
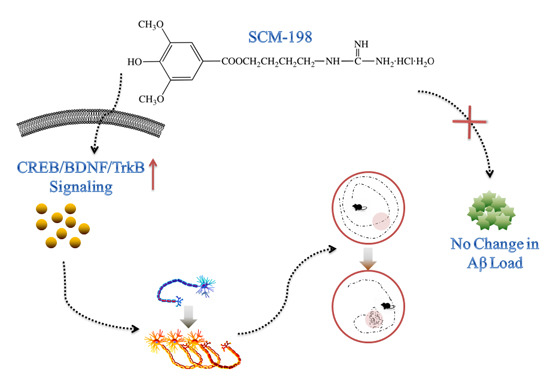
SCM-198 Ameliorates Cognitive Deficits, Promotes Neuronal Survival and Enhances CREB/BDNF/TrkB Signaling without Affecting Aβ Burden in AβPP/PS1 Mice

Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
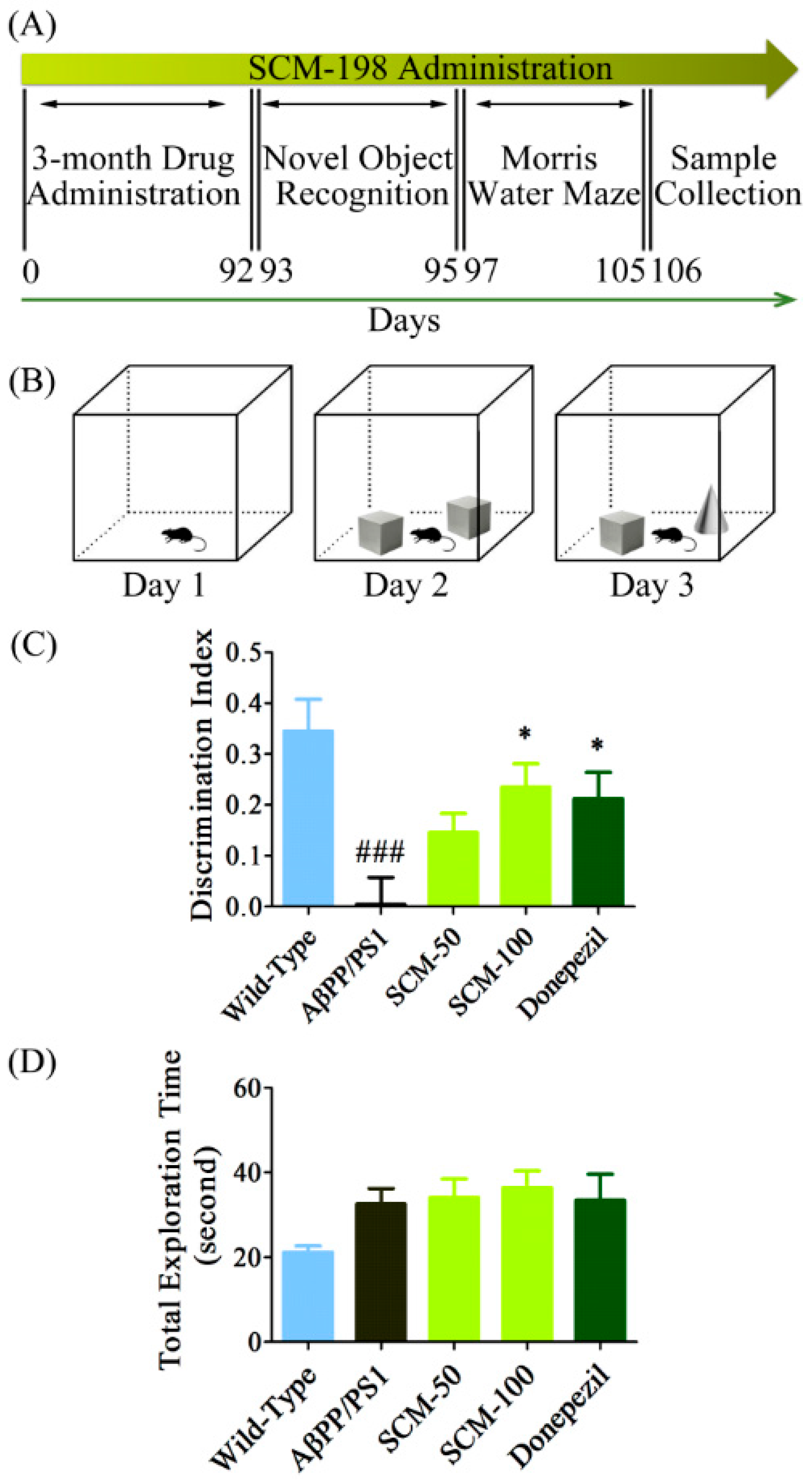
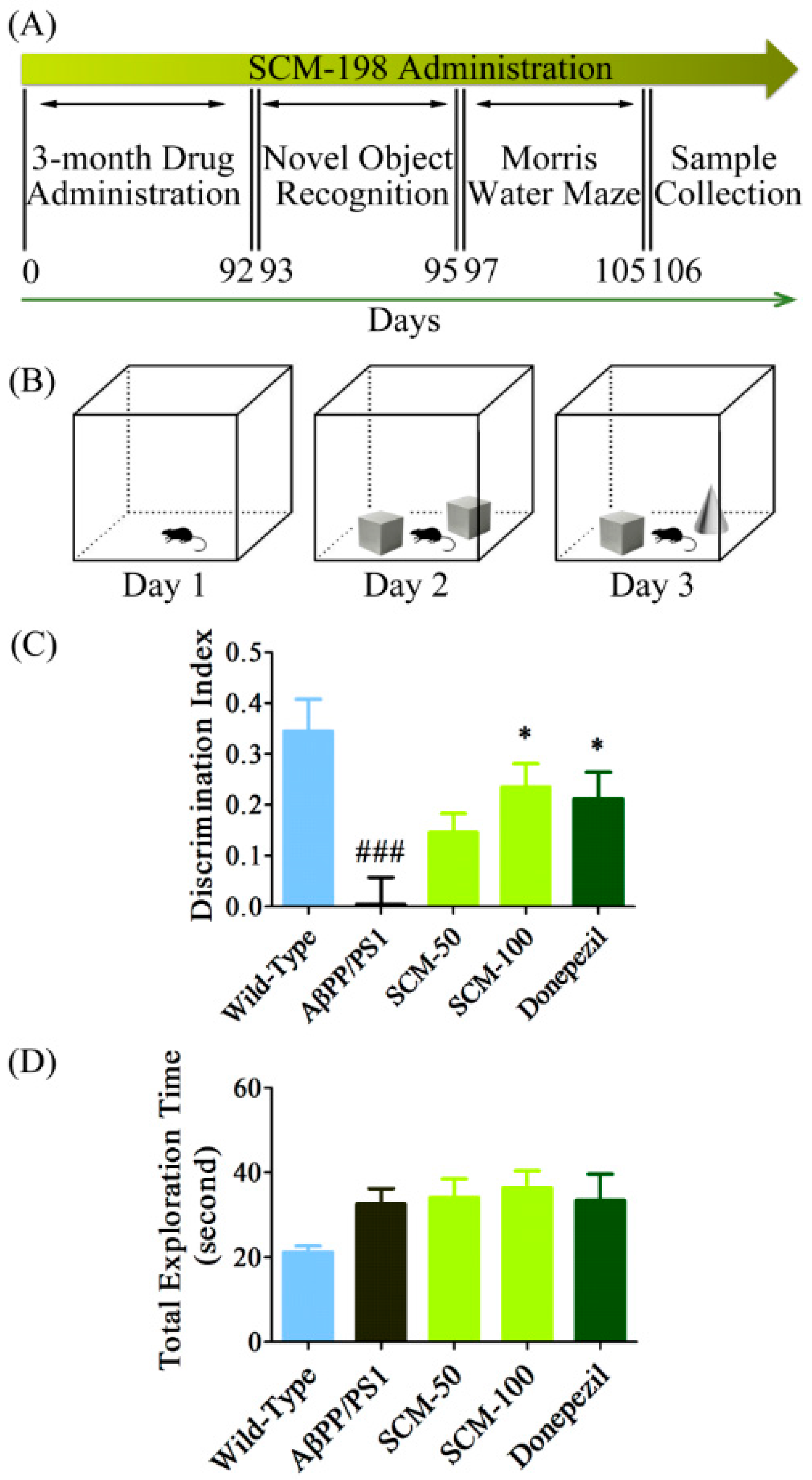
2.1. SCM-198 Rescued Recognition Memory Deficits in AβPP/PS1 Mice in NOR Test
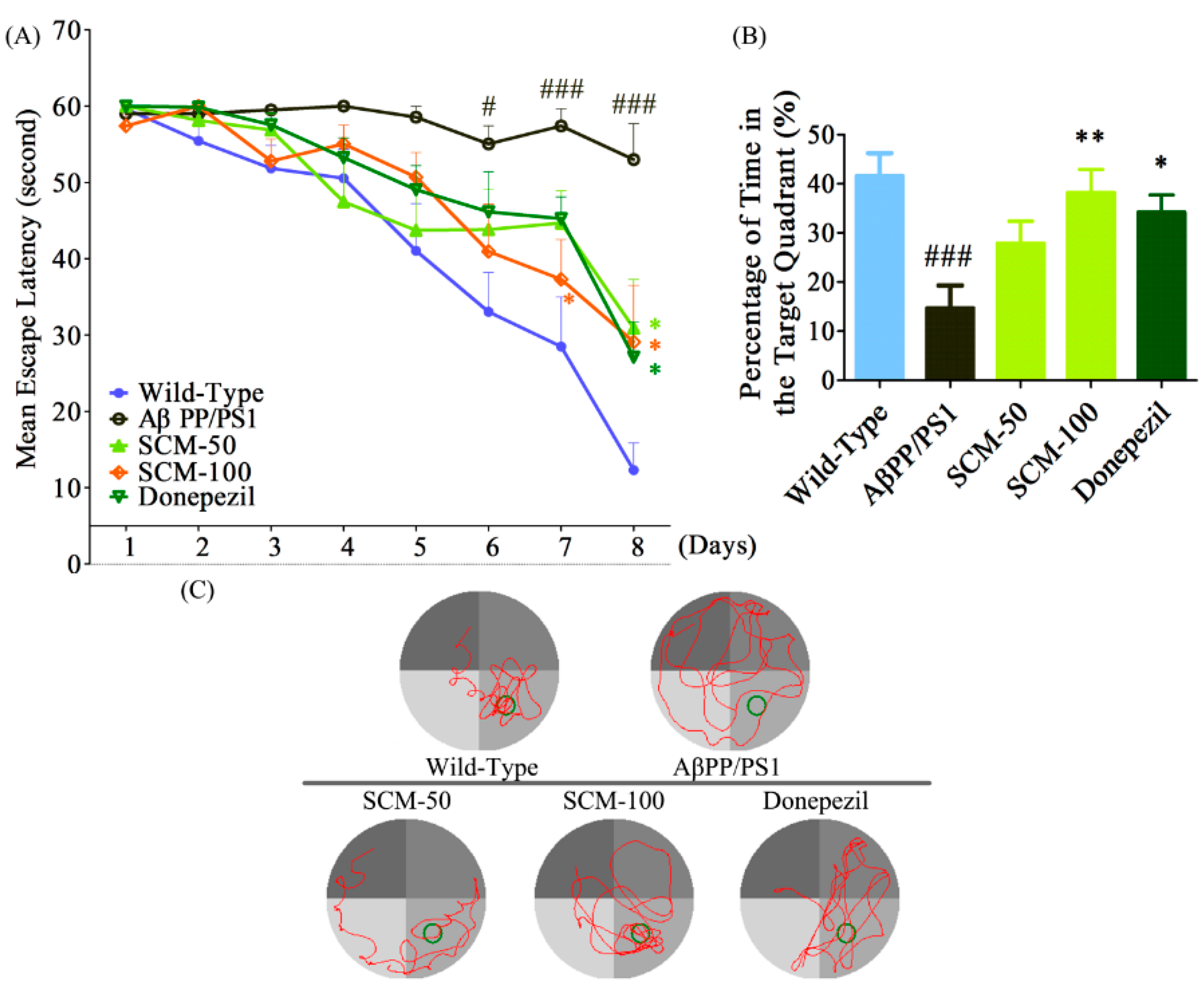
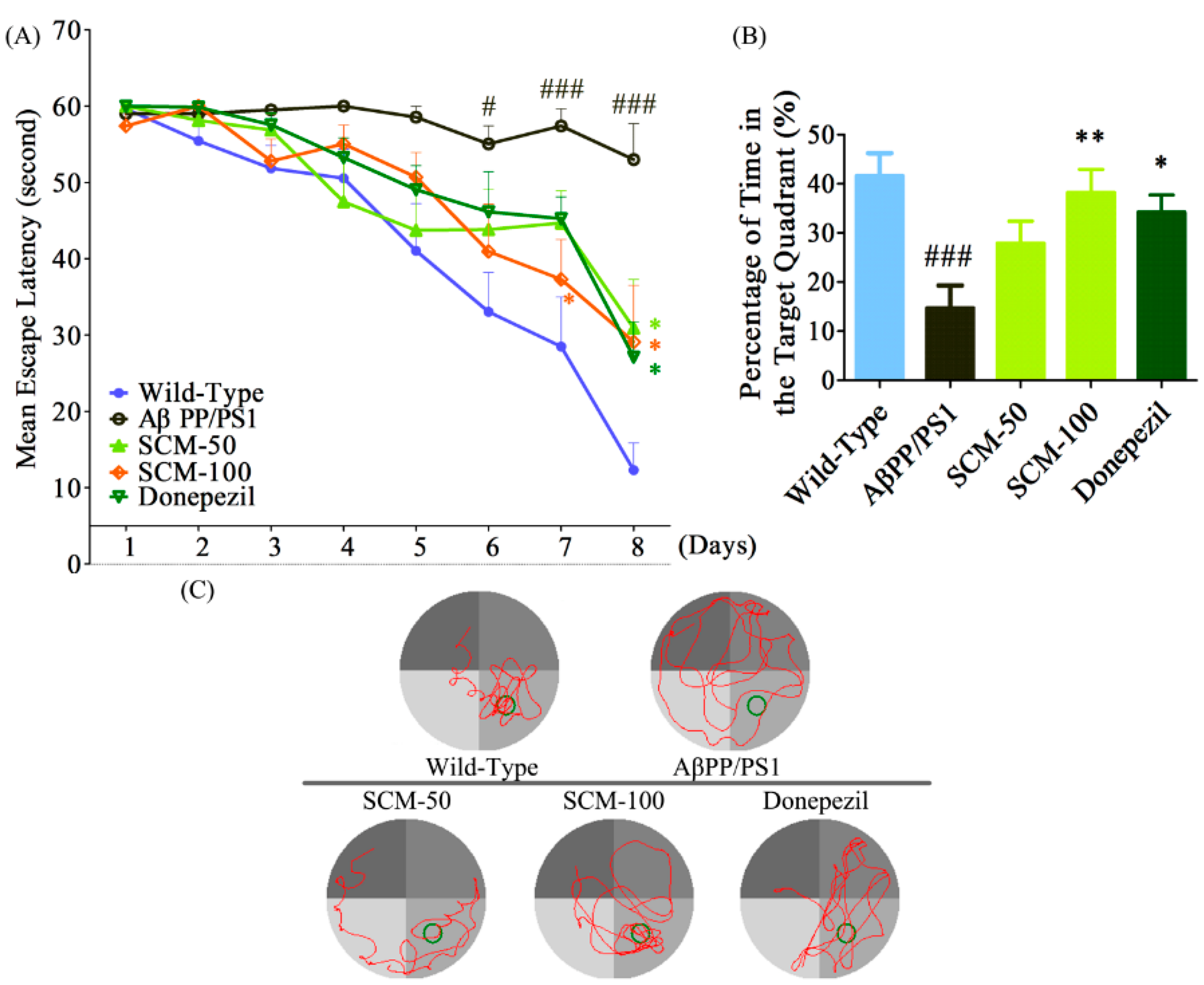
2.2. SCM-198 Alleviated Spatial Memory Deficits in AβPP/PS1 Mice in MWM Test
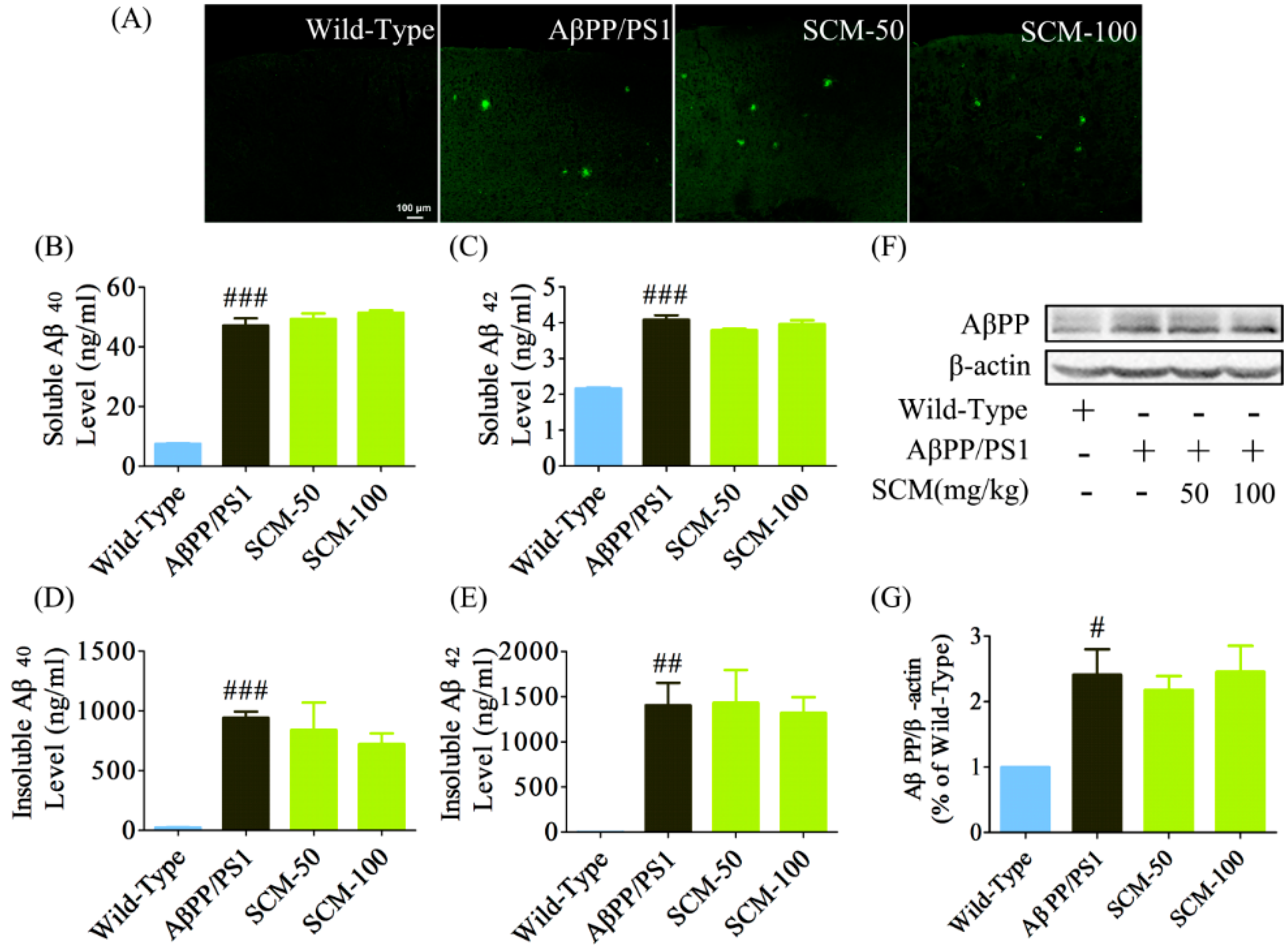
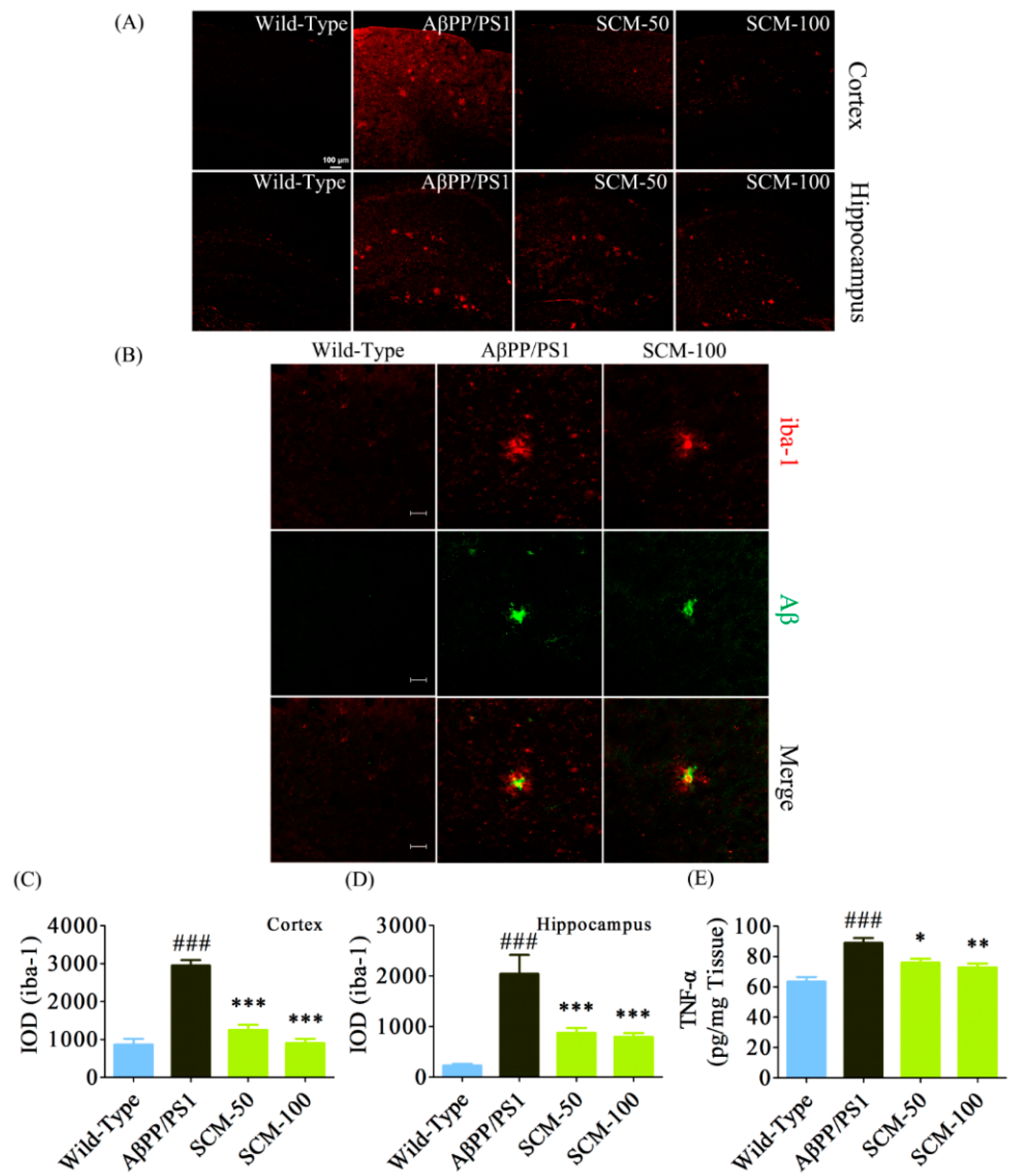
2.3. Long-Term SCM-198 Treatment Did Not Affect Aβ Burden

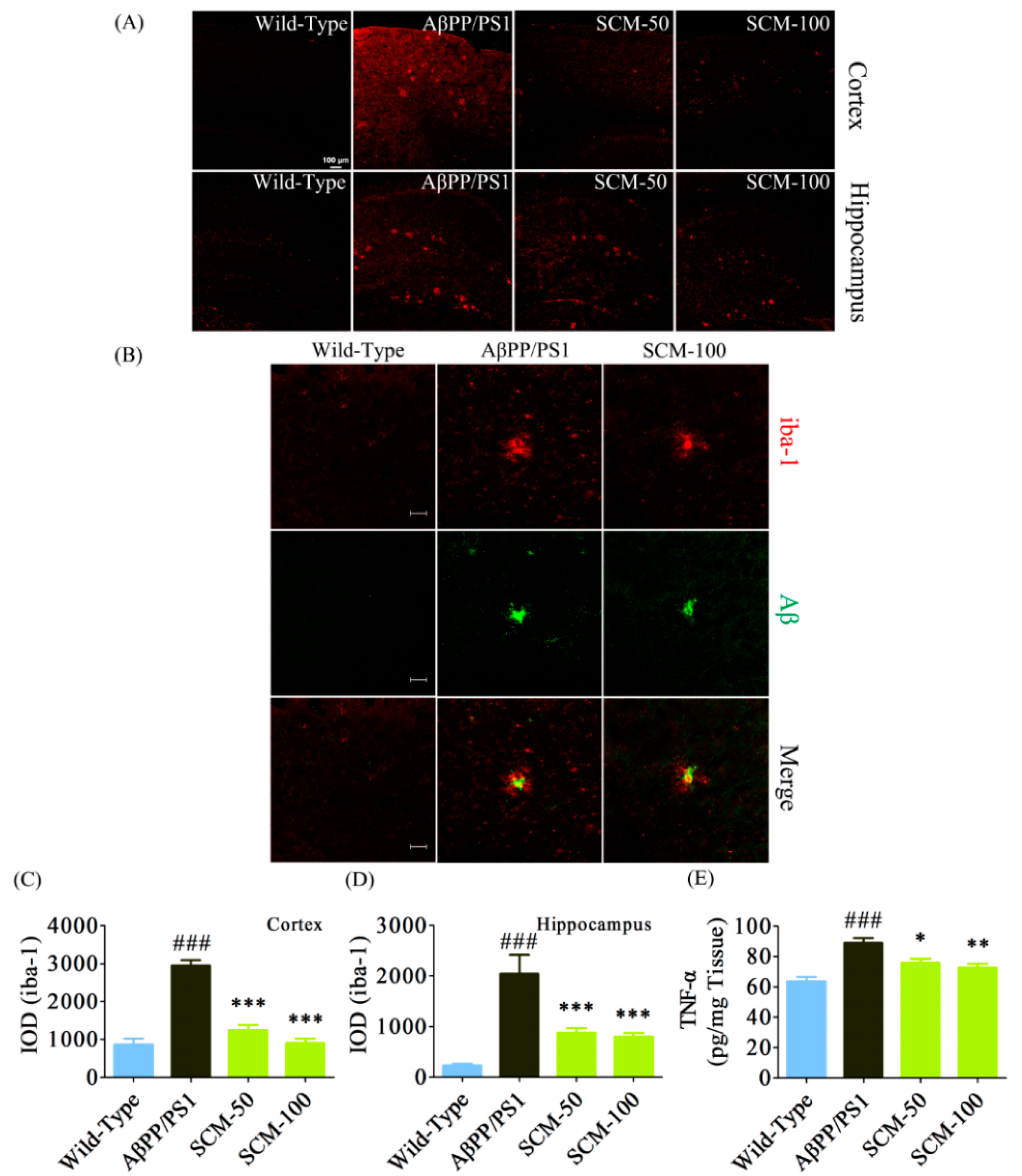
2.4. SCM-198 Alleviated Excessive Microglial Activation and Decreased TNF-α Level in AβPP/PS1 Mice
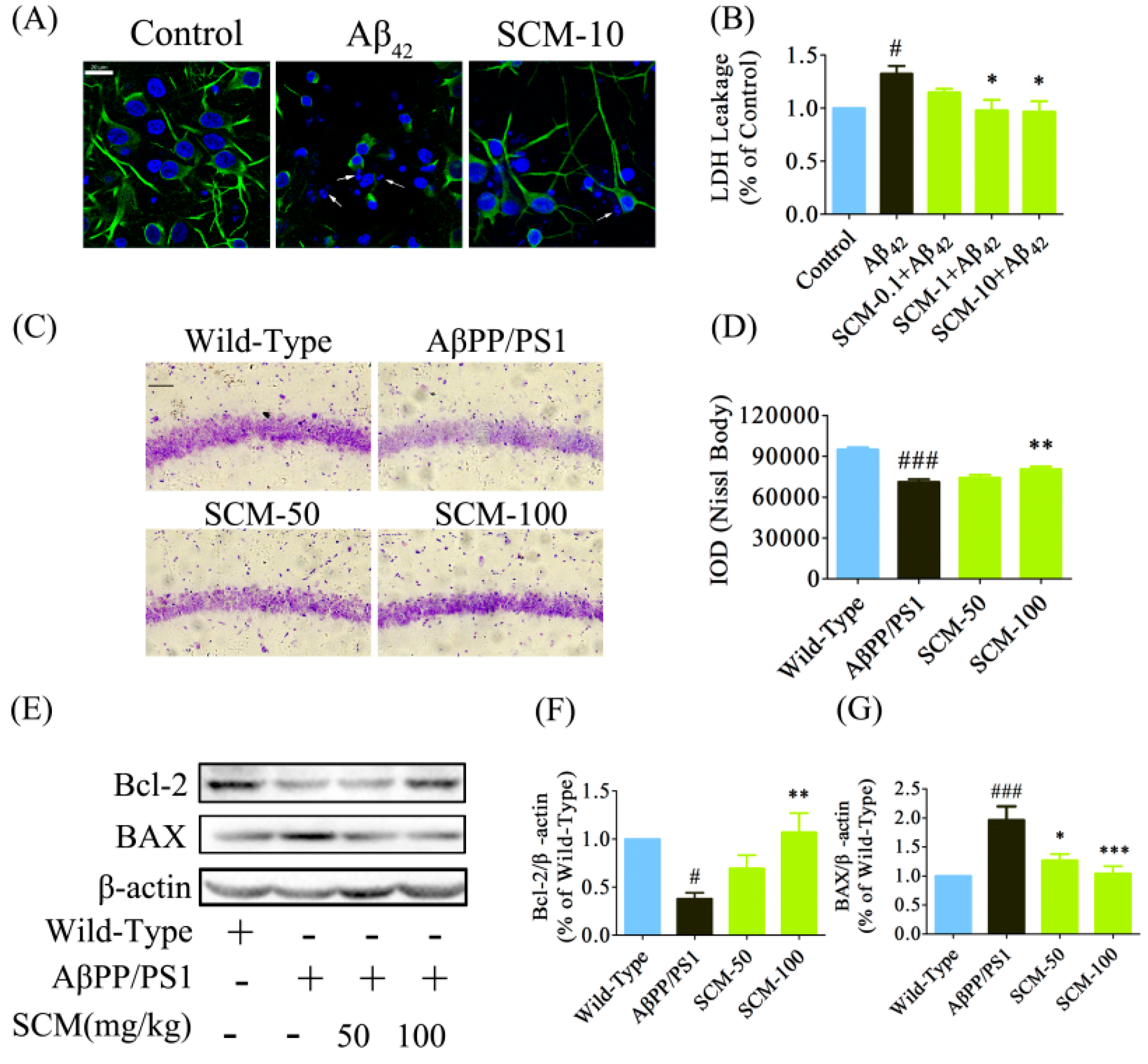
2.5. SCM-198 Promoted Neuronal Survival both in Vitro and in Vivo
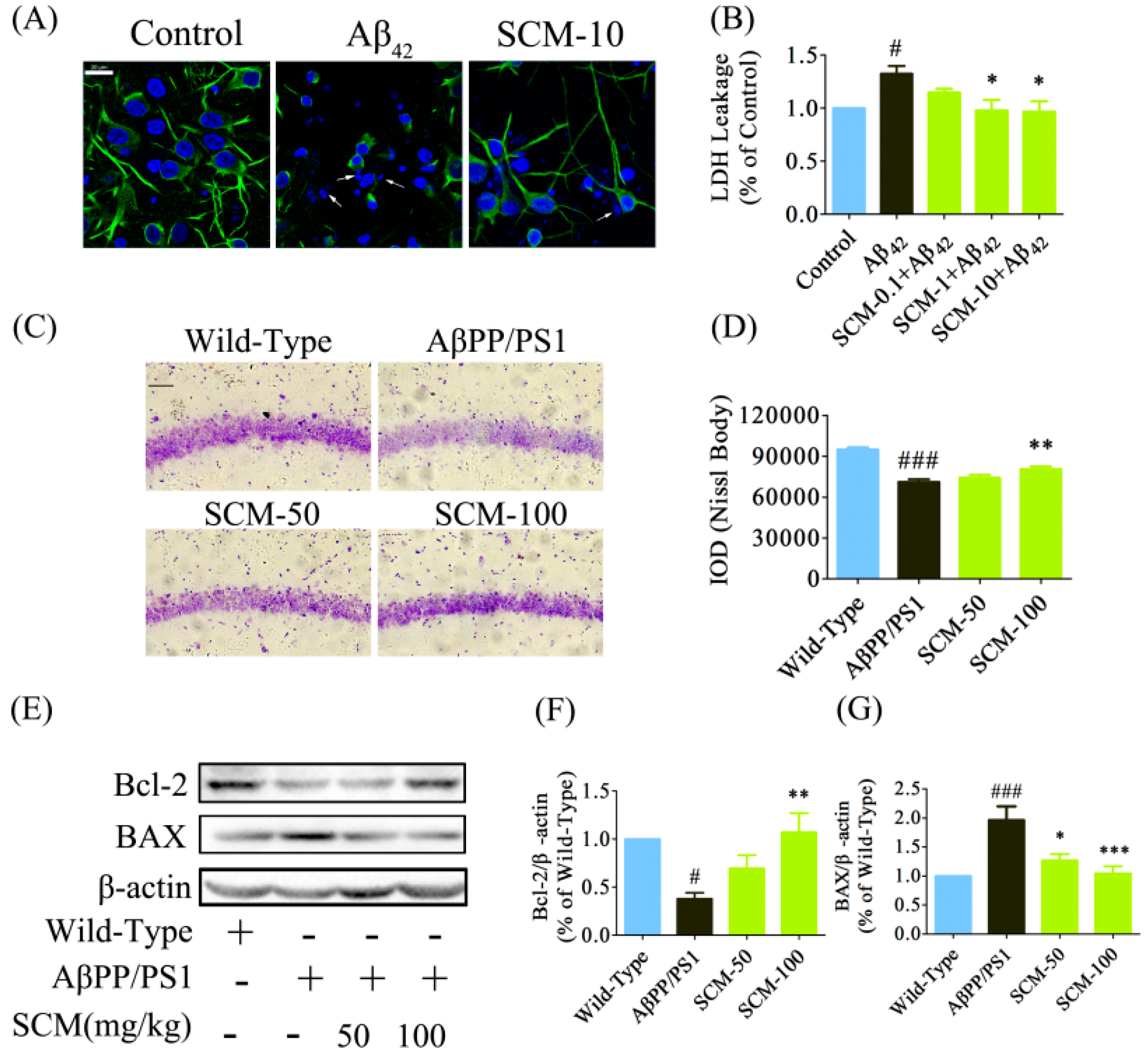
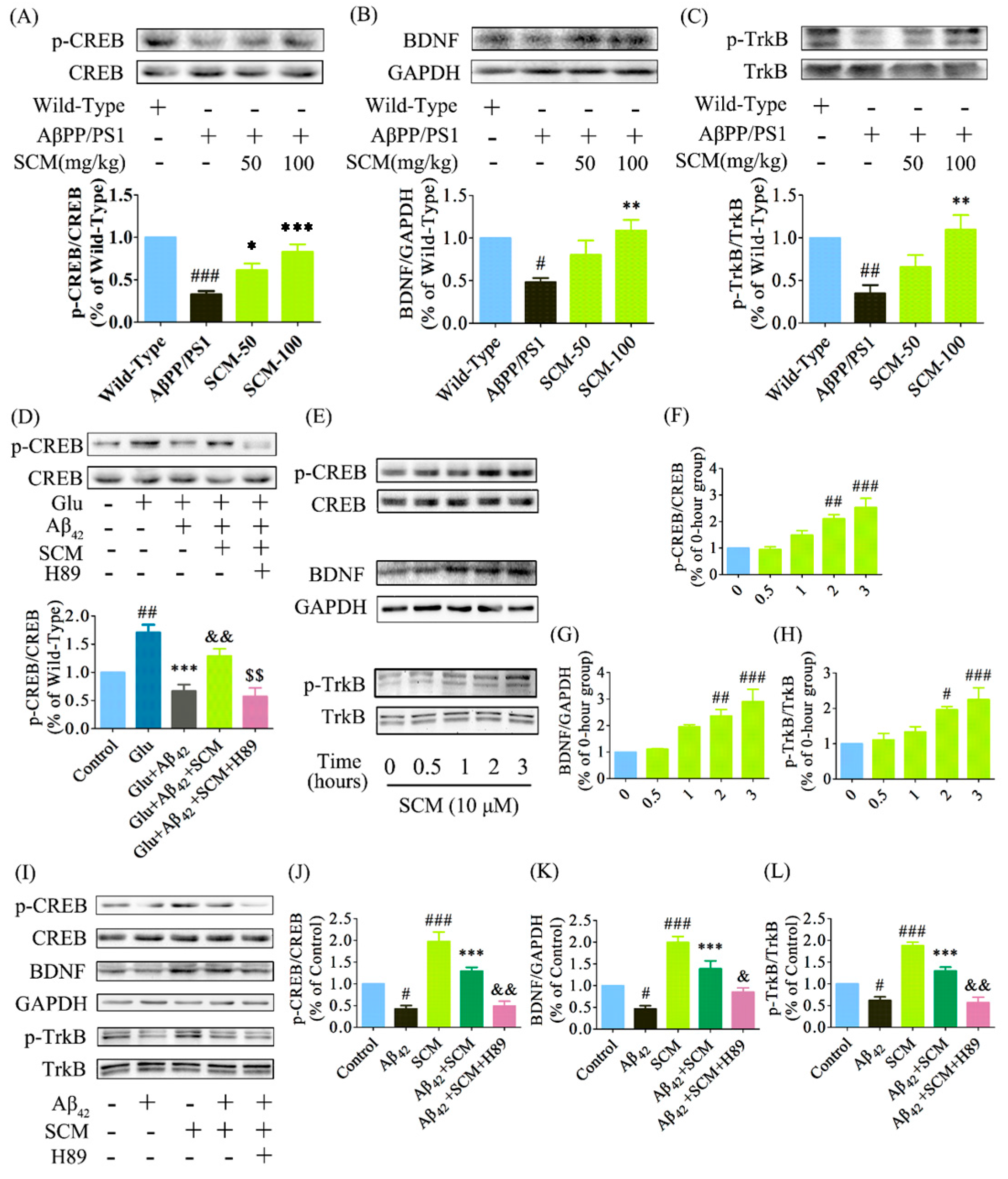
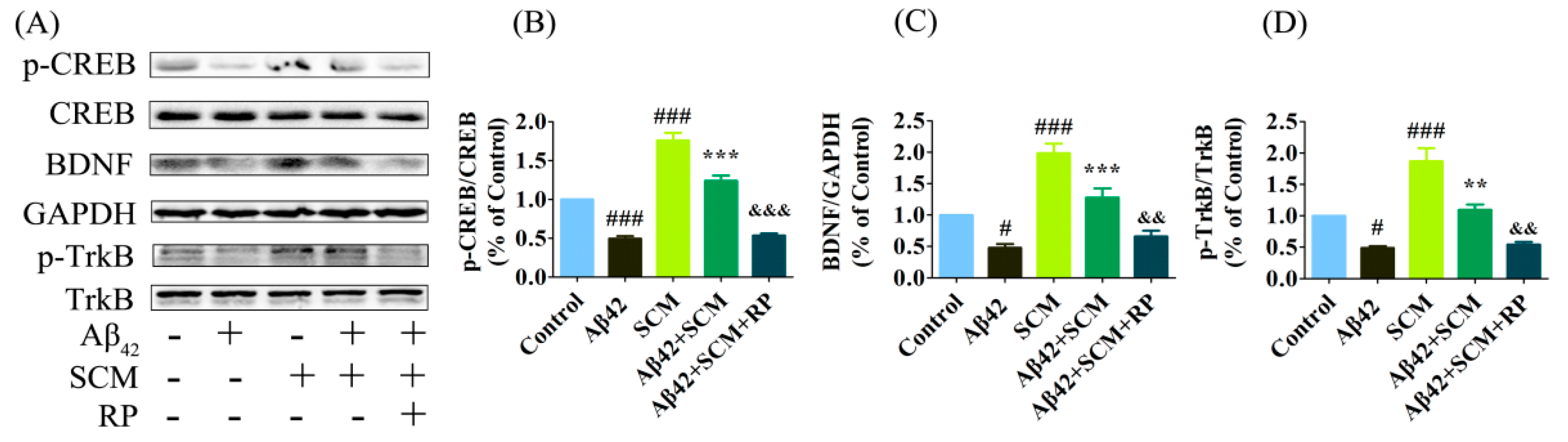
2.6. SCM-198 Enhanced CREB/BDNF/TrkB Signaling both In Vivo and In Vitro
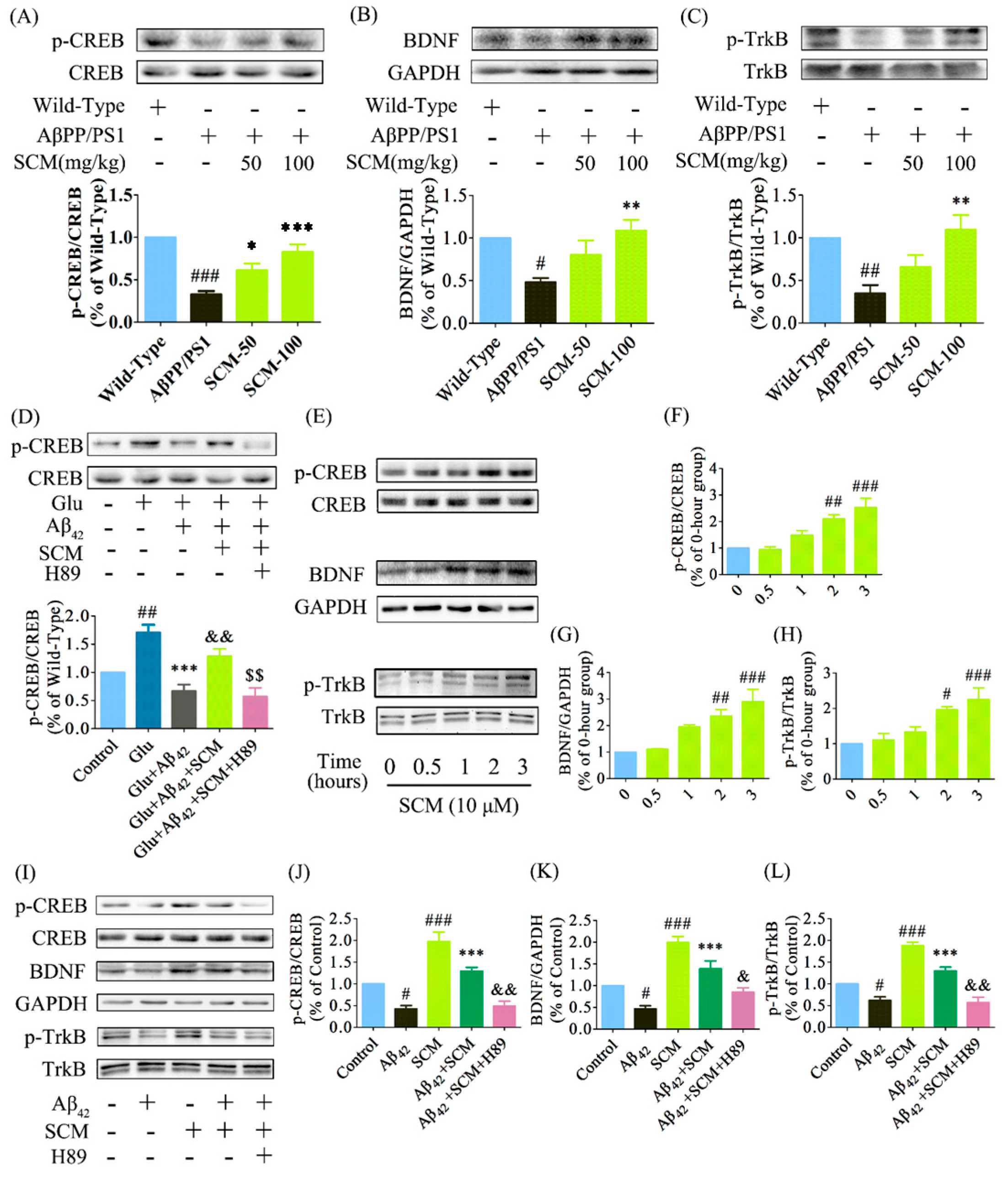
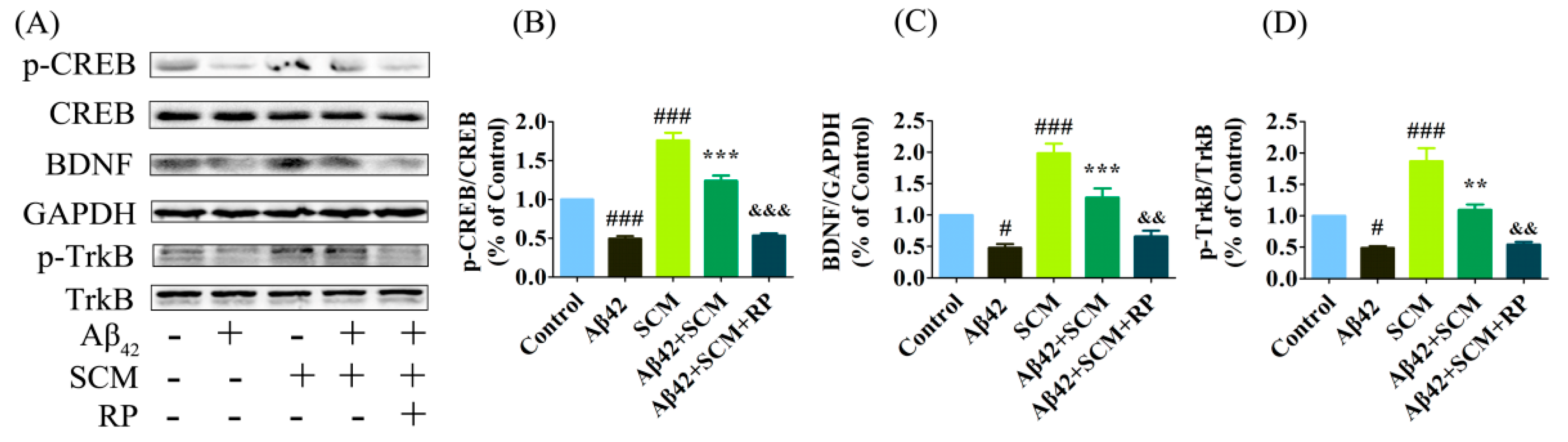
3. Discussion
4. Experimental Section
4.1. Regents
4.2. Animals and Drug Administration
4.3. Novel Object Recognition (NOR) Test
4.4. Morris Water Maze (MWM) Test
4.5. Quantification of Aβ40 and Aβ42 by Enzyme-Linked Immunosorbent Assay (ELISA)
4.6. Western Blot Analysis
4.7. Fluorescent Immunohistochemistry (IHC) and Nissl Staining
