
Comparative Analysis of the Brassica napus Root and Leaf Transcript Profiling in Response to Drought Stress

Abstract
:1. Introduction
2. Results and Discussion
2.1. Results
2.1.1. Transcriptome Sequencing of Drought Stress

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Items | CKR | DSR | CKL | DSL |
|---|---|---|---|---|
| Total Raw Reads | 11,957,048 | 12,476,345 | 12,338,528 | 12,054,260 |
| Reads Containing N | 22 | 29 | 28 | 22 |
| Only Adaptors | 115,945 | 81,690 | 84,941 | 68,814 |
| Low Quality Reads | 77,646 | 85,765 | 84,565 | 79,619 |
| Clean Reads | 11,763,435 | 12,308,861 | 12,168,994 | 11,905,805 |
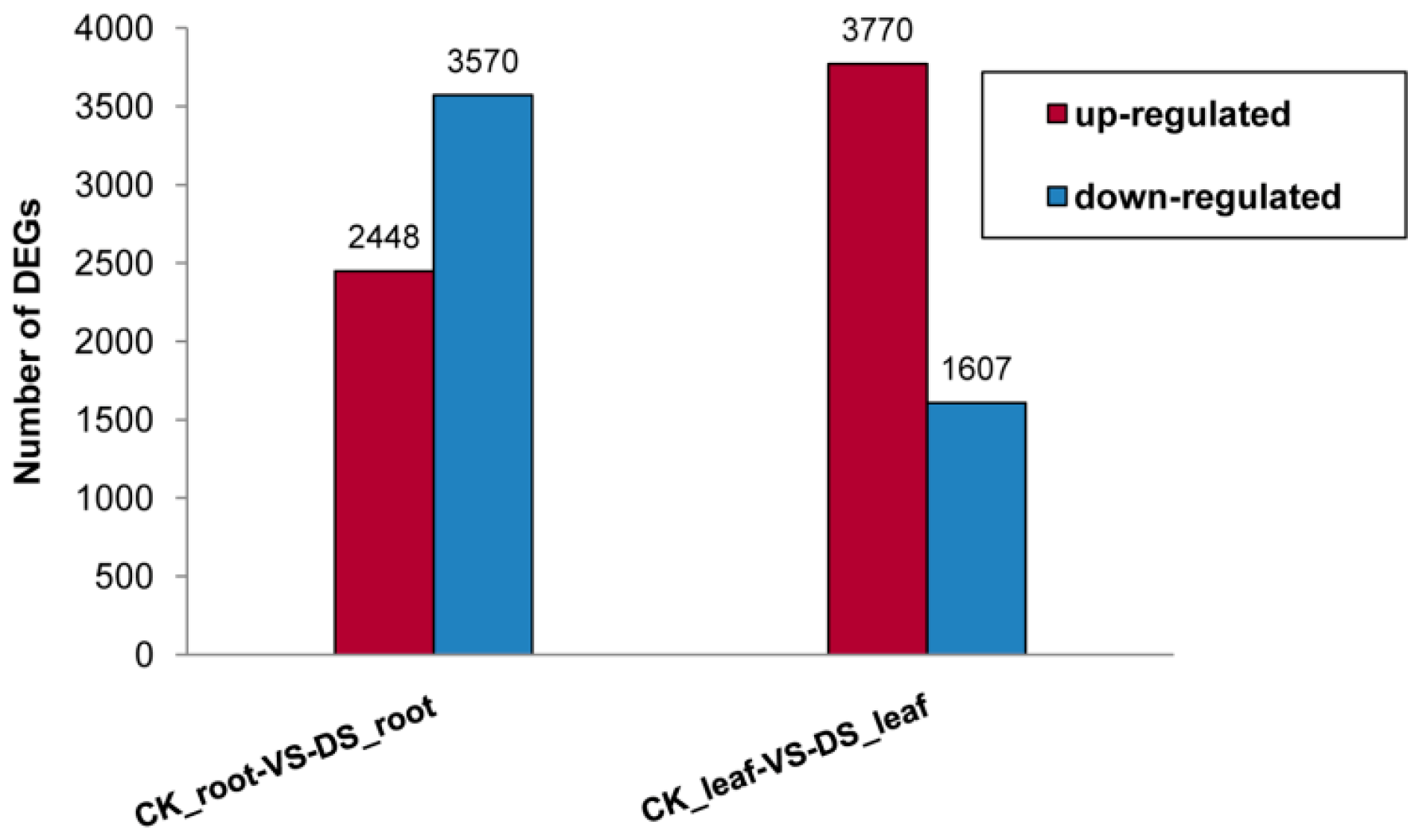
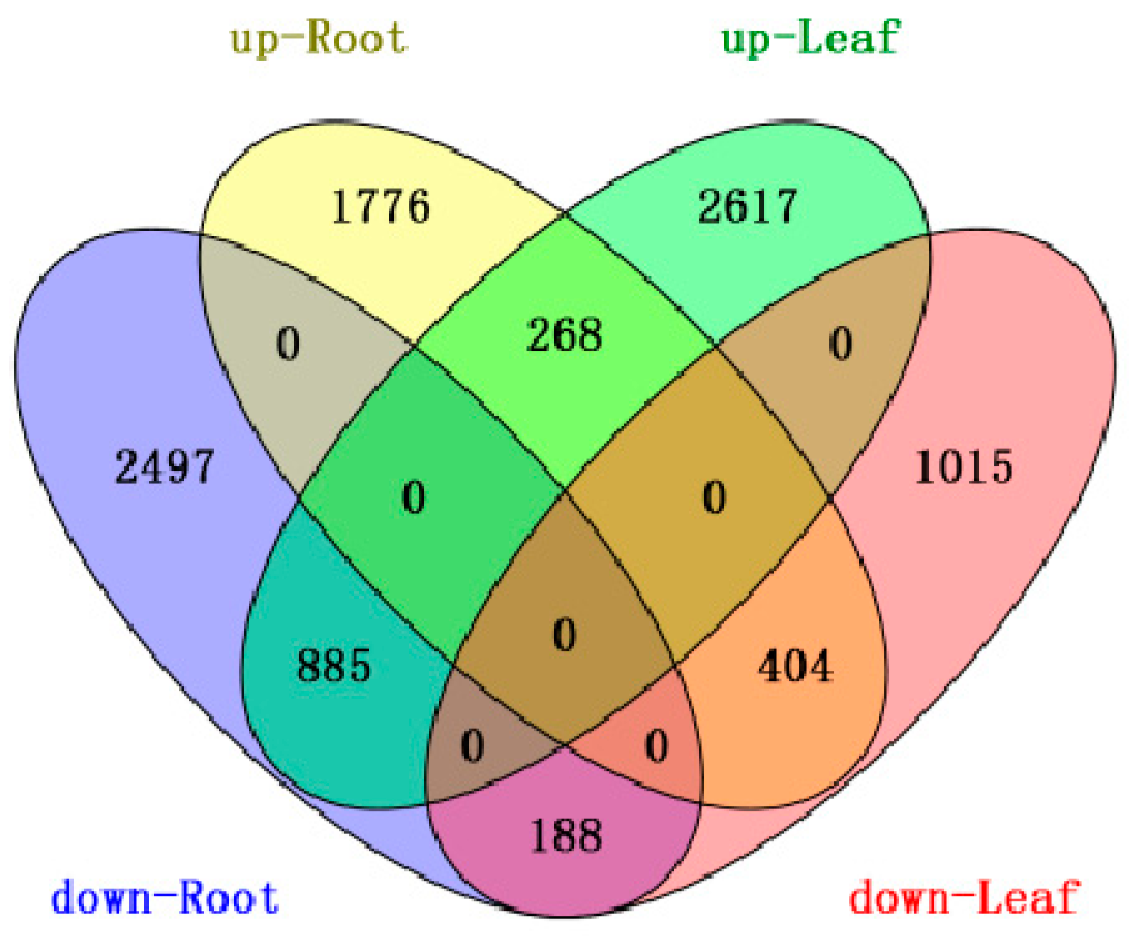
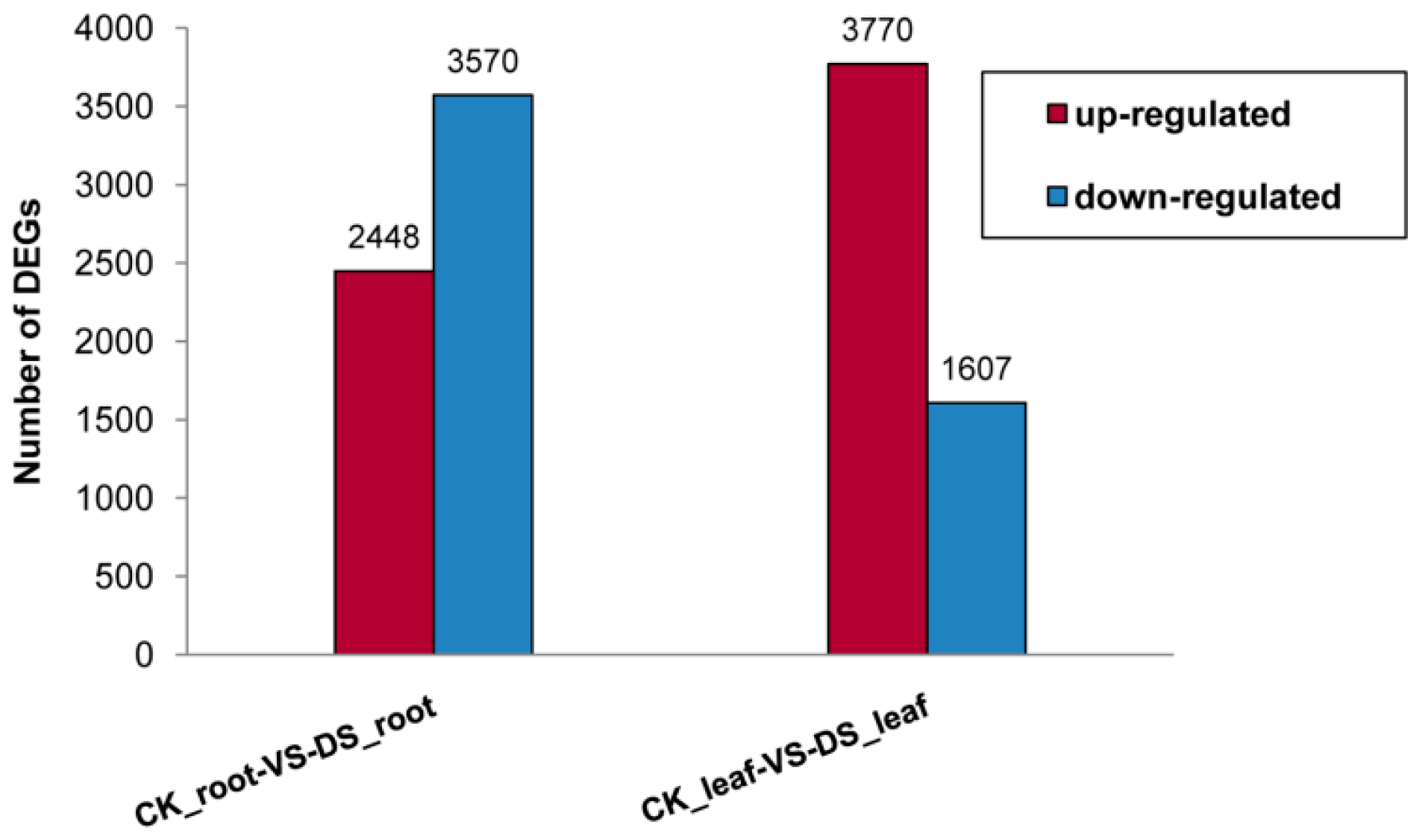
2.1.2. Differential Gene Expression between the Root and Leaf Libraries
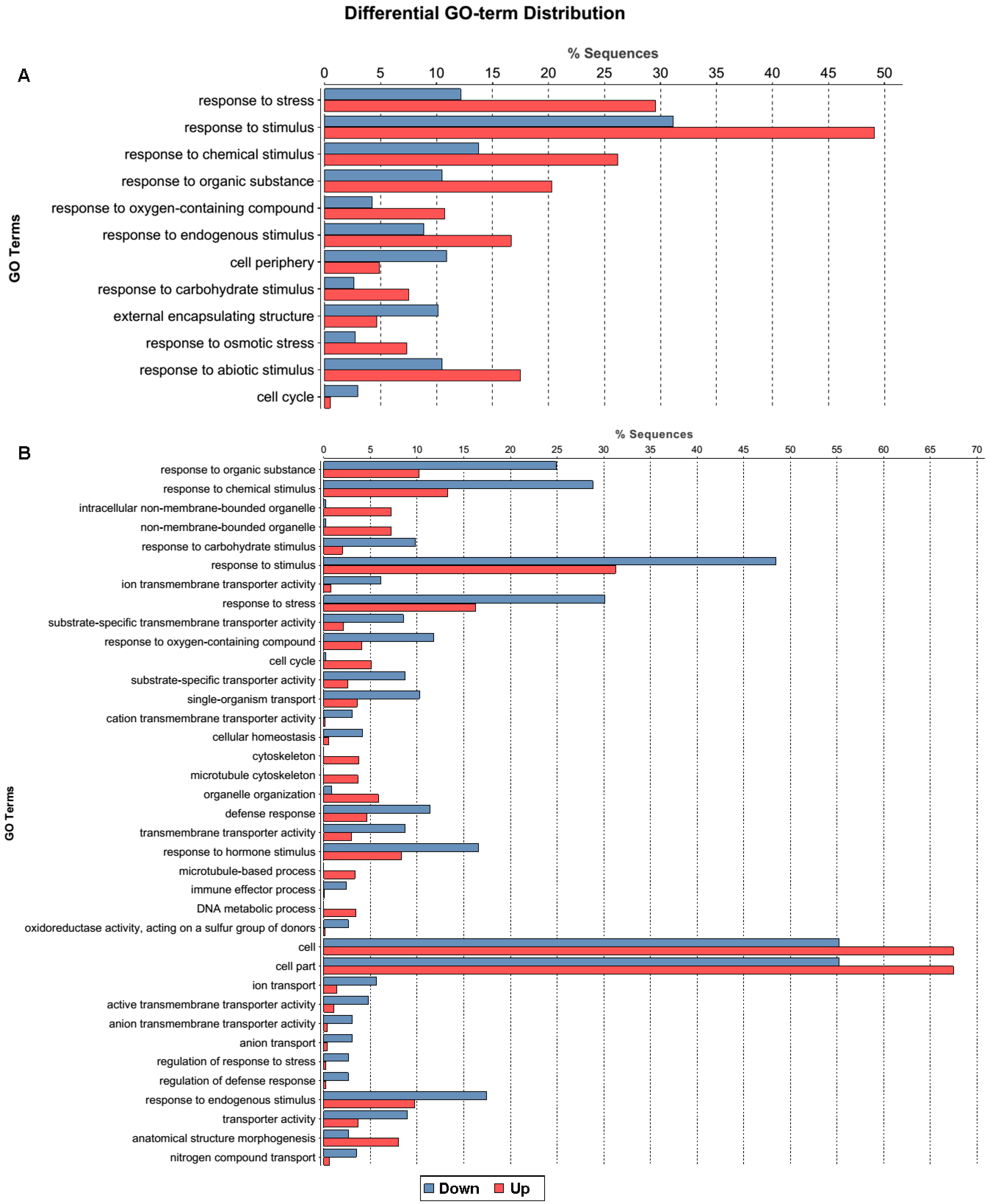
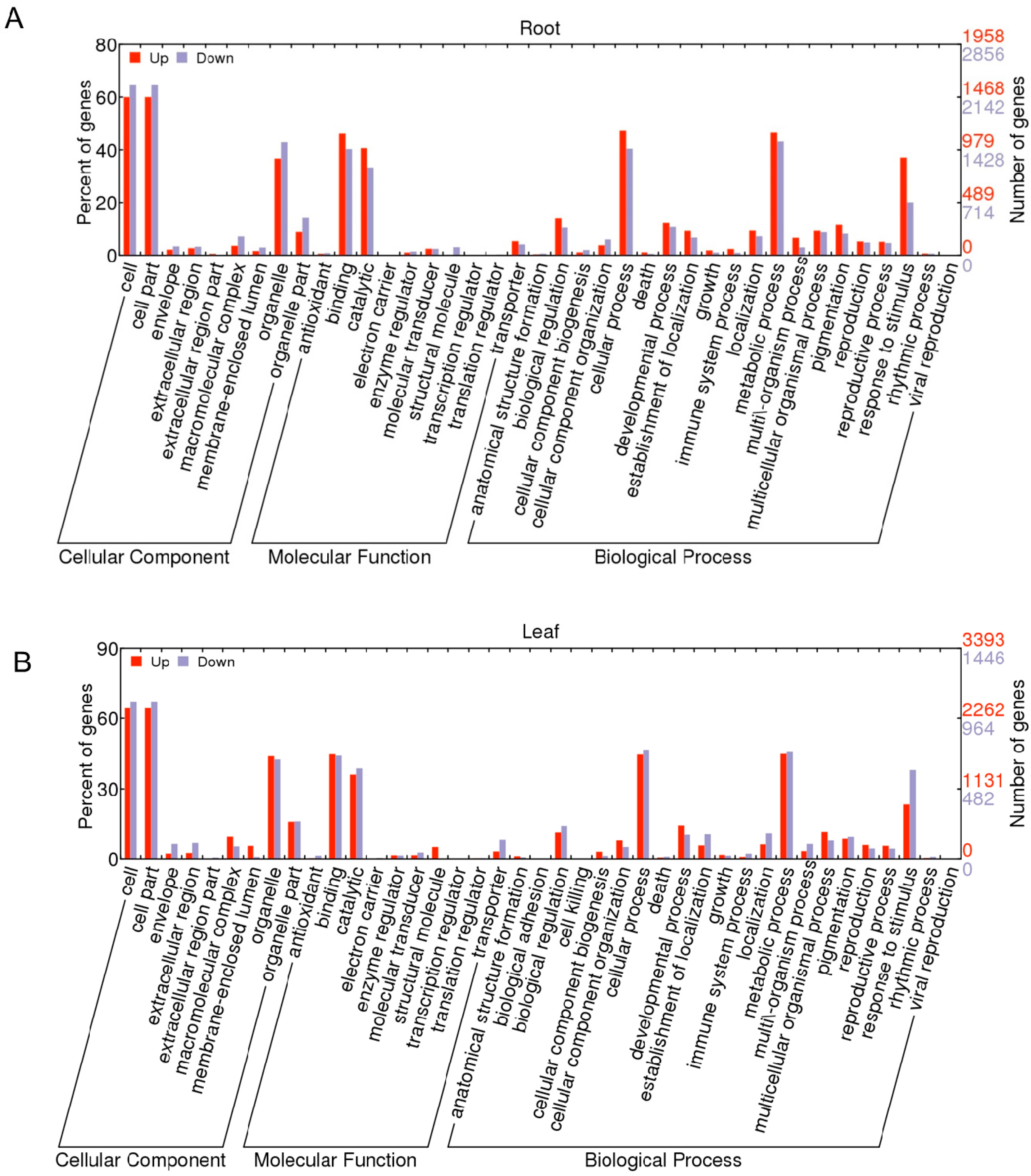
2.1.3. Gene Ontology Function Analysis of DEGs

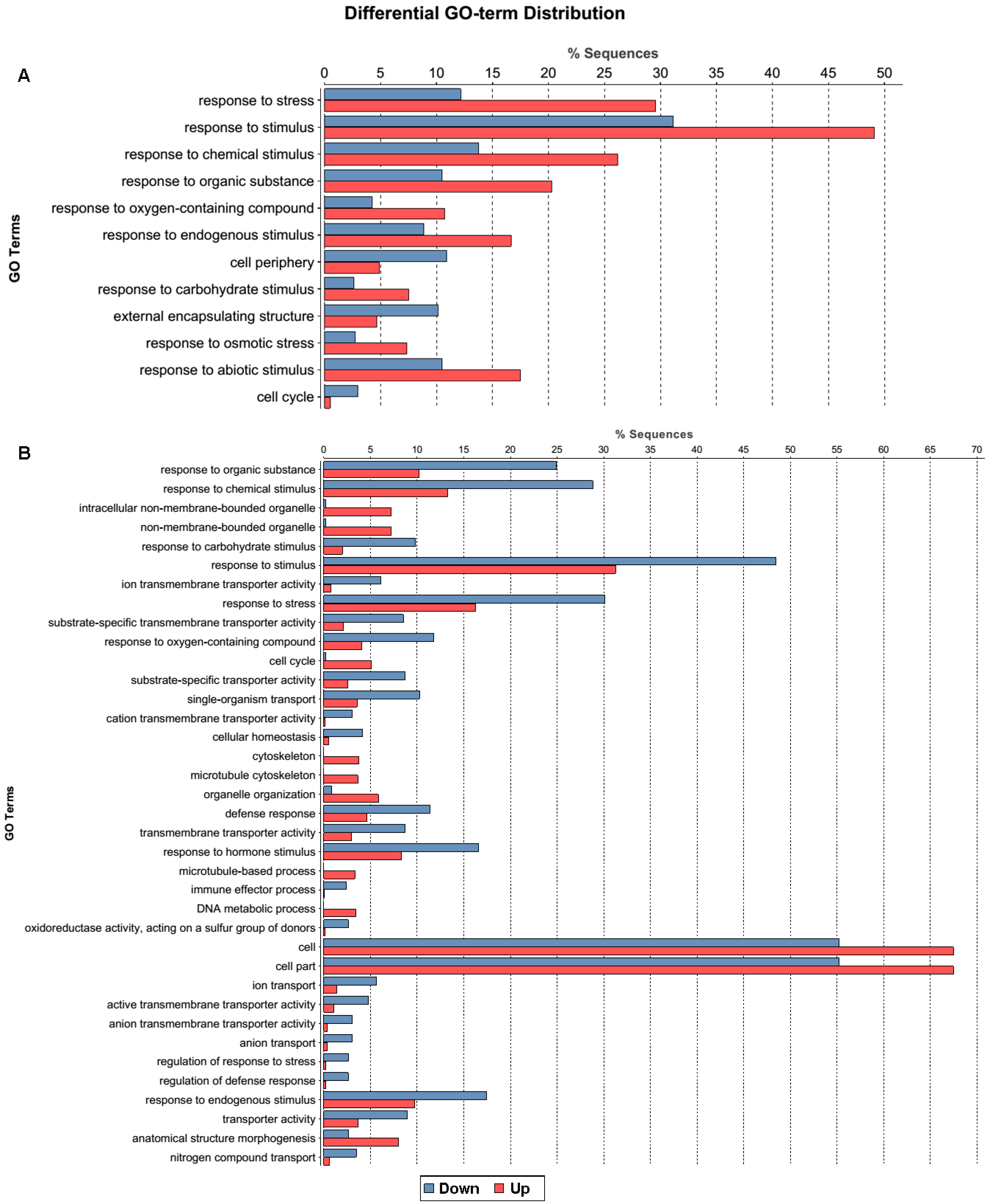
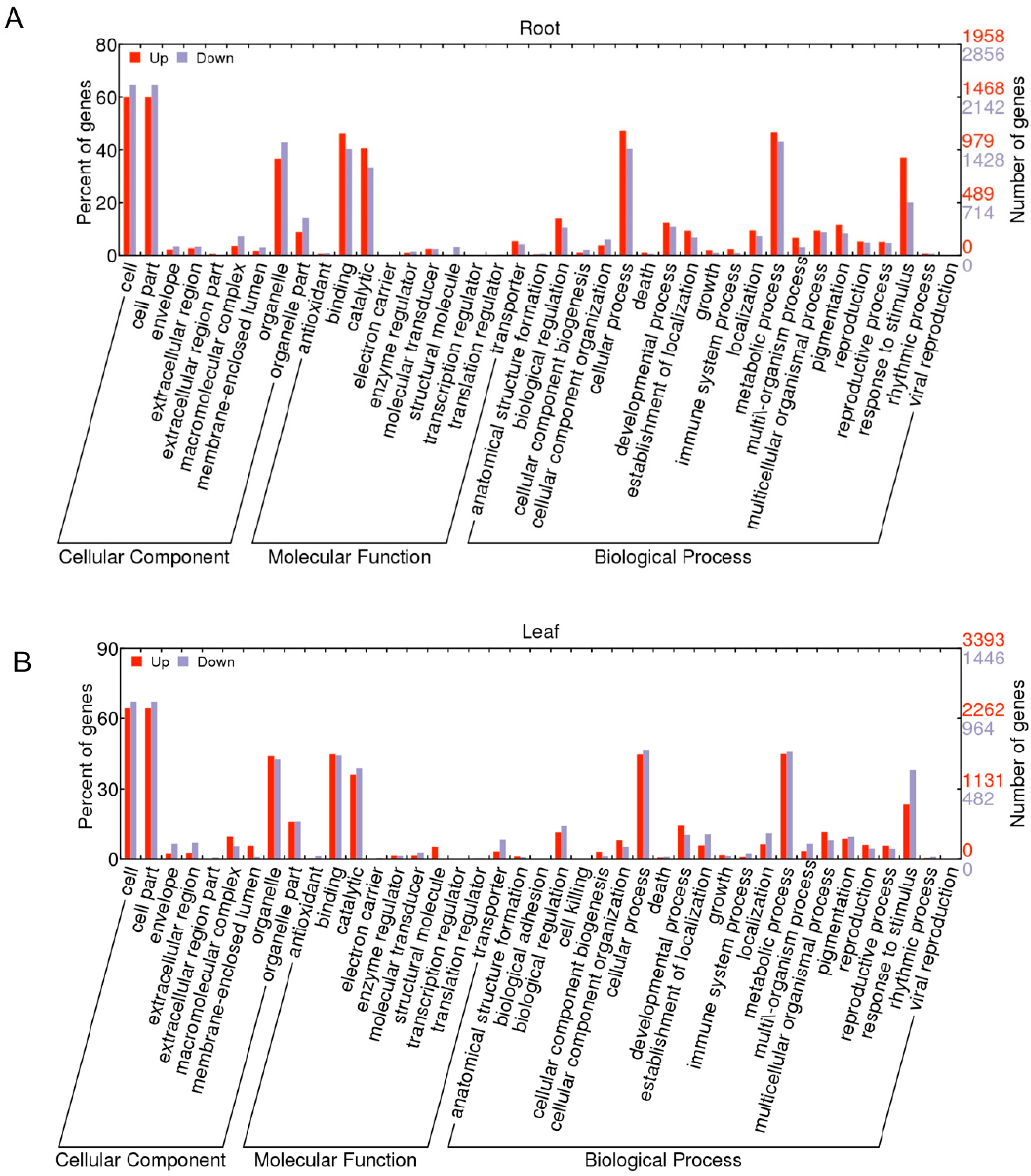
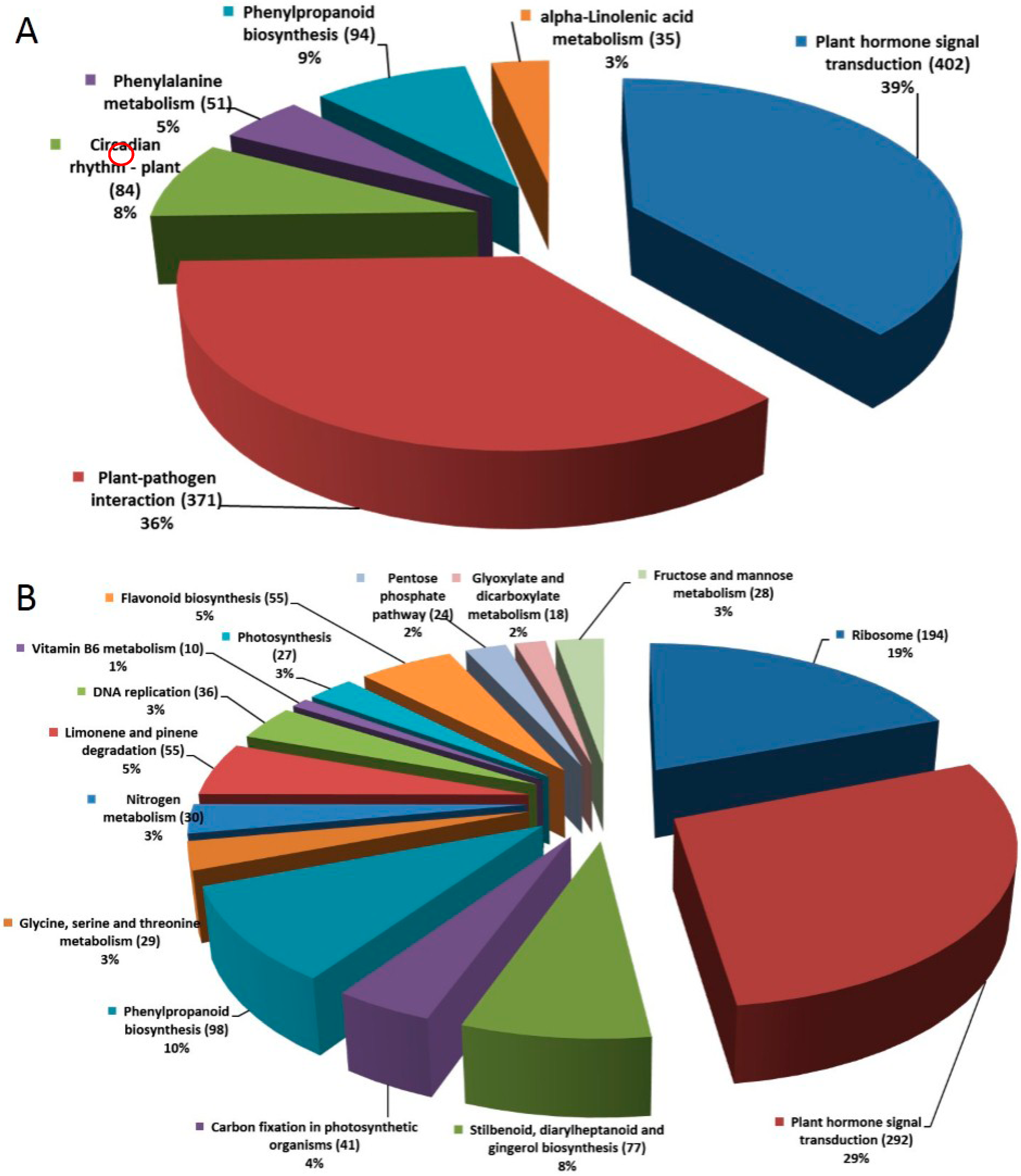
2.1.4. Pathway Analysis of the DEGs

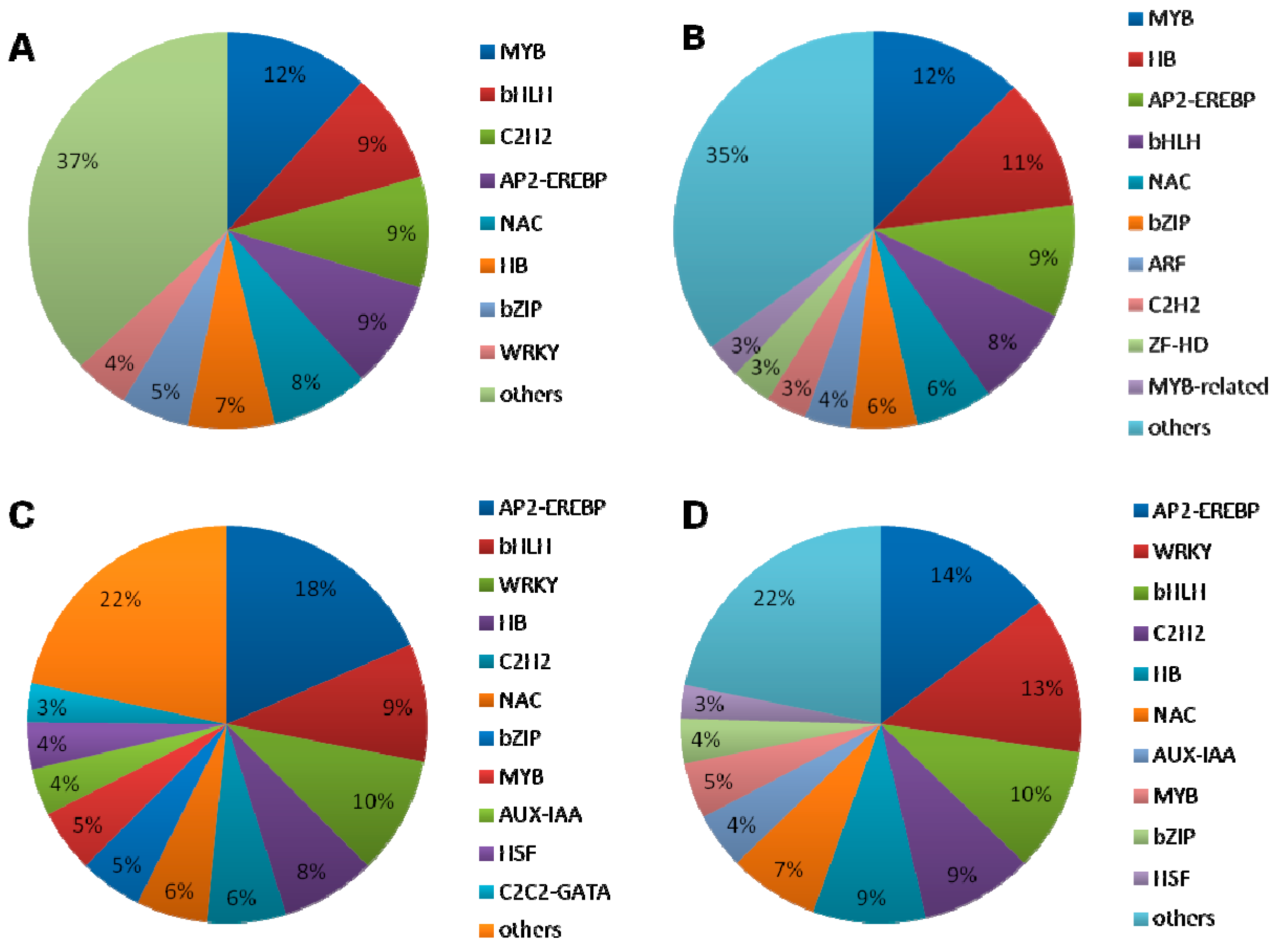
2.1.5. DEGs Encoding Transcription Factors
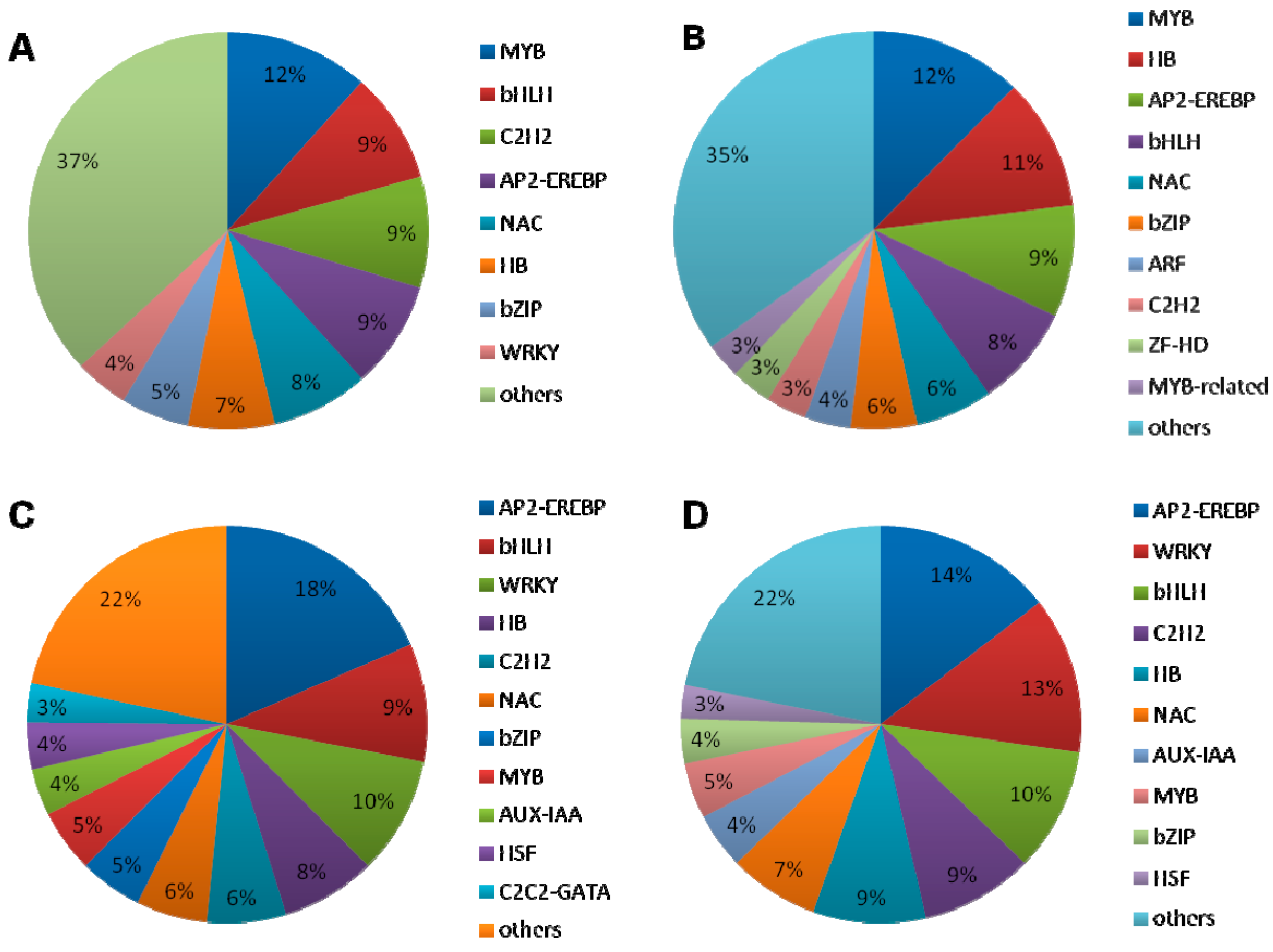
2.1.6. Interaction between the Transcription Factors and Hormone-Related Genes
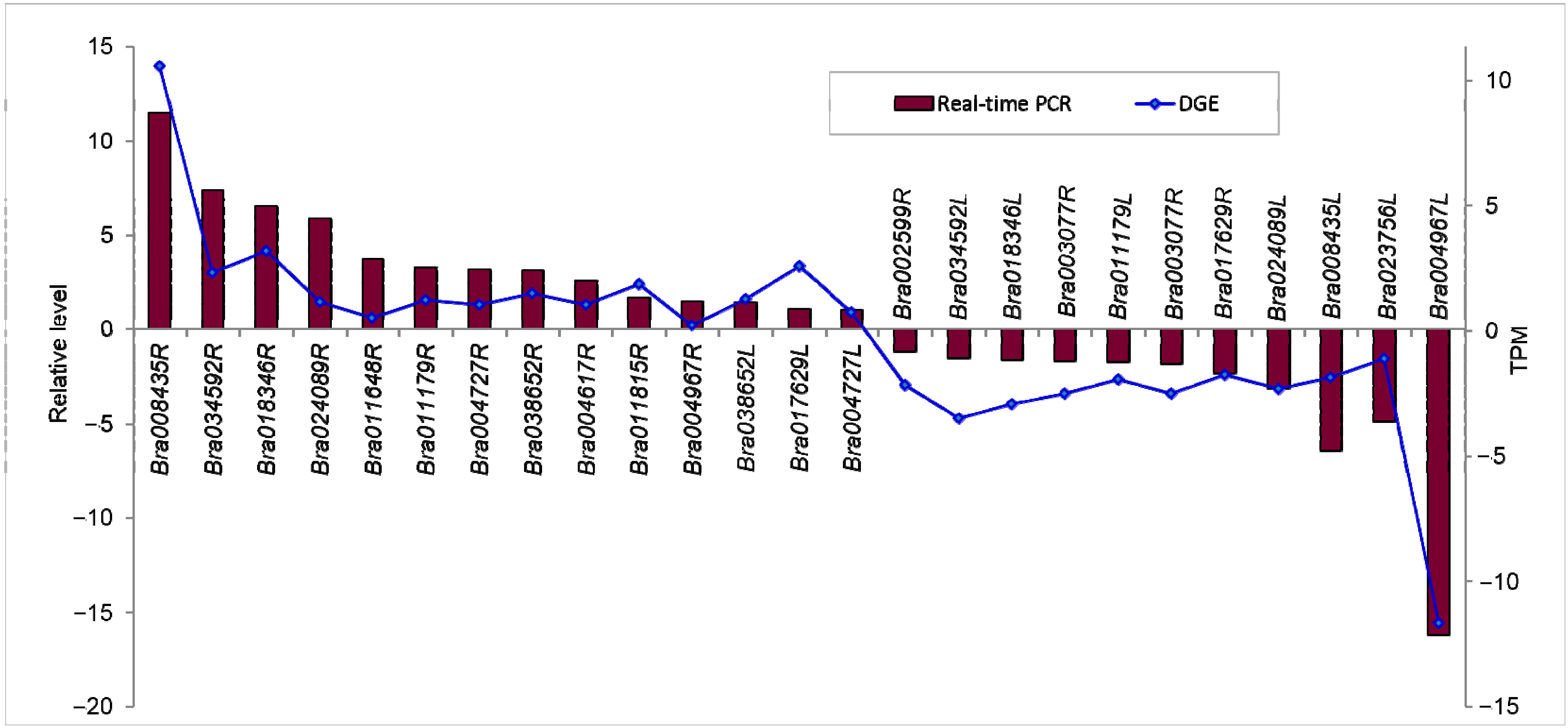
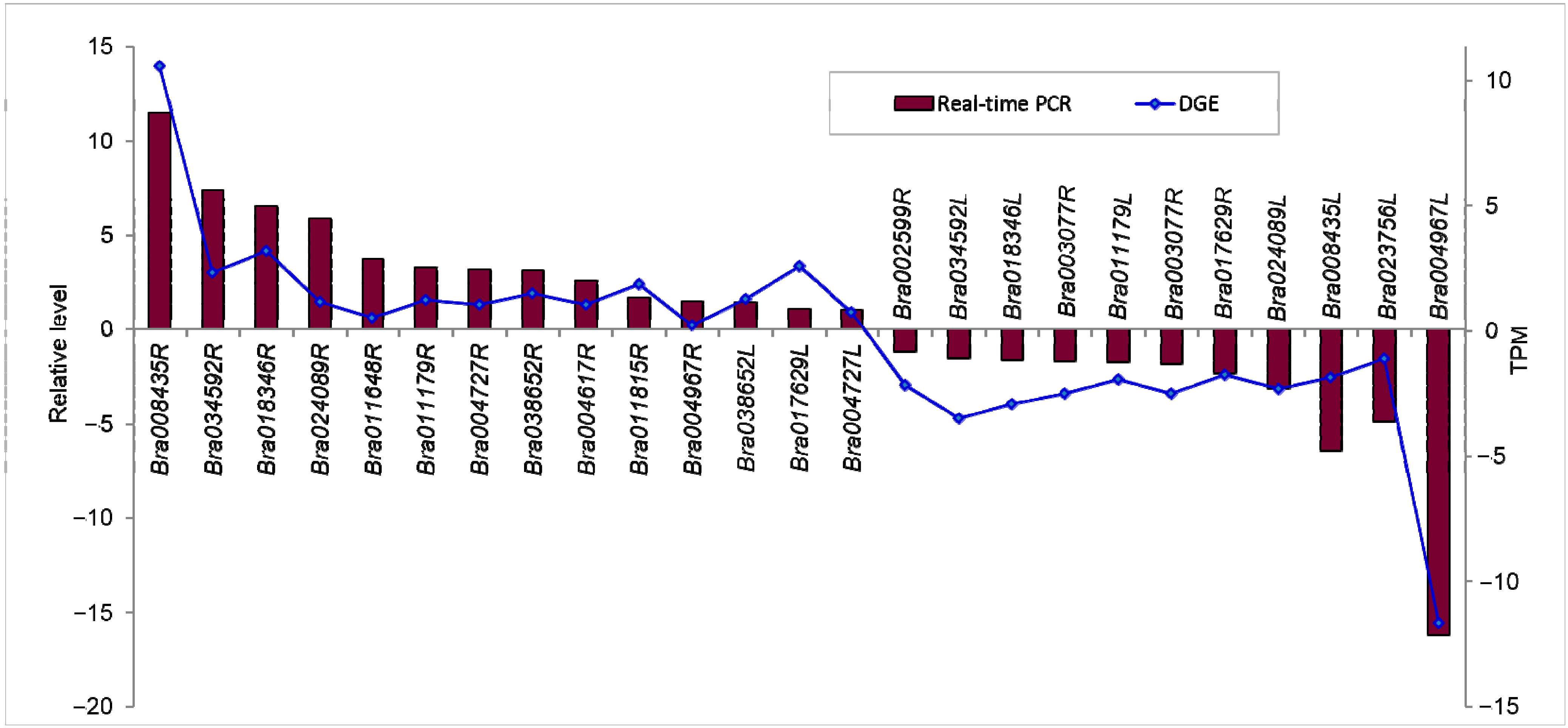
2.1.7. Confirmation of Tag-Mapped Genes by qRT-PCR (Quantitative Real-Time PCR)
2.2. Discussion
2.2.1. Different Regulation Patterns between Root and Leaf Based on Transcriptome Profiles
2.2.2. TFs Functional Protein-Protein Interactions in Hormone Transduction Pathway
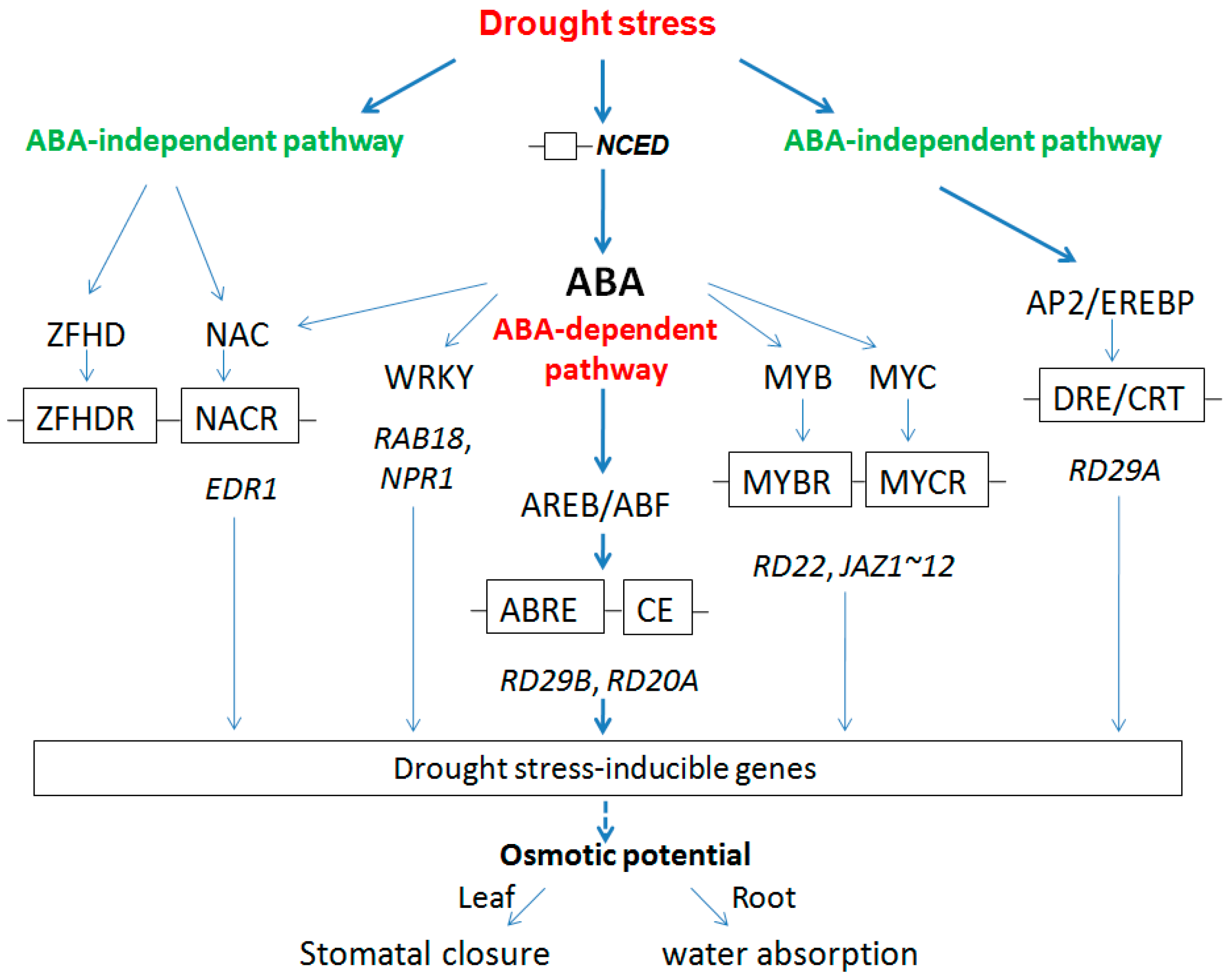
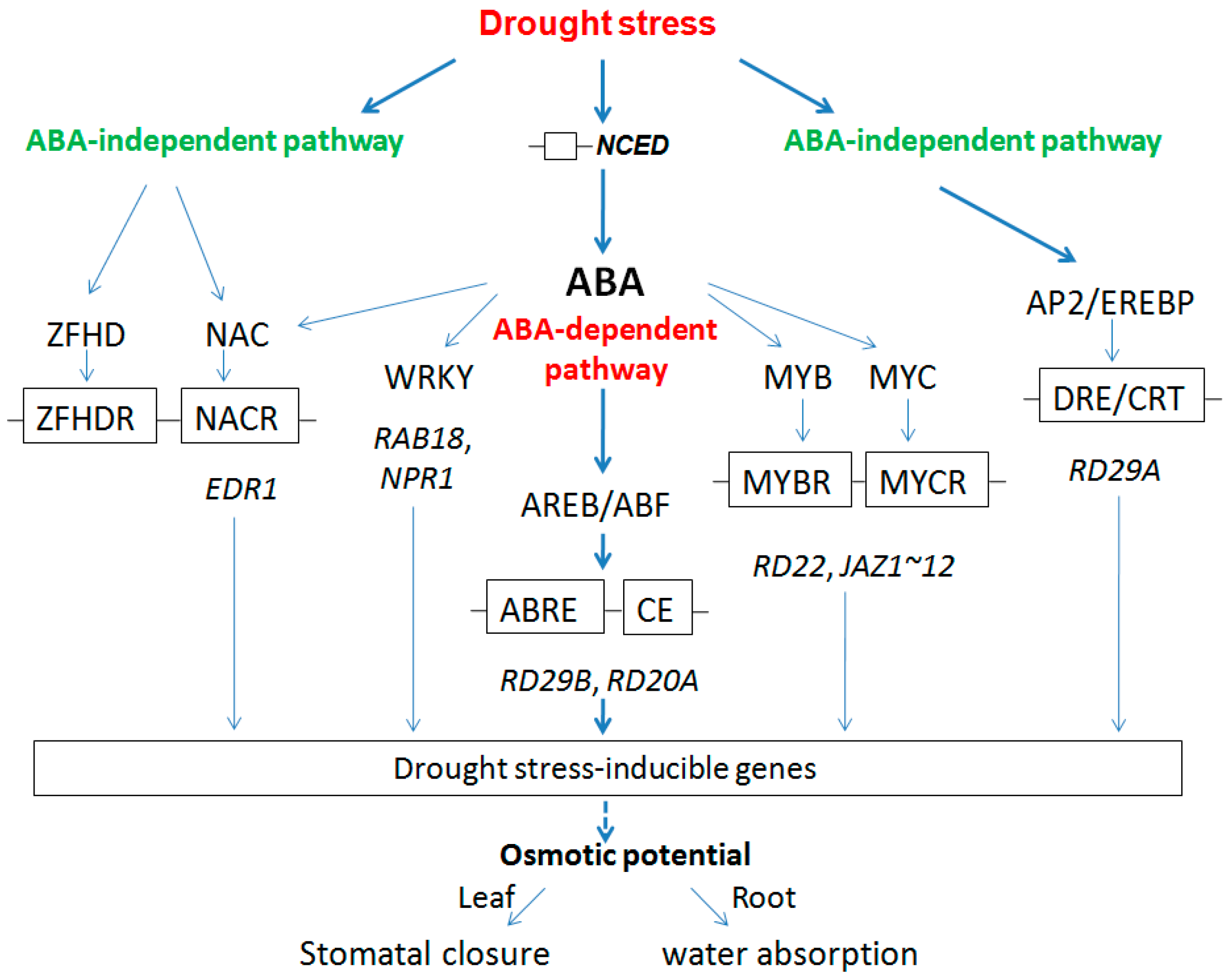
2.2.3. Regulation Model for Root and Leaf under Drought Stress
3. Experimental Section
3.1. Plant Materials and Stress Conditions
3.2. RNA Isolation and Illumina Sequencing Results
3.3. Gene Expression Profiling RNA-Seq Analysis
3.4. qRT-PCR Analysis
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Lobell, D.B.; Schlenker, W.; Costa-Roberts, J. Climate trends and global crop production since 1980. Science 2011, 333, 616–620. [Google Scholar] [CrossRef] [PubMed]
- Khalili, M.; Aboughadareh, A.P.; Naghavi, M.R.; Talebzadeh, S.J. Response of spring Canola (Brassica napus L.) genotypes to water deficit stress. Int. J. Agric. Crop Sci. 2012, 4, 1579–1586. [Google Scholar]
- Müller, T.; Lentzsch, P.; Müller, M.E.H. Carbohydrate dynamics in leaves of rapeseed (Brassica napus) under drought. J. Agron. Crop Sci. 2012, 198, 207–217. [Google Scholar] [CrossRef]
- Ahuja, I.; de Vos, R.C.H.; Bones, A.M.; Hall, R.D. Plant molecular stress responses face climate change. Trends Plant Sci. 2010, 15, 664–674. [Google Scholar] [CrossRef] [PubMed]
- Moumeni, A.; Satoh, K.; Kondoh, H.; Asano, T.; Hosaka, A.; Venuprasad, R.; Serraj, R.; Kumar, A.; Leung, H.; Kikuchi, S. Comparative analysis of root transcriptome profiles of two pairs of drought-tolerant and susceptible rice near-isogenic lines under different drought stress. BMC Plant Biol. 2011, 11, 174. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.K. Salt and drought stress signal transduction in plants. Annu. Rev. Plant Biol. 2002, 53, 247–273. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Li, Z.F.; Xiong, L.M. A plant microRNA regulates the adaptation of roots to drought stress. FEBS Lett. 2012, 586, 1742–1747. [Google Scholar] [CrossRef] [PubMed]
