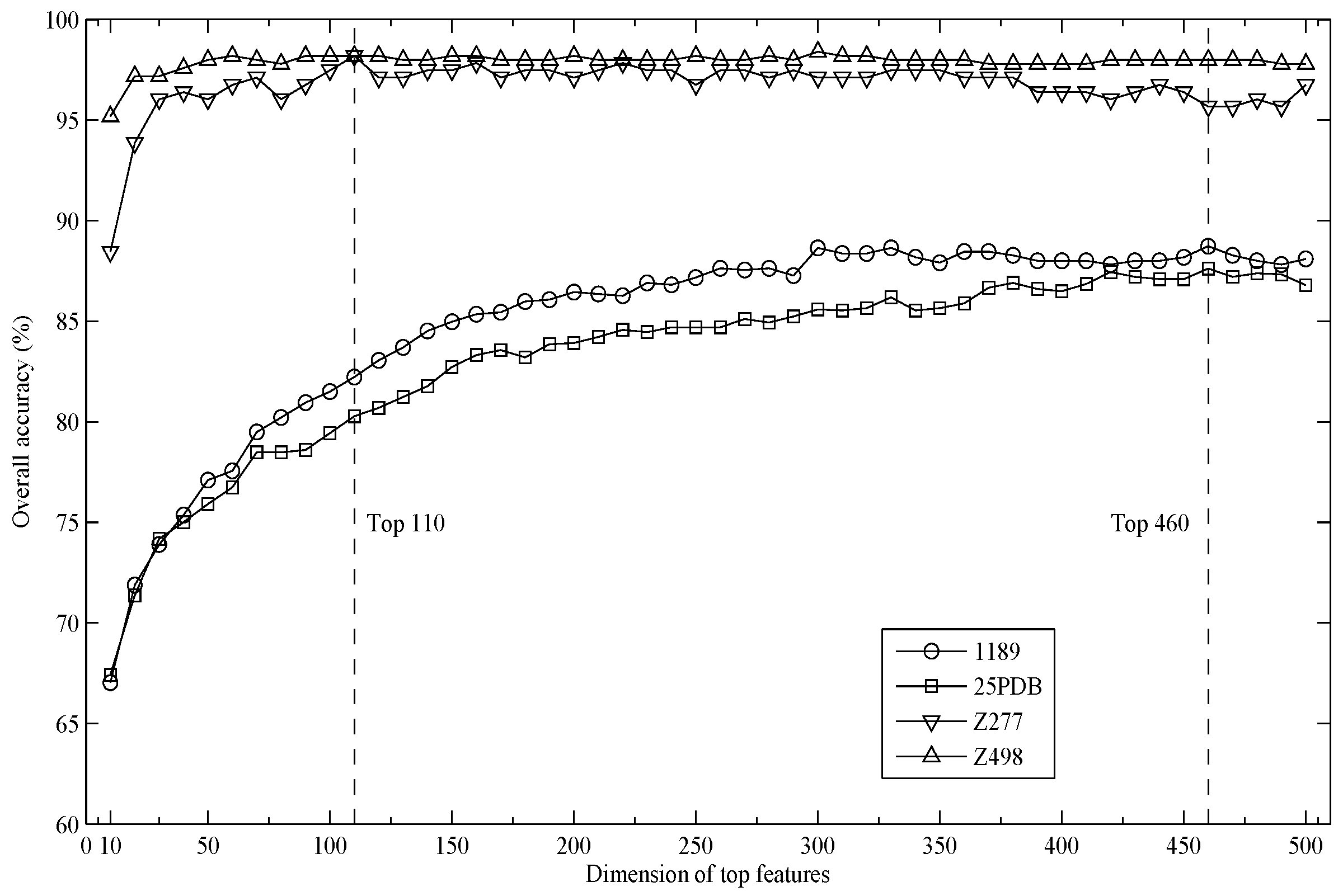
2.2. Performance Comparison with Existing Methods
| Method | Prediction Accuracy (%) | ||||
|---|---|---|---|---|---|
| All-α | All-β | α/β | α + β | Overall | |
| Neural network [7] | 68.6 | 85.2 | 86.4 | 56.9 | 74.7 |
| Component coupled [23] | 84.3 | 82.0 | 81.5 | 67.7 | 79.1 |
| LogitBoost [19] | 81.4 | 88.5 | 92.6 | 72.3 | 84.1 |
| IGA-SVM [10] | 84.3 | 88.5 | 92.6 | 70.7 | 84.5 |
| CWT-PCA-SVM [27] | 85.7 | 90.2 | 87.7 | 80.1 | 85.9 |
| Markov-SVM [42] | 90.0 | 85.2 | 86.4 | 81.5 | 85.9 |
| SVM fusion [21] | 85.7 | 90.2 | 93.8 | 80.0 | 87.7 |
| AAC-PSSM-AC [30] | 88.6 | 95.1 | 97.5 | 81.5 | 91.0 |
| Our method | 97.1 | 98.4 | 97.5 | 96.9 | 97.5 |
| Method | Prediction Accuracy (%) | ||||
|---|---|---|---|---|---|
| All-α | All-β | α/β | α + β | Overall | |
| Neural network [7] | 86.0 | 96.0 | 88.2 | 86.0 | 89.2 |
| Component-coupled [23] | 93.5 | 88.9 | 90.4 | 84.5 | 89.2 |
| SVM fusion [21] | 99.1 | 96.0 | 80.9 | 91.5 | 91.4 |
| Markov-SVM [42] | 91.6 | 94.4 | 96.3 | 91.5 | 93.6 |
| IGA-SVM [10] | 96.3 | 93.6 | 97.8 | 89.2 | 94.2 |
| LogitBoost [19] | 92.6 | 96.0 | 97.1 | 93.0 | 94.8 |
| CWT-PCA-SVM [27] | 94.4 | 96.8 | 97.0 | 92.3 | 95.2 |
| AAC-PSSM-AC [30] | 94.4 | 96.8 | 97.8 | 93.8 | 95.8 |
| Our method | 98.1 | 100 | 98.5 | 97.7 | 98.6 |
| Method | Prediction Accuracy (%) | ||||
|---|---|---|---|---|---|
| All-α | All-β | α/β | α + β | Overall | |
| AADP-PSSM [31] | 69.1 | 83.7 | 85.6 | 35.7 | 70.7 |
| AAC-PSSM-AC [30] | 80.7 | 86.4 | 81.4 | 45.2 | 74.6 |
| Comb_11,10,6 1 [22] | 80.2 | 83.6 | 85.4 | 44.6 | 74.8 |
| SCPRED [9] | 89.1 | 86.7 | 89.6 | 53.8 | 80.6 |
| LCC-PSSM [29] | 89.2 | 88.8 | 85.6 | 58.5 | 81.2 |
| RKS-PPSC [43] | 89.2 | 86.7 | 82.6 | 65.6 | 81.3 |
| MODAS [33] | 92.3 | 87.1 | 87.9 | 65.4 | 83.5 |
| PSSM-SPINE-S [34] | 98.2 | 91.5 | 83.8 | 72.2 | 86.3 |
| Our method | 94.2 | 93.2 | 92.5 | 83.0 | 90.9 |
| Method | Prediction Accuracy (%) | ||||
|---|---|---|---|---|---|
| All-α | All-β | α/β | α + β | Overall | |
| AADP-PSSM [31] | 83.3 | 78.1 | 76.3 | 54.4 | 72.9 |
| AAC-PSSM-AC [30] | 85.3 | 81.7 | 73.7 | 55.3 | 74.1 |
| Comb_11,10,6 1 [22] | 86.1 | 80.8 | 80.6 | 60.1 | 76.7 |
| LCC-PSSM [29] | 91.7 | 80.8 | 79.8 | 64.0 | 79.0 |
| SCPRED [9] | 92.6 | 80.1 | 74.0 | 71.0 | 79.7 |
| MODAS [33] | 92.3 | 83.7 | 81.2 | 68.3 | 81.4 |
| RKS-PPSC [43] | 92.8 | 83.3 | 85.8 | 70.1 | 82.9 |
| PSSM-SPINE-S [34] | 96.8 | 93.7 | 90.1 | 87.0 | 92.2 |
| Our method | 94.8 | 92.3 | 87.0 | 86.4 | 90.3 |
3. Materials and Methods
3.1. Datasets
| Dataset | All-α | All-β | α/β | α + β | Total |
|---|---|---|---|---|---|
| Z277 | 70 | 61 | 81 | 65 | 277 |
| Z498 | 107 | 126 | 136 | 129 | 498 |
| 1189 | 223 | 294 | 334 | 241 | 1092 |
| 25PDB | 443 | 443 | 346 | 441 | 1673 |
3.2. Protein Sequence Representation