4.8. Measurements of Tumor Necrosis Factor-α (TNF-α) and Lactate Dehydrogenase (LDH) Activity
4.9. Aβ42 Preparation, Primary Cortical Neuron Culture and in Vitro Drug Treatment
4.10. Statistical Analysis
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Huang, Y.; Mucke, L. Alzheimer mechanisms and therapeutic strategies. Cell 2012, 148, 1204–1222. [Google Scholar]
- Raina, P.; Santaguida, P.; Ismaila, A.; Patterson, C.; Cowan, D.; Levine, M.; Booker, L.; Oremus, M. Effectiveness of cholinesterase inhibitors and memantine for treating dementia: Evidence review for a clinical practice guideline. Ann Intern. Med. 2008, 148, 379–385. [Google Scholar]
- Doody, R.; Raman, R.; Farlow, M.; Iwatsubo, T.; Vellas, B.; Joffe, S.; Kieburtz, K.; He, F.; Sun, X.; Thomas, R.; et al. A phase 3 trial of semagacestat for treatment of alzheimer’s disease. N. Engl. J. Med. 2013, 369, 341–350. [Google Scholar]
- Doody, R.; Thomas, R.; Farlow, M.; Iwatsubo, T.; Vellas, B.; Joffe, S.; Kieburtz, K.; Raman, R.; Sun, X.; Aisen, P.; et al. Phase 3 trials of solanezumab for mild-to-moderate alzheimer’s disease. N. Engl. J. Med. 2014, 370, 311–321. [Google Scholar] [PubMed]
- Salloway, S.; Sperling, R.; Fox, N.; Blennow, K.; Klunk, W.; Raskind, M.; Sabbagh, M.; Honig, L.; Porsteinsson, A.; Ferris, S.; et al. Two phase 3 trials of bapineuzumab in mild-to-moderate alzheimer’ disease. N. Engl. J. Med. 2014, 370, 322–333. [Google Scholar] [PubMed]