- Bartels, D.; Sunkar, R. Drought and Salt Tolerance in Plants. Crit. Rev. Plant Sci. 2005, 24, 23–58. [Google Scholar] [CrossRef]
- Mahajan, S.; Tuteja, N. Cold, salinity and drought stresses: An overview. Arch. Biochem. Biophys. 2005, 444, 139–158. [Google Scholar] [CrossRef] [PubMed]
- Fujita, Y.; Fujita, M.; Shinozaki, K.; Yamaguchi-Shinozaki, K. ABA-mediated transcriptional regulation in response to osmotic stress in plants. J. Plant Res. 2011, 124, 509–525. [Google Scholar] [CrossRef] [PubMed]
- Shinozaki, K.; Yamaguchi-Shinozaki, K. Gene networks involved in drought stress response and tolerance. J. Exp. Bot. 2007, 58, 221–227. [Google Scholar] [CrossRef] [PubMed]
- Cutler, S.R.; Rodriguez, P.L.; Finkelstein, R.R.; Abrams, S.R. Abscisic acid: Emergence of a core signaling network. Annu. Rev. Plant Biol. 2010, 61, 651–679. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Szostkiewicz, I.; Korte, A.; Moes, D.; Yang, Y.; Christmann, A.; Grill, E. Regulators of PP2C phosphatase activity function as abscisic acid sensors. Science 2009, 324, 1064–1068. [Google Scholar] [CrossRef] [PubMed]
- Park, S.Y.; Fung, P.; Nishimura, N.; Jensen, D.R.; Fujii, H.; Zhao, Y.; Lumba, S.; Santiago, J.; Rodrigues, A.; Chow, T.F.; et al. Abscisic acid inhibits type 2C protein phosphatases via the PYR/PYL family of START proteins. Science 2009, 324, 1068–1071. [Google Scholar] [CrossRef] [PubMed]
- Fujii, H.; Zhu, J.K. Arabidopsis mutant deficient in 3 abscisic acid-activated protein kinases reveals critical roles in growth, reproduction, and stress. Proc. Natl. Acad. Sci. USA 2009, 106, 8380–8385. [Google Scholar] [CrossRef] [PubMed]
- Daszkowska-Golec, A.; Szarejko, I. Open or close the gate-stomata action under the control of phytohormones in drought stress conditions. Front. Plant Sci. 2013, 4, 138. [Google Scholar] [CrossRef] [PubMed]
- Udvardi, M.K.; Kakar, K.; Wandrey, M.; Montanari, O.; Murray, J.; Andriankaja, A.; Zhang, J.Y.; Benedito, V.; Hofer, J.M.; Chueng, F.; et al. Legume transcription factors: Global regulators of plant development and response to the environment. Plant Physiol. 2007, 144, 538–549. [Google Scholar] [CrossRef] [PubMed]
- Riaño-Pachón, D.M.; Ruzicic, S.; Dreyer, I.; Mueller-Roeber, B. PlnTFDB: An integrative plant transcription factor database. BMC Bioinform. 2007, 8, 42. [Google Scholar] [CrossRef] [PubMed]
- Alves, M.S.; Dadalto, S.P.; Gonçalves, A.B.; de Souza, G.B.; Barros, V.A.; Fietto, L.G. Transcription factor functional protein-protein interactions in plant defense responses. Proteomes 2014, 2, 85–106. [Google Scholar] [CrossRef]
- Zhu, Q.; Zhang, J.T.; Gao, X.S.; Tong, J.H.; Xiao, L.T.; Li, W.B.; Zhang, H.X. The Arabidopsis AP2/ERF transcription factor RAP2.6 participates in ABA, salt and osmotic stress responses. Gene 2010, 457, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Rushton, D.L.; Tripathi, P.; Rabara, R.C.; Lin, J.; Ringler, P.; Boken, A.K.; Langum, T.J.; Smidt, L.; Boomsma, D.D.; Emme, N.J.; et al. WRKY transcription factors: Key components in abscisic acid signalling. Plant Biotechnol. J. 2012, 10, 2–11. [Google Scholar] [CrossRef] [PubMed]
- Sakamoto, H.; Maruyama, K.; Sakuma, Y.; Meshi, T.; Iwabuchi, M.; Shinozaki, K.; Yamaguchi-Shinozaki, K. Arabidopsis Cys2/His2-type zinc-finger proteins function as transcription repressors under drought, cold, and high-salinity stress conditions. Plant Physiol. 2004, 136, 2734–2746. [Google Scholar] [CrossRef] [PubMed]