3.3. Recursive Feature Selection
3.4. Support Vector Machine
3.5. Cross-Validation and Performance Evaluation
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Leung, C.H.; Chan, D.S.; He, H.Z.; Cheng, Z.; Yang, H.; Ma, D.L. Luminescent detection of DNA-binding proteins. Nucleic Acids Res. 2012, 40, 941–955. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.; Brewer, M.K.; Mashburn, C.B.; Lou, D.; Bondada, V.; Graham, B.; Geddes, J.W. Calpain 5 is highly expressed in the central nervous system (CNS), carries dual nuclear localization signals, and is associated with nuclear promyelocytic leukemia protein bodies. J. Biol. Chem. 2014, 289, 19383–19394. [Google Scholar] [CrossRef] [PubMed]
- Leung, K.H.; He, B.; Yang, C.; Leung, C.H.; Wang, H.M.; Ma, D.L. Development of an aptamer-based sensing platform for metal ions, proteins, and small molecules through terminal deoxynucleotidyl transferase induced G-quadruplex formation. ACS Appl. Mater. Interfaces 2015, 7, 24046–24052. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.; Gao, W.; Tian, Z.; Yang, C.; Lu, L.; Mergny, J.-L.; Leung, C.-H.; Ma, D.-L. Luminescence switch-on detection of protein tyrosine kinase-7 using a G-quadruplex-selective probe. Chem. Sci. 2015, 6, 4284–4290. [Google Scholar] [CrossRef]
- Levitt, M.; Chothia, C. Structural patterns in globular proteins. Nature 1976, 261, 552–558. [Google Scholar] [CrossRef] [PubMed]
- Chou, K.C. Progress in protein structural class prediction and its impact to bioinformatics and proteomics. Curr. Protein Pept. Sci. 2005, 6, 423–436. [Google Scholar] [CrossRef] [PubMed]
- Cai, Y.D.; Zhou, G.P. Prediction of protein structural classes by neural network. Biochimie 2000, 82, 783–785. [Google Scholar] [CrossRef]
- Cai, Y.D.; Liu, X.J.; Xu, X.; Zhou, G.P. Support vector machines for predicting protein structural class. BMC Bioinformatics 2001, 2, 3. [Google Scholar] [CrossRef] [PubMed]
- Kurgan, L.; Cios, K.; Chen, K. Scpred: Accurate prediction of protein structural class for sequences of twilight-zone similarity with predicting sequences. BMC Bioinform. 2008, 9, 226. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.C.; Zhou, X.B.; Lin, Y.R.; Zou, X.Y. Prediction of protein structure class by coupling improved genetic algorithm and support vector machine. Amino Acids 2008, 35, 581–590. [Google Scholar] [CrossRef] [PubMed]
- Zheng, X.; Li, C.; Wang, J. An information-theoretic approach to the prediction of protein structural class. J. Comput. Chem. 2010, 31, 1201–1206. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.L.; Ding, Y.S.; Chou, K.C. Prediction protein structural classes with pseudo-amino acid composition: Approximate entropy and hydrophobicity pattern. J. Theor. Biol. 2008, 250, 186–193. [Google Scholar] [CrossRef] [PubMed]