- Malm, T.; Iivonen, H.; Goldsteins, G.; Keksa-Goldsteine, V.; Ahtoniemi, T.; Kanninen, K.; Salminen, A.; Auriola, S.; van Groen, T.; Tanila, H.; et al. Pyrrolidine dithiocarbamate activates akt and improves spatial learning in app/ps1 mice without affecting beta-amyloid burden. J. Neurosci. 2007, 27, 3712–3721. [Google Scholar] [PubMed]
- Dodart, J.; Bales, K.; Gannon, K.; Greene, S.; DeMattos, R.; Mathis, C.; DeLong, C.; Wu, S.; Wu, X.; Holtzman, D.; et al. Immunization reverses memory deficits without reducing brain a beta burden in alzheimer’s disease model. Nat. Neurosci. 2002, 5, 452–457. [Google Scholar] [PubMed]
- Snyder, E.; Nong, Y.; Almeida, C.; Paul, S.; Moran, T.; Choi, E.; Nairn, A.; Salter, M.; Lombroso, P.; Gouras, G.; et al. Regulation of nmda receptor trafficking by amyloid-beta. Nat. Neurosci. 2005, 8, 1051–1058. [Google Scholar] [PubMed]
- Saura, C.; Valero, J. The role of creb signaling in alzheimer’s disease and other cognitive disorders. Rev. Neurosci. 2011, 22, 153–169. [Google Scholar] [PubMed]
- Vitolo, O.; Sant'Angelo, A.; Costanzo, V.; Battaglia, F.; Arancio, O.; Shelanski, M. Amyloid beta-peptide inhibition of the pka/creb pathway and long-term potentiation: Reversibility by drugs that enhance camp signaling. Proc. Natl. Acad. Sci. USA 2002, 99, 13217–13221. [Google Scholar] [PubMed]
- Bollen, E.; Vanmierlo, T.; Akkerman, S.; Wouters, C.; Steinbusch, H.M.; Prickaerts, J. 7,8-dihydroxyflavone improves memory consolidation processes in rats and mice. Behav. Brain Res. 2013, 257, 8–12. [Google Scholar] [PubMed]
- Meng, C.; He, Z.; Xing, D. Low-level laser therapy rescues dendrite atrophy via upregulating bdnf expression: Implications for alzheimer’s disease. J. Neurosci. 2013, 33, 13505–13517. [Google Scholar] [PubMed]