- Dodig, D.; Zorić, M.; Kandić, V.; Perović, D.; Šurlan-Momirović, G. Comparison of responses to drought stress of 100 wheat accessions and landraces to identify opportunities for improving wheat drought resistance. Plant Breed. 2012, 131, 369–379. [Google Scholar] [CrossRef]
- Xiong, L.; Wang, R.G.; Mao, G.; Koczan, J.M. Identification of drought tolerance determinants by genetic analysis of root response to drought stress and abscisic Acid. Plant Physiol. 2006, 142, 1065–1074. [Google Scholar] [CrossRef] [PubMed]
- Hou, X.; Xie, K.B.; Yao, J.L.; Qi, Z.Y.; Xiong, L.Z. A homolog of human ski-interacting protein in rice positively regulates cell viability and stress tolerance. Proc. Natl. Acad. Sci. USA 2009, 106, 6410–6415. [Google Scholar] [CrossRef] [PubMed]
- Hu, H.H.; Dai, M.Q.; Yao, J.L.; Xiao, B.Z.; Li, X.H.; Zhang, Q.F.; Xiong, L.Z. Overexpressing a NAM, ATAF, and CUC (NAC) transcription factor enhances drought resistance and salt tolerance in rice. Proc. Natl. Acad. Sci. USA 2006, 103, 12987–12992. [Google Scholar] [CrossRef] [PubMed]
- Schnurbusch, T.; Collins, N.C.; Eastwood, R.F.; Sutton, T.; Jefferies, S.P.; Langridge, P. Fine mapping and targeted SNP survey using rice wheat gene colinearity in the region of the Bo1 boron toxicity tolerance locus of bread wheat. Theor. Appl. Genet. 2007, 115, 451–461. [Google Scholar] [CrossRef] [PubMed]
- Ismail, A.M.; Thomson, M.J.; Ching, J.-H.; Septiningsih, E.; Wissuwa, M.; Heuer, S.; Singh, R.K.; Gregorio, G.B.; Mackill, D.J. Introgression of QTLs associated with tolerance to abiotic stresses in rice using marker-assisted breeding. In Proceedings of Plant and Animal Genome XVI, San Diego, CA, USA, 12–16 January,2008; Scherago International: Jersey City, NJ, USA, 2008. W233. p. 55. [Google Scholar]
- Ducrocq, S.; Madur, D.; Veyrieras, J.B.; Camus-Kulandaivelu, L.; Kloiber-Maitz, M.; Presterl, T.; Ouzunova, M.; Manicacci, D.; Charcosset, A. Key impact of Vgt1 on flowering time adaptation in maize: Evidence from association mapping and ecogeographical information. Genetics 2008, 178, 2433–2437. [Google Scholar] [CrossRef] [PubMed]
- Harris, K.; Subudhi, P.K.; Borrell, A.; Jordan, D.; Rosenow, D.; Nguyen, H.; Klein, P.; Klein, R.; Mullet, J. Sorghum stay-green QTL individually reduce post-flowering drought-induced leaf senescence. J. Exp. Bot. 2007, 58, 327–338. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.H. Identification and QTL Analysis of Drought Tolerant Traits in Brassica Napus L. Master’s Thesis, Southwestern University, Chongqing, China, 2011. [Google Scholar]
- Hall, N.M.; Griffiths, H.; Corlett, J.A.; Jones, H.G.; Lynn, J.; King, G.J. Relationships between water-use traits and photosynthesis in Brassica oleracea resolved by quantitative genetic analysis. Plant Breed. 2005, 124, 557–564. [Google Scholar] [CrossRef]
- Park, S.H.; Chung, P.J.; Juntawong, P.; Bailey-Serres, J.; Kim, Y.S.; Jung, H.; Bang, S.W.; Kim, Y.K.; Do, Choi. Y.; Kim, J.K. Posttranscriptional control of photosynthetic mRNA decay under stress conditions requires 3ʹ and 5ʹ untranslated regions and correlates with differential polysome association in rice. Plant Physiol. 2012, 159, 1111–1124. [Google Scholar] [CrossRef] [PubMed]