- Shen, H.B.; Yang, J.; Liu, X.J.; Chou, K.C. Using supervised fuzzy clustering to predict protein structural classes. Biochem. Biophys. Res. Commun. 2005, 334, 577–581. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.X.; Yuan, Z. How good is prediction of protein structural class by the component-coupled method? Proteins 2000, 38, 165–175. [Google Scholar] [CrossRef]
- Kurgan, L.A.; Homaeian, L. Prediction of structural classes for protein sequences and domains—Impact of prediction algorithms, sequence representation and homology, and test procedures on accuracy. Pattern Recogn. 2006, 39, 2323–2343. [Google Scholar] [CrossRef]
- Kurgan, L.; Chen, K. Prediction of protein structural class for the twilight zone sequences. Biochem. Biophys. Res. Commun. 2007, 357, 453–460. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y.F.; Liu, S.; Zhang, L.D.; Qin, J.; Wang, J.; Tang, K.X. Prediction of protein structural class with rough sets. BMC Bioinform. 2006, 7, 20. [Google Scholar] [CrossRef] [PubMed]
- Cai, Y.D.; Feng, K.Y.; Lu, W.C.; Chou, K.C. Using logitboost classifier to predict protein structural classes. J. Theor. Biol. 2006, 238, 172–176. [Google Scholar] [CrossRef] [PubMed]
- Feng, K.Y.; Cai, Y.D.; Chou, K.C. Boosting classifier for predicting protein domain structural class. Biochem. Biophys. Res. Commun. 2005, 334, 213–217. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Lu, L.; Feng, K.; Li, W.; Song, J.; Zheng, L.; Yuan, Y.; Zeng, Z.; Lu, W.; Cai, Y. Multiple classifier integration for the prediction of protein structural classes. J. Comput. Chem. 2009, 30, 2248–2254. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Zhou, X.; Tian, Y.; Zou, X.; Cai, P. Predicting protein structural class with pseudo-amino acid composition and support vector machine fusion network. Anal. Biochem. 2006, 357, 116–121. [Google Scholar] [CrossRef] [PubMed]
- Dehzangi, A.; Paliwal, K.; Sharma, A.; Dehzangi, O.; Sattar, A. A combination of feature extraction methods with an ensemble of different classifiers for protein structural class prediction problem. IEEE/ACM Trans. Comput. Biol. Bioinform. 2013, 10, 564–575. [Google Scholar] [CrossRef] [PubMed]
- Zhou, G.P. An intriguing controversy over protein structural class prediction. J. Protein Chem. 1998, 17, 729–738. [Google Scholar] [CrossRef] [PubMed]
- Chou, K.C. A key driving force in determination of protein structural classes. Biochem. Biophys. Res. Commun. 1999, 264, 216–224. [Google Scholar] [CrossRef] [PubMed]