- Liu, X.; Pan, L.; Deng, H.; Xiong, Q.; Wu, D.; Huang, G.; Gong, Q.; Zhu, Y. Leonurine (scm-198) attenuates myocardial fibrotic response via inhibition of nadph oxidase 4. Free Radic. Biol. Med. 2013, 54, 93–104. [Google Scholar] [PubMed]
- Loh, K.; Qi, J.; Tan, B.; Liu, X.; Wei, B.; Zhu, Y. Leonurine protects middle cerebral artery occluded rats through antioxidant effect and regulation of mitochondrial function. Stroke 2010, 41, 2661–2668. [Google Scholar] [PubMed]
- Shi, X.; Hong, Z.; Liu, H.; Zhang, Y.; Zhu, Y. Neuroprotective effects of scm198 on 6-hydroxydopamine-induced behavioral deficit in rats and cytotoxicity in neuronal sh-sy5y cells. Neurochem. Int. 2011, 58, 851–860. [Google Scholar] [PubMed]
- Hong, Z.; Shi, X.; Zhu, K.; Wu, T.; Zhu, Y. Scm-198 inhibits microglial overactivation and attenuates a beta(1-40)-induced cognitive impairments in rats via jnk and ne-kappa b pathways. J. Neuroinflamm. 2014, 11, 1–15. [Google Scholar]
- Bevins, R.; Besheer, J. Object recognition in rats and mice: A one-trial non-matching-to-sample learning task to study “recognition memory”. Nat. Protoc. 2006, 1, 1306–1311. [Google Scholar] [PubMed]
- Halassa, M.; Florian, C.; Fellin, T.; Munoz, J.; Lee, S.; Abel, T.; Haydon, P.; Frank, M. Astrocytic modulation of sleep homeostasis and cognitive consequences of sleep loss. Neuron 2009, 61, 213–219. [Google Scholar] [PubMed]