- Wei, W.; Qi, X.; Wang, L.; Zhang, Y.; Hua, W.; Li, D.; Lv, H.; Zhang, X. Characterization of the sesame (Sesamum indicum L.) global transcriptome using Illumina paired-end sequencing and development of EST-SSR markers. BMC Genom. 2011, 12, 451. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Provart, N.J.; Glazebrook, J.; Katagiri, F.; Chang, H.S.; Eulgem, T.; Mauch, F.; Luan, S.; Zou, G.; Whitham, S.A.; et al. Expression profile matrix of Arabidopsis transcription factor genes suggests their putative functions in response to environmental stresses. Plant Cell 2002, 14, 559–574. [Google Scholar] [CrossRef] [PubMed]
- Rasmussen, S.; Barah, P.; Suarez-Rodriguez, M.C.; Bressendorff, S.; Friis, P.; Costantino, P.; Bones, A.M.; Nielsen, H.B.; Mundy, J. Transcriptome responses to combinations of stresses in Arabidopsis. Plant Physiol. 2013, 161, 1783–1794. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, J.; Fu, J.; Gou, M.; Huai, J.; Liu, Y.; Jian, M.; Huang, Q.; Guo, X.; Dong, Z.; Wang, H.; et al. Genome-wide transcriptome analysis of two maize inbred lines under drought stress. Plant Mol. Biol. 2010, 72, 407–421. [Google Scholar] [CrossRef] [PubMed]
- Mohammadi, M.; Kav, N.N.; Deyholos, M.K. Transcript expression profile of water-limited roots of hexaploid wheat (Triticum aestivum 'Opata'). Genome 2008, 51, 357–367. [Google Scholar] [PubMed]
- Aprile, A.; Mastrangelo, A.M.; De Leonardis, A.M.; Galiba, G.; Roncaglia, E.; Ferrari, F.; De Bellis, L.; Turchi, L.; Giuliano, G.; Cattivelli, L. Transcriptional profiling in response to terminal drought stress reveals differential responses along the wheat genome. BMC Genom. 2009, 10, 279. [Google Scholar] [CrossRef] [PubMed]
- Talamè, V.; Ozturk, N.Z.; Bohnert, H.J.; Tuberosa, R. Barley transcript profiles under dehydration shock and drought stress treatments. A comparative analysis. J. Exp. Bot. 2007, 58, 229–240. [Google Scholar] [CrossRef] [PubMed]
- Gao, R.H.; Duan, K.; Guo, G.M.; Du, Z.Z.; Chen, Z.W.; Li, L.; He, T.; Lu, R.J.; Huang, J.H. Comparative transcriptional profiling of two contrasting barley genotypes under salinity stress during the seedling stage. Plant Physiol. 2013, 139, 822–835. [Google Scholar] [CrossRef] [PubMed]
- Micheletto, S.; Rodriguez-Uribe, L.; Hernandez, R.; Richins, R.D.; Curry, J.; O’Connell, M.A. Comparative transcript profiling in roots of Phaseolus acutifolius and P. vulgaris under water deficit stress. Plant Sci. 2007, 173, 510–520. [Google Scholar] [CrossRef]
- Huang, L.; Zhang, F.; Zhang, F.; Wang, W.; Zhou, Y.; Fu, B.; Li, Z. Comparative transcriptome sequencing of tolerant rice introgression line and its parents in response to drought stress. BMC Genom. 2014, 15, 1026. [Google Scholar] [CrossRef] [PubMed]
- Long, W.; Zou, X.; Zhang, X. Transcriptome Analysis of canola (Brassica napus) under salt stress at the germination stage. PLoS ONE 2015, 10, e0116217. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Q.S. Drought-related gene expression analysis during drought stress in Rapeseed (Brassica napus. L). Master’s Thesis, Oil Crops Research Institute Chinese Academy of Agricultural Sciences, Wuhan, China, 2011. [Google Scholar]
- Li, Z. Evaluation of drought tolerance in varieties of rapeseed (Brassica napus L.) and role of exogenous GA3. Master’s Thesis, Oil Crops Research Institute Chinese Academy of Agricultural Sciences, Wuhan, China, 2010. [Google Scholar]
- Cammue, B.P; Broekaert, W.F.; Kellens, J.T.; Raikhel, N.V.; Peumans, W.J. Stress-induced accumulation of wheat germ agglutinin and abscisic acid in roots of wheat seedlings. Plant Physiol. 1989, 91, 1432–1435. [Google Scholar] [CrossRef] [PubMed]
