
Trehalose Analogues: Latest Insights in Properties and Biocatalytic Production

Abstract
:
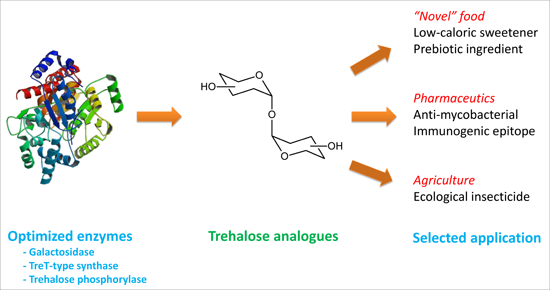{kind=link}
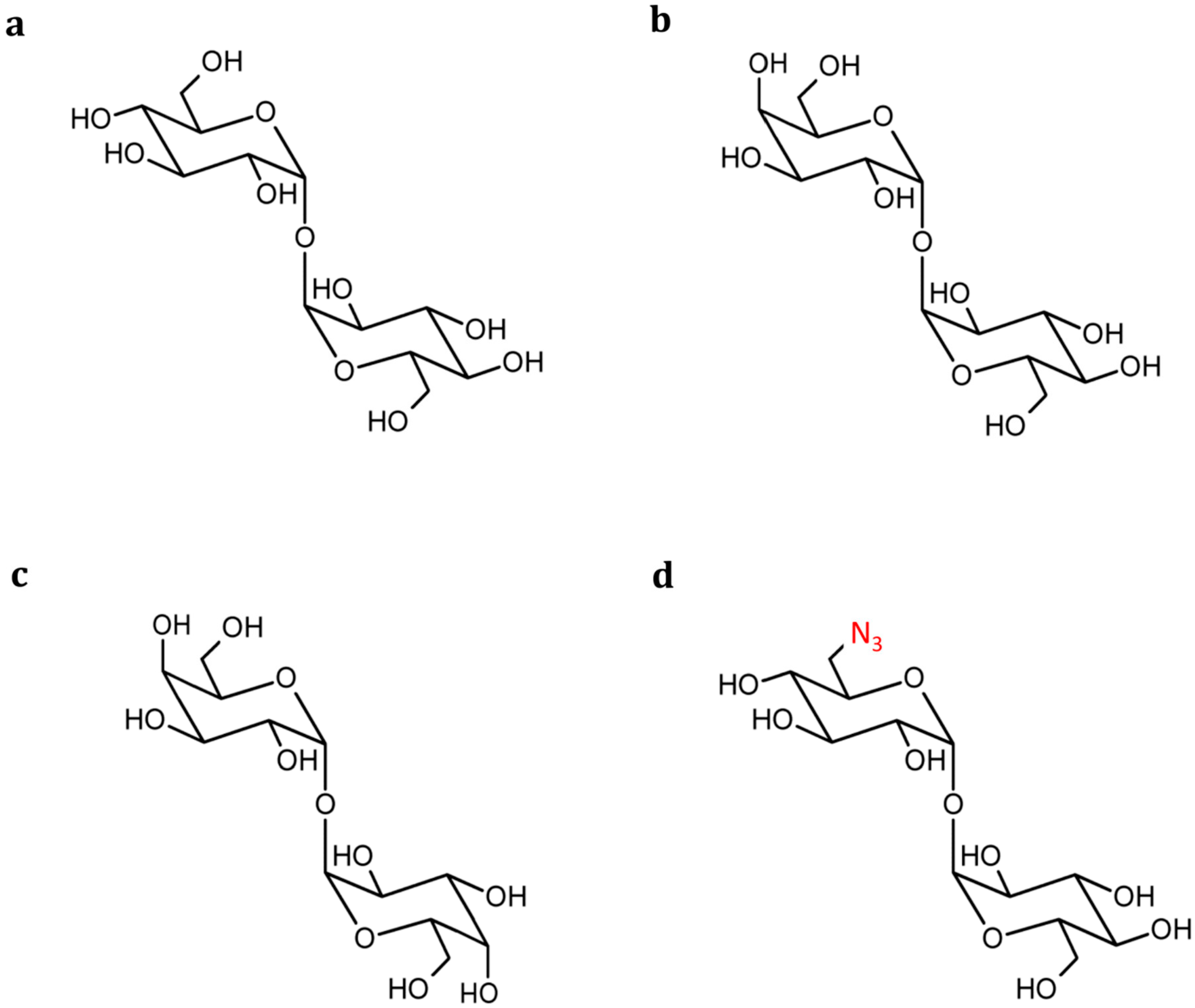{kind=link}
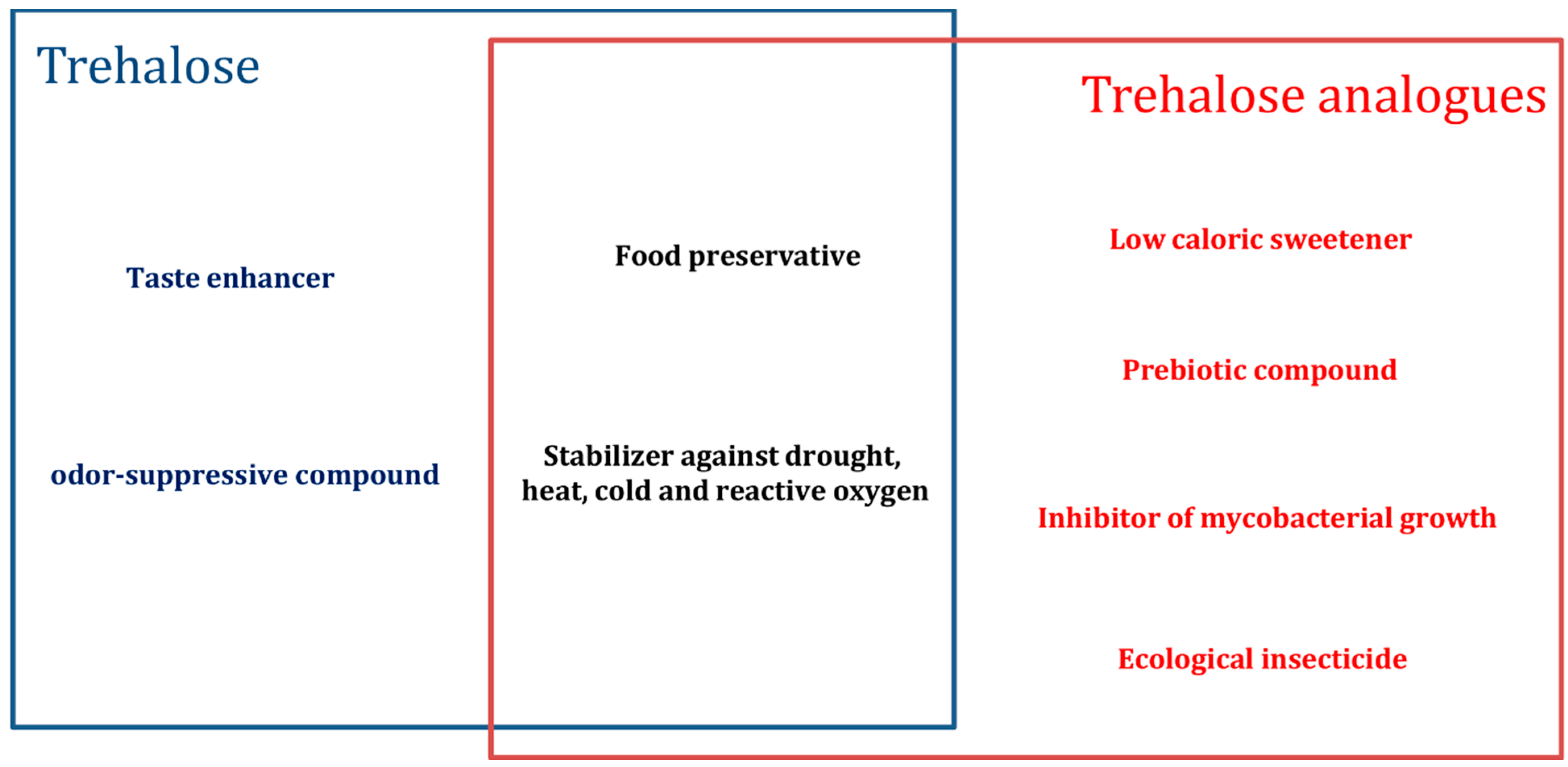{kind=link}
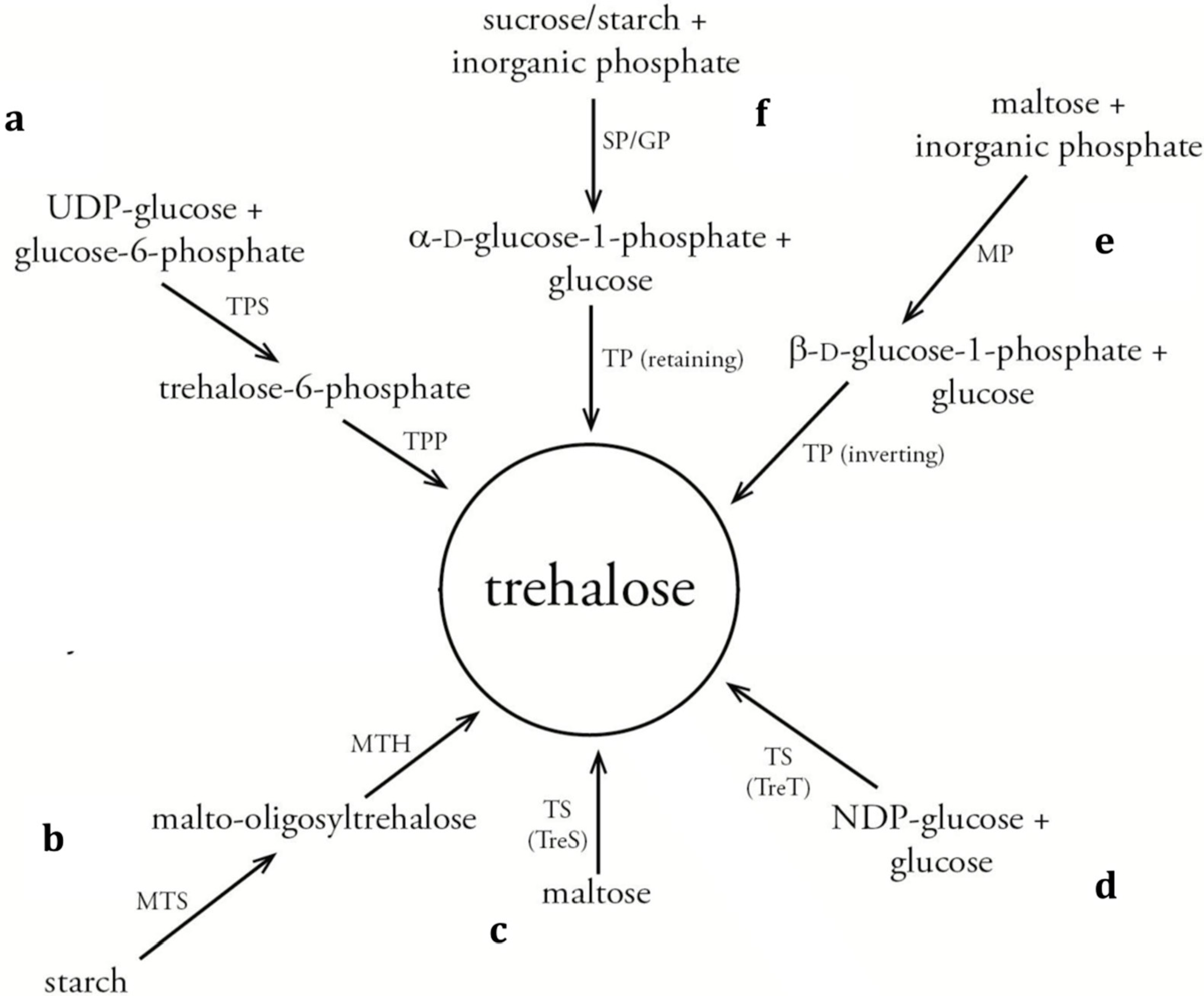