- Trinchese, F.; Liu, S.; Battaglia, F.; Walter, S.; Mathews, P.; Arancio, O. Progressive age-related development of alzheimer-like pathology in app/ps1 mice. Ann. Neurol. 2004, 55, 801–814. [Google Scholar] [PubMed]
- Yiu, A.; Rashid, A.; Josselyn, S. Increasing creb function in the ca1 region of dorsal hippocampus rescues the spatial memory deficits in a mouse model of alzheimer’s disease. Neuropsychopharmacology 2011, 36, 2169–2186. [Google Scholar] [CrossRef] [PubMed]
- Pugazhenthi, S.; Wang, M.; Pham, S.; Sze, C.; Eckman, C. Downregulation of creb expression in alzheimer’s brain and in a beta-treated rat hippocampal neurons. Mol. Neurodegener. 2011, 6, 60. [Google Scholar] [CrossRef] [PubMed]
- Broadbent, N.; Squire, L.; Clark, R. Spatial memory, recognition memory, and the hippocampus. Proc. Natl. Acad. Sci. USA 2004, 101, 14515–14520. [Google Scholar] [CrossRef] [PubMed]
- LaFerla, F.; Green, K.; Oddo, S. Intracellular amyloid-beta in alzheimer’s disease. Nat. Rev. Neurosci. 2007, 8, 499–509. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Thompson, R.; Zhang, H.; Xu, H. App processing in alzheimer’s disease. Mol. Brain 2011, 4, 3. [Google Scholar] [CrossRef] [PubMed]
- Walsh, D.; Selkoe, D. Deciphering the molecular basis of memory failure in alzheimer’s disease. Neuron 2004, 44, 181–193. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto-Sasaki, M.; Ozawa, H.; Saito, T.; Rosler, M.; Riederer, P. Impaired phosphorylation of cyclic amp response element binding protein in the hippocampus of dementia of the alzheimer type. Brain Res. 1999, 824, 300–303. [Google Scholar] [CrossRef]