- Audic, S.; Claverie, J.M. The significance of digital gene expression profiles. Genome Res. 1997, 7, 986–995. [Google Scholar] [PubMed]
- Du, Z.; Zhou, X.; Ling, Y.; Zhang, Z.H.; Su, Z. agriGO: A GO analysis toolkit for the agricultural community. Nucleic Acids Res. 2010, 38, W64–W70. [Google Scholar] [CrossRef] [PubMed]
- Conesa, A.; Götz, S.; García-Gómez, J.M.; Terol, J.; Talón, M.; Robles, M. Blast2GO: A universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 2005, 21, 3674–3676. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Araki, M.; Goto, S.; Hattori, M.; Hirakawa, M.; Itoh, M.; Katayama, T.; Kawashima, S.; Okuda, S.; Tokimatsu, T.; et al. KEGG for linking genomes to life and the environment. Nucleic Acids Res. 2008, 36, 480–484. [Google Scholar] [CrossRef] [PubMed]
- Xiong, L.M.; Schumaker, K.S.; Zhu, J.K. Cell signaling during cold, drought, and salt stress. Plant Cell 2002, 14, S165–S183. [Google Scholar] [PubMed]
- Shen, W.; Gómez-Cadenas, A.; Routly, E.L.; Ho, T.H.; Simmonds, J.A.; Gulick, P.J. The salt stress-inducible protein kinase gene, Esi47, from the salt-tolerant wheatgrass Lophopyrum elongatum is involved in plant hormone signaling. Plant Physiol. 2001, 125, 1429–1441. [Google Scholar] [CrossRef] [PubMed]
- Cramer, G.R.; Urano, K.; Delrot, S.; Pezzotti, M.; Shinozaki, K. Effects of abiotic stress on plants: A systems biology perspective. BMC Plant Biol. 2011, 11, 163. [Google Scholar] [CrossRef] [PubMed]
- Vranová, E.; Atichartpongkul, S.; Villarroel, R.; Van Montagu, M.; Inzé, D.; van Camp, W. Comprehensive analysis of gene expression in Nicotiana tabacum leaves acclimated to oxidative stress. Proc. Natl. Acad. Sci. USA 2002, 99, 10870–10875. [Google Scholar] [CrossRef] [PubMed]
- Atkinson, N.J.; Urwin, P.E. The interaction of plant biotic and abiotic stresses: From genes to the field. J. Exp. Bot. 2012, 63, 3523–3543. [Google Scholar] [CrossRef] [PubMed]
- Nakashima, K.; Yamaguchi-Shinozaki, K.; Shinozaki, K. The transcriptional regulatory network in the drought response and its crosstalk in abiotic stress responses including drought, cold, and heat. Front. Plant Sci. 2014, 5, 170. [Google Scholar] [CrossRef] [PubMed]
- Lippuner, V.; Cyert, M.S.; Gasser, C.S. Two classes of plant cDNA clones differentially complement yeast calcineurin mutants and increase salt tolerance of wild-type yeast. J. Biol. Chem. 1996, 271, 12859–12866. [Google Scholar] [PubMed]
- Mehrtens, F.; Kranz, H.; Bednarek, P.; Weisshaar, B. The Arabidopsis transcription factor MYB12 is a flavonol-specific regulator of phenylpropanoid biosynthesis. Plant Physiol. 2005, 138, 1083–1096. [Google Scholar] [CrossRef] [PubMed]
- Sablowski, R.W.; Moyano, E.; Culianez-Macia, F.A.; Schuch, W.; Martin, C.; Bevan, M. A flower-specific Myb protein activates transcription of phenylpropanoid biosynthetic genes. EMBO J. 1994, 13, 128–137. [Google Scholar] [PubMed]
- Zhang, J.H.; Jia, W.S.; Yang, J.C.; Ismail, A.M. Role of ABA in integrating plant responses to drought and salt stresses. Field Crops Res. 2006, 97, 111–119. [Google Scholar] [CrossRef]
- Mitra, J. Genetics and genetic improvement of drought resistance in crop plants. Curr. Sci. 2001, 80, 758–763. [Google Scholar]
- Steudle, E. Water uptake by roots: Effects of water deficit. J. Exp. Bot. 2000, 51, 1531–1542. [Google Scholar] [CrossRef] [PubMed]
- Jones, H.G. Stomatal control of photosynthesis and transpiration. J. Exp. Bot. 1998, 49, 387–398. [Google Scholar] [CrossRef]