- Chou, K.C. Prediction of protein cellular attributes using pseudo-amino acid composition. Proteins 2001, 43, 246–255. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.L.; Ding, Y.S. Using pseudo amino acid composition and binary-tree support vector machines to predict protein structural classes. Amino Acids 2007, 33, 623–629. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.C.; Zhou, X.B.; Dai, Z.; Zou, X.Y. Prediction of protein structural classes by Chou’s pseudo amino acid composition: Approached using continuous wavelet transform and principal component analysis. Amino Acids 2009, 37, 415–425. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.; Kurgan, L.A.; Ruan, J.S. Prediction of protein structural class using novel evolutionary collocation-based sequence representation. J. Comput. Chem. 2008, 29, 1596–1604. [Google Scholar] [CrossRef] [PubMed]
- Ding, S.; Yan, S.; Qi, S.; Li, Y.; Yao, Y. A protein structural classes prediction method based on PSI-BLAST profile. J. Theor. Biol. 2014, 353, 19–23. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Geng, X.; Zheng, X.; Li, R.; Wang, J. Accurate prediction of protein structural class using auto covariance transformation of PSI-BLAST profiles. Amino Acids 2012, 42, 2243–2249. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Zheng, X.; Wang, J. Prediction of protein structural class for low-similarity sequences using support vector machine and PSI-BLAST profile. Biochimie 2010, 92, 1330–1334. [Google Scholar] [CrossRef] [PubMed]
- Kurgan, L.A.; Zhang, T.; Zhang, H.; Shen, S.Y.; Ruan, J.S. Secondary structure-based assignment of the protein structural classes. Amino Acids 2008, 35, 551–564. [Google Scholar] [CrossRef] [PubMed]
- Mizianty, M.J.; Kurgan, L. Modular prediction of protein structural classes from sequences of twilight-zone identity with predicting sequences. BMC Bioinform. 2009, 10, 414. [Google Scholar] [CrossRef] [PubMed]
- Dehzangi, A.; Paliwal, K.; Lyons, J.; Sharma, A.; Sattar, A. Proposing a highly accurate protein structural class predictor using segmentation-based features. BMC Genom. 2014, 15, S2. [Google Scholar] [CrossRef] [PubMed]
- Nieto, J.J.; Torres, A.; Georgiou, D.N.; Karakasidis, T.E. Fuzzy polynucleotide spaces and metrics. Bull. Math. Biol. 2006, 68, 703–725. [Google Scholar] [CrossRef] [PubMed]
- Georgiou, D.N.; Karakasidis, T.E.; Nieto, J.J.; Torres, A. Use of fuzzy clustering technique and matrices to classify amino acids and its impact to Chou’s pseudo amino acid composition. J. Theor. Biol. 2009, 257, 17–26. [Google Scholar] [CrossRef] [PubMed]
- Georgiou, D.N.; Karakasidis, T.E.; Nieto, J.J.; Torres, A. A study of entropy/clarity of genetic sequences using metric spaces and fuzzy sets. J. Theor. Biol. 2010, 267, 95–105. [Google Scholar] [CrossRef] [PubMed]
- Du, P.; Gu, S.; Jiao, Y. Pseaac-general: Fast building various modes of general form of Chou’s pseudo-amino acid composition for large-scale protein datasets. Int. J. Mol. Sci. 2014, 15, 3495–3506. [Google Scholar] [CrossRef] [PubMed]
- Qiu, W.R.; Xiao, X.; Chou, K.C. Irspot-tncpseaac: Identify recombination spots with trinucleotide composition and pseudo amino acid components. Int. J. Mol. Sci. 2014, 15, 1746–1766. [Google Scholar] [CrossRef] [PubMed]