- Bitner, R.S. Cyclic amp response element-binding protein (creb) phosphorylation: A mechanistic marker in the development of memory enhancing alzheimer’s disease therapeutics. Biochem. Pharmacol. 2012, 83, 705–714. [Google Scholar] [CrossRef] [PubMed]
- Tao, X.; Finkbeiner, S.; Arnold, D.; Shaywitz, A.; Greenberg, M. Ca2+ influx regulates bdnf transcription by a creb family transcription factor-dependent mechanism. Neuron 1998, 20, 709–726. [Google Scholar] [CrossRef]
- Shi, Y.Q.; Huang, T.W.; Chen, L.M.; Pan, X.D.; Zhang, J.; Zhu, Y.G.; Chen, X.C. Ginsenoside rg1 attenuates amyloid-beta content, regulates pka/creb activity, and improves cognitive performance in samp8 mice. J. Alzheimers Dis. 2010, 19, 977–989. [Google Scholar] [PubMed]
- Lu, B.; Nagappan, G.; Guan, X.; Nathan, P.; Wren, P. Bdnf-based synaptic repair as a disease-modifying strategy for neurodegenerative diseases. Nat. Rev. Neurosci. 2013, 14, 401–416. [Google Scholar] [CrossRef] [PubMed]
- Lu, B.; Pang, P.T.; Woo, N.H. The yin and yang of neurotrophin action. Nat. Rev. Neurosci. 2005, 6, 603–614. [Google Scholar] [CrossRef] [PubMed]
- Devi, L.; Ohno, M. 7,8-dihydroxyflavone, a small-molecule trkb agonist, reverses memory deficits and bace1 elevation in a mouse model of alzheimer’s disease. Neuropsychopharmacology 2012, 37, 434–444. [Google Scholar] [CrossRef] [PubMed]
- Peng, S.; Wuu, J.; Mufson, E.J.; Fahnestock, M. Precursor form of brain-derived neurotrophic factor and mature brain-derived neurotrophic factor are decreased in the pre-clinical stages of alzheimer’s disease. J. Neurochem. 2005, 93, 1412–1421. [Google Scholar] [CrossRef] [PubMed]