- Kramer, P.J.; Boyer, J.S. Water Relations of Plants and Soils; Academic Press: New York, NY, USA, 1995; pp. 167–200. [Google Scholar]
- Steudle, E.; Peterson, C.A. How does water get through roots? J. Exp. Bot. 1998, 49, 775–788. [Google Scholar] [CrossRef]
- Collins, N.C.; Tardieu, F.; Tuberosa, R. Quantitative trait loci and crop performance under abiotic stress: Where do we stand? Plant Physiol. 2008, 147, 469–486. [Google Scholar] [CrossRef] [PubMed]
- Rudd, S. Expressed sequence tags: Alternative or complement to whole genome sequences? Trends Plant Sci. 2003, 8, 321–329. [Google Scholar] [CrossRef]
- Ziolkowski, P.A.; Kaczmarek, M.; Babula, D.; Sadowski, J. Genome evolution in Arabidopsis/Brassica: Conservation and divergence of ancient rearranged segments and their breakpoints. Plant J. 2006, 47, 63–74. [Google Scholar] [CrossRef] [PubMed]
- Hoskins, R.A.; Smith, C.D.; Carlson, J.W.; Carvalho, A.B.; Halpern, A.; Kaminker, J.S.; Kennedy, C.; Mungall, C.J.; Sullivan, B.A.; Sutton, G.G.; et al. Heterochromatic sequences in a Drosophila whole-genome shotgun assembly. Genome Biol. 2002, 3. [Google Scholar] [CrossRef] [Green Version]
- Whittle, C.A.; Malik, M.R.; Li, R.; Krochko, J.E. Comparative transcript analyses of the ovule, microspore, and mature pollen in Brassica napus. Plant Mol. Biol. 2010, 72, 279–299. [Google Scholar] [CrossRef] [PubMed]
- Harrison, M.A. Cross-talk between phytohormone signaling pathways under optimal and stressful conditions. In Phytohormones and Abiotic Stress Tolerance in Plants; Nafees, A.K., Ed.; Springer-Verlag GmbH: Berlin, Germany, 2012; pp. 49–76. [Google Scholar]
- Piskurewicz, U.; Tureckova, V.; Lacombe, E.; Lopez-Molina, L. Far-red light inhibits germination through DELLA-dependent stimulation of ABA synthesis and ABI3 activity. EMBO J. 2009, 28, 2259–2271. [Google Scholar] [CrossRef] [PubMed]
- Teng, S.; Rognoni, S.; Bentsink, L.; Smeekens, S. The Arabidopsis GSQ5/DOG1 Cvi allele is induced by the ABA-mediated sugar signalling pathway, and enhances sugar sensitivity by stimulating ABI4 expression. Plant J. 2008, 55, 372–381. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Takemiya, A.; Kinoshita, T.; Shimazaki, K. Nitric oxide inhibits blue light-specific stomatal opening via abscisic acid signaling pathways in Vicia guard cells. Plant Cell Physiol. 2007, 48, 715–723. [Google Scholar] [CrossRef] [PubMed]
- Schroeder, J.I.; Allen, G.J.; Hugouvieux, V.; Kwak, J.M.; Waner, D. Guard cell signal transduction. Annu. Rev. Plant Physiol. Plant Mol. Biol. 2001, 52, 627–658. [Google Scholar] [CrossRef] [PubMed]
- Nemhauser, J.L.; Hong, F.; Chory, J. Different plant hormones regulate similar processes through largely nonoverlapping transcriptional responses. Cell 2006, 126, 467–475. [Google Scholar] [CrossRef] [PubMed]
- Huang, D.; Wu, W.; Abrams, S.R.; Cutler, A.J. The relationship of drought-related gene expression in Arabidopsis thaliana to hormonal and environmental factors. J. Exp. Bot. 2008, 59, 2991–3007. [Google Scholar] [CrossRef] [PubMed]
- Pandey, S.P.; Somssich, I.E. The role of WRKY transcription factors in plant immunity. Plant Physiol. 2009, 150, 1648–1655. [Google Scholar] [CrossRef] [PubMed]
- Sakamoto, H.; Araki, T.; Meshi, T.; Iwabuchi, M. Expression of a subset of the Arabidopsis Cys2/His2-type zinc-finger protein gene family under water stress. Gene 2000, 248, 23–32. [Google Scholar] [CrossRef]