- Huang, Q.; You, Z.; Zhang, X.; Zhou, Y. Prediction of protein-protein interactions with clustered amino acids and weighted sparse representation. Int. J. Mol. Sci. 2015, 16, 10855–10869. [Google Scholar] [CrossRef] [PubMed]
- Jones, D.T. Protein secondary structure prediction based on position-specific scoring matrices. J. Mol. Biol. 1999, 292, 195–202. [Google Scholar] [CrossRef] [PubMed]
- Qin, Y.F.; Wang, C.H.; Yu, X.Q.; Zhu, J.; Liu, T.G.; Zheng, X.Q. Predicting protein structural class by incorporating patterns of over-represented k-mers into the general form of Chou’s PseAAC. Protein Pept. Lett. 2012, 19, 388–397. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.Y.; Peng, Z.L.; Chen, X. Prediction of protein structural classes for low-homology sequences based on predicted secondary structure. BMC Bioinform. 2010, 11, S9. [Google Scholar] [CrossRef] [PubMed]
- Faraggi, E.; Zhang, T.; Yang, Y.; Kurgan, L.; Zhou, Y. Spine x: Improving protein secondary structure prediction by multistep learning coupled with prediction of solvent accessible surface area and backbone torsion angles. J. Comput. Chem. 2012, 33, 259–267. [Google Scholar] [CrossRef] [PubMed]
- Altschul, S.F.; Madden, T.L.; Schaffer, A.A.; Zhang, J.; Zhang, Z.; Miller, W.; Lipman, D.J. Gapped blast and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 1997, 25, 3389–3402. [Google Scholar] [CrossRef] [PubMed]
- Guyon, I.; Weston, J.; Barnhill, S.; Vapnik, V. Gene selection for cancer classification using support vector machines. Mach. Learn. 2002, 46, 389–422. [Google Scholar] [CrossRef]
- Vapnik, V. Statistical Learning Theory; Wiley: New York, NY, USA, 1998. [Google Scholar]
- Chang, C.C.; Lin, C.J. Libsvm: A library for support vector machines. ACM Trans. Intell. Syst. Technol. 2001. [Google Scholar] [CrossRef]
- Chou, K.C.; Zhang, C.T. Prediction of protein structural classes. Crit. Rev. Biochem. Mol. Biol. 1995, 30, 275–349. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, T.; Qin, Y.; Wang, Y.; Wang, C. Prediction of Protein Structural Class Based on Gapped-Dipeptides and a Recursive Feature Selection Approach. Int. J. Mol. Sci. 2016, 17, 15. https://doi.org/10.3390/ijms17010015
Liu T, Qin Y, Wang Y, Wang C. Prediction of Protein Structural Class Based on Gapped-Dipeptides and a Recursive Feature Selection Approach. International Journal of Molecular Sciences. 2016; 17(1):15. https://doi.org/10.3390/ijms17010015
Chicago/Turabian StyleLiu, Taigang, Yufang Qin, Yongjie Wang, and Chunhua Wang. 2016. "Prediction of Protein Structural Class Based on Gapped-Dipeptides and a Recursive Feature Selection Approach" International Journal of Molecular Sciences 17, no. 1: 15. https://doi.org/10.3390/ijms17010015