- Ding, Y.; Qiao, A.; Wang, Z.; Goodwin, J.; Lee, E.; Block, M.; Allsbrook, M.; McDonald, M.; Fan, G. Retinoic acid attenuates beta-amyloid deposition and rescues memory deficits in an alzheimer’s disease transgenic mouse model. J. Neurosci. 2008, 28, 11622–11634. [Google Scholar] [CrossRef] [PubMed]
- Nagakura, A.; Shitaka, Y.; Yarimizu, J.; Matsuoka, N. Characterization of cognitive deficits in a transgenic mouse model of alzheimer’s disease and effects of donepezil and memantine. Eur. J. Pharmacol. 2013, 703, 53–61. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.; Xue, G.; Wang, S.; Zhang, L.; Shi, C.; Xie, X. Novel object recognition as a facile behavior test for evaluating drug effects in a beta pp/ps1 alzheimer’s disease mouse model. J. Alzheimers Dis. 2012, 31, 801–812. [Google Scholar] [PubMed]
- Dong, H.; Yuede, C.; Coughlan, C.; Murphy, K.; Csernansky, J. Effects of donepezil on amyloid-beta and synapse density in the tg2576 mouse model of alzheimer’s disease. Brain Res. 2009, 1303, 169–178. [Google Scholar] [CrossRef] [PubMed]
- Su, D.; Zhao, Y.; Wang, B.; Xu, H.; Li, W.; Chen, J.; Wang, X. Isoflurane-induced spatial memory impairment in mice is prevented by the acetylcholinesterase inhibitor donepezil. PLos ONE 2011, 6, e27632. [Google Scholar] [CrossRef] [PubMed]
- Morishima-Kawashima, M.; Oshima, N.; Ogata, H.; Yamaguchi, H.; Yoshimura, M.; Sugihara, S.; Ihara, Y. Effect of apolipoprotein e allele epsilon 4 on the initial phase of amyloid beta-protein accumulation in the human brain. Am. J. Pathol. 2000, 157, 2093–2099. [Google Scholar] [CrossRef]