- Seo, P.J.; Xiang, F.; Qiao, M.; Park, J.Y.; Lee, Y.N.; Kim, S.G.; Lee, Y.H.; Park, W.J.; Park, C.M. The MYB96 transcription factor mediates abscisic acid signaling during drought stress response in Arabidopsis. Plant Physiol. 2009, 151, 275–289. [Google Scholar] [CrossRef] [PubMed]
- Sasaki-Sekimoto, Y.; Jikumaru, Y.; Obayashi, T.; Saito, H.; Masuda, S.; Kamiya, Y.; Ohta, H.; Shirasu, K. Basic helix-loop-helix transcription factors JASMONATE-ASSOCIATED MYC2-LIKE1 (JAM1), JAM2, and JAM3 are negative regulators of jasmonate responses in Arabidopsis. Plant Physiol. 2013, 163, 291–304. [Google Scholar] [CrossRef] [PubMed]
- Coego, A.; Ramirez, V.; Gil, M.J.; Flors, V.; Mauch-Mani, B.; Vera, P. An Arabidopsis homeodomain transcription factor, OVEREXPRESSOR OF CATIONIC PEROXIDASE 3, mediates resistance to infection by necrotrophic pathogens. Plant Cell 2005, 17, 2123–2137. [Google Scholar] [CrossRef] [PubMed]
- Dubos, C.; Stracke, R.; Grotewold, E.; Weisshaar, B.; Martin, C.; Lepiniec, L. MYB transcription factors in Arabidopsis. Trends Plant Sci. 2010, 15, 573–581. [Google Scholar] [CrossRef] [PubMed]
- Raffaele, S.; Vailleau, F.; Léger, A.; Joubès, J.; Miersch, O.; Huard, C.; Blée, E.; Mongrand, S.; Domergue, F.; Roby, D. A MYB transcription factor regulates very-long-chain fatty acid biosynthesis for activation of the hypersensitive cell death response in Arabidopsis. Plant Cell 2008, 20, 752–767. [Google Scholar] [CrossRef] [PubMed]
- Cominelli, E.; Galbiati, M.; Vavasseur, A.; Conti, L.; Sala, T.; Vuylsteke, M.; Leonhardt, N.; Dellaporta, S.L.; Tonelli, C. A guard-cell-specific MYB transcription factor regulates stomatal movements and plant drought tolerance. Curr. Biol. 2005, 15, 1196–1200. [Google Scholar] [CrossRef] [PubMed]
- Abe, H.; Urao, T.; Ito, T.; Seki, M.; Shinozaki, K.; Yamaguchi-Shinozaki, K. Arabidopsis AtMYC2 (bHLH) and AtMYB2 (MYB) function as transcriptional activators in abscisic acid signaling. Plant Cell 2003, 15, 63–78. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi-Shinozaki, K.; Shinozaki, K. Transcriptional regulatory networks in cellular responses and tolerance to dehydration and cold stresses. Annu. Rev. Plant Biol. 2006, 57, 781–803. [Google Scholar] [CrossRef] [PubMed]
- Mengiste, T.; Chen, X.; Salmeron, J.; Dietrich, R. The BOTRYTIS SUSCEPTIBLE1 gene encodes an R2R3MYB transcription factor protein that is required for biotic and abiotic stress responses in Arabidopsis. Plant Cell 2003, 15, 2551–2565. [Google Scholar] [CrossRef] [PubMed]
- Fujimoto, S.Y.; Ohta, M.; Usui, A.; Shinshi, H.; Ohme-Takagi, M. Arabidopsis ethylene-responsive element binding factors act as transcriptional activators or repressors of GCC box-mediated gene expression. Plant Cell 2000, 12, 393–404. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.Q.; Yan, L.; Wu, Z.; Mei, C.; Lu, K.; Yu, Y.T.; Liang, S.; Zhang, X.F.; Wang, X.F.; Zhang, D.P. Cooperation of three WRKY-domain transcription factors WRKY18, WRKY40, and WRKY60 in repressing two ABA-responsive genes ABI4 and ABI5 in Arabidopsis. J. Exp. Bot. 2012, 63, 6371–6392. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Liang, G.; Yu, D. Activated expression of WRKY57 confers drought tolerance in Arabidopsis. Mol. Plant 2012, 5, 1375–1388. [Google Scholar] [CrossRef] [PubMed]
- Endo, A.; Sawada, Y.; Takahashi, H.; Okamoto, M.; Ikegami, K.; Koiwai, H.; Seo, M.; Toyomasu, T.; Mitsuhashi, W.; Shinozaki, K.; et al. Drought induction of Arabidopsis 9-cis-epoxycarotenoid dioxygenase occurs in vascular parenchyma cells. Plant Physiol. 2008, 147, 1984–1993. [Google Scholar] [CrossRef] [PubMed]