{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction

2. Trehalose and Its Analogues: Diversity, Function and Applications

3. Biocatalytic Synthesis Routes for Trehalose Analogues

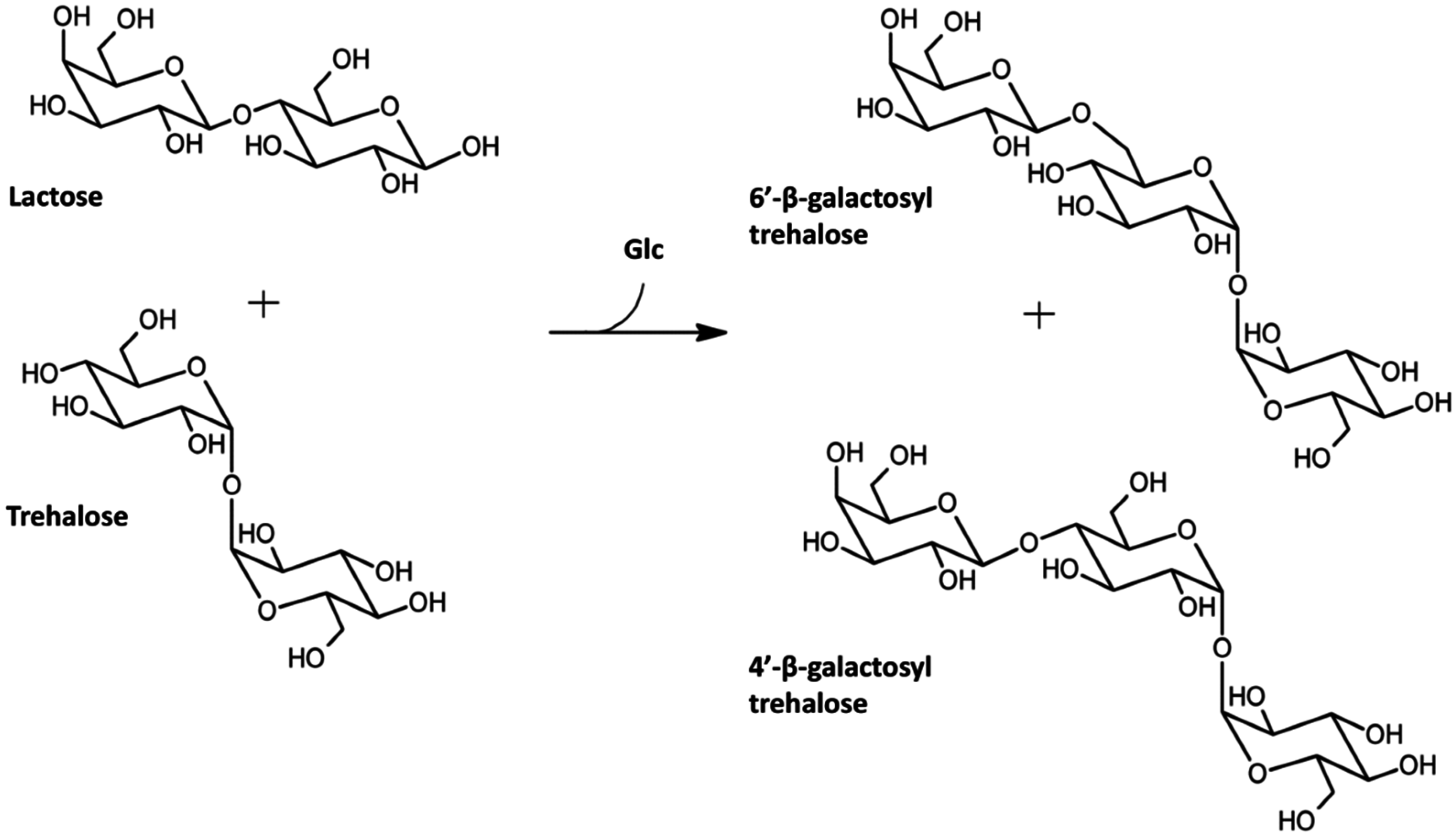
3.1. Galactosidase-Catalyzed Production