- Goodrich-Hunsaker, N.J.; Hunsaker, M.R.; Kesner, R.P. Dissociating the role of the parietal cortex and dorsal hippocampus for spatial information processing. Behav. Neurosci. 2005, 119, 1307–1315. [Google Scholar] [CrossRef] [PubMed]
- Foundas, A.L.; Leonard, C.M.; Mahoney, S.M.; Agee, O.F.; Heilman, K.M. Atrophy of the hippocampus, parietal cortex, and insula in alzheimer’s disease: A volumetric magnetic resonance imaging study. Neuropsychiatry Neuropsychol. Behav. Neurol. 1997, 10, 81–89. [Google Scholar] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hong, Z.-Y.; Yu, S.-S.; Wang, Z.-J.; Zhu, Y.-Z. SCM-198 Ameliorates Cognitive Deficits, Promotes Neuronal Survival and Enhances CREB/BDNF/TrkB Signaling without Affecting Aβ Burden in AβPP/PS1 Mice. Int. J. Mol. Sci. 2015, 16, 18544-18563. https://doi.org/10.3390/ijms160818544
Hong Z-Y, Yu S-S, Wang Z-J, Zhu Y-Z. SCM-198 Ameliorates Cognitive Deficits, Promotes Neuronal Survival and Enhances CREB/BDNF/TrkB Signaling without Affecting Aβ Burden in AβPP/PS1 Mice. International Journal of Molecular Sciences. 2015; 16(8):18544-18563. https://doi.org/10.3390/ijms160818544
Chicago/Turabian StyleHong, Zhen-Yi, Shuang-Shuang Yu, Zhi-Jun Wang, and Yi-Zhun Zhu. 2015. "SCM-198 Ameliorates Cognitive Deficits, Promotes Neuronal Survival and Enhances CREB/BDNF/TrkB Signaling without Affecting Aβ Burden in AβPP/PS1 Mice" International Journal of Molecular Sciences 16, no. 8: 18544-18563. https://doi.org/10.3390/ijms160818544