- Ozhuner, E.; Eldem, V.; Ipek, A.; Okay, S.; Sakcali, S.; Zhang, B.; Boke, H.; Unver, T. Boron stress responsive microRNAs and their targets in barley. PLoS ONE 2013, 8, e59543. [Google Scholar] [CrossRef] [PubMed]
- Tran, L.S.; Nakashima, K.; Sakuma, Y.; Simpson, S.D.; Fujita, Y.; Maruyama, K.; Fujita, M.; Seki, M.; Shinozaki, K.; Yamaguchi-Shinozaki, K. Isolation and functional analysis of Arabidopsis stress-inducible NAC transcription factors that bind to a drought-responsive cis-element in the early responsive to dehydration stress 1 promoter. Plant Cell 2004, 16, 2481–2498. [Google Scholar] [CrossRef] [PubMed]
- Niu, F.; Wang, B.; Wu, F.; Yan, J.; Li, L.; Wang, C.; Wang, Y.; Yang, B.; Jiang, Y.Q. Canola (Brassica napus L.) NAC103 transcription factor gene is a novel player inducing reactive oxygen species accumulation and cell death in plants. Biochem. Biophys. Res. Commun. 2014, 454, 30–35. [Google Scholar] [CrossRef] [PubMed]
- Zhong, H.; Guo, Q.Q.; Chen, L.; Ren, F.; Wang, Q.Q.; Zheng, Y.; Li, X.B. Two Brassica napus genes encoding NAC transcription factors are involved in response to high-salinity stress. Plant Cell Rep. 2012, 31, 1991–2003. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi-Shinozaki, K.; Shinozaki, K. Organization of cis-acting regulatory elements in osmotic- and cold-stress-responsive promoters. Trends Plant Sci. 2005, 10, 88–94. [Google Scholar] [CrossRef] [PubMed]
- AC’t Hoen, P.A.; Ariyurek, Y.; Thygesen, H.H.; Vreugdenhil, E.; Vossen, R.H.; de Menezes, R.X.; Boer, J.M.; van Ommen, G.J.; den Dunnen, J.T. Deep sequencing-based expression analysis shows major advances in robustness, resolution and inter-lab portability over five microarray platforms. Nucleic Acids Res. 2008, 36, e141. [Google Scholar] [CrossRef] [PubMed]
- Yan, X.H.; Dong, C.H.; Yu, J.Y.; Liu, W.H.; Jiang, C.H.; Liu, J.; Hu, Q.; Fang, X.P.; Wei, W.H. Transcriptome profile analysis of young floral buds of fertile and sterile plants from the self-pollinated offspring of the hybrid between novel restorer line NR1 and Nsa CMS line in Brassica napus. BMC Genom. 2013, 14, 26. [Google Scholar] [CrossRef] [PubMed]
- Morrissy, A.S.; Morin, R.D.; Delaney, A.; Zeng, T.; McDonald, H.; Jones, S.; Zhao, Y.; Hirst, M.; Marra, M.A. Next-generation tag sequencing for cancer gene expression profiling. Genome Res. 2009, 19, 1825–1835. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, C.; Zhang, X.; Zhang, K.; An, H.; Hu, K.; Wen, J.; Shen, J.; Ma, C.; Yi, B.; Tu, J.; et al. Comparative Analysis of the Brassica napus Root and Leaf Transcript Profiling in Response to Drought Stress. Int. J. Mol. Sci. 2015, 16, 18752-18777. https://doi.org/10.3390/ijms160818752
Liu C, Zhang X, Zhang K, An H, Hu K, Wen J, Shen J, Ma C, Yi B, Tu J, et al. Comparative Analysis of the Brassica napus Root and Leaf Transcript Profiling in Response to Drought Stress. International Journal of Molecular Sciences. 2015; 16(8):18752-18777. https://doi.org/10.3390/ijms160818752
Chicago/Turabian StyleLiu, Chunqing, Xuekun Zhang, Ka Zhang, Hong An, Kaining Hu, Jing Wen, Jinxiong Shen, Chaozhi Ma, Bin Yi, Jinxing Tu, and et al. 2015. "Comparative Analysis of the Brassica napus Root and Leaf Transcript Profiling in Response to Drought Stress" International Journal of Molecular Sciences 16, no. 8: 18752-18777. https://doi.org/10.3390/ijms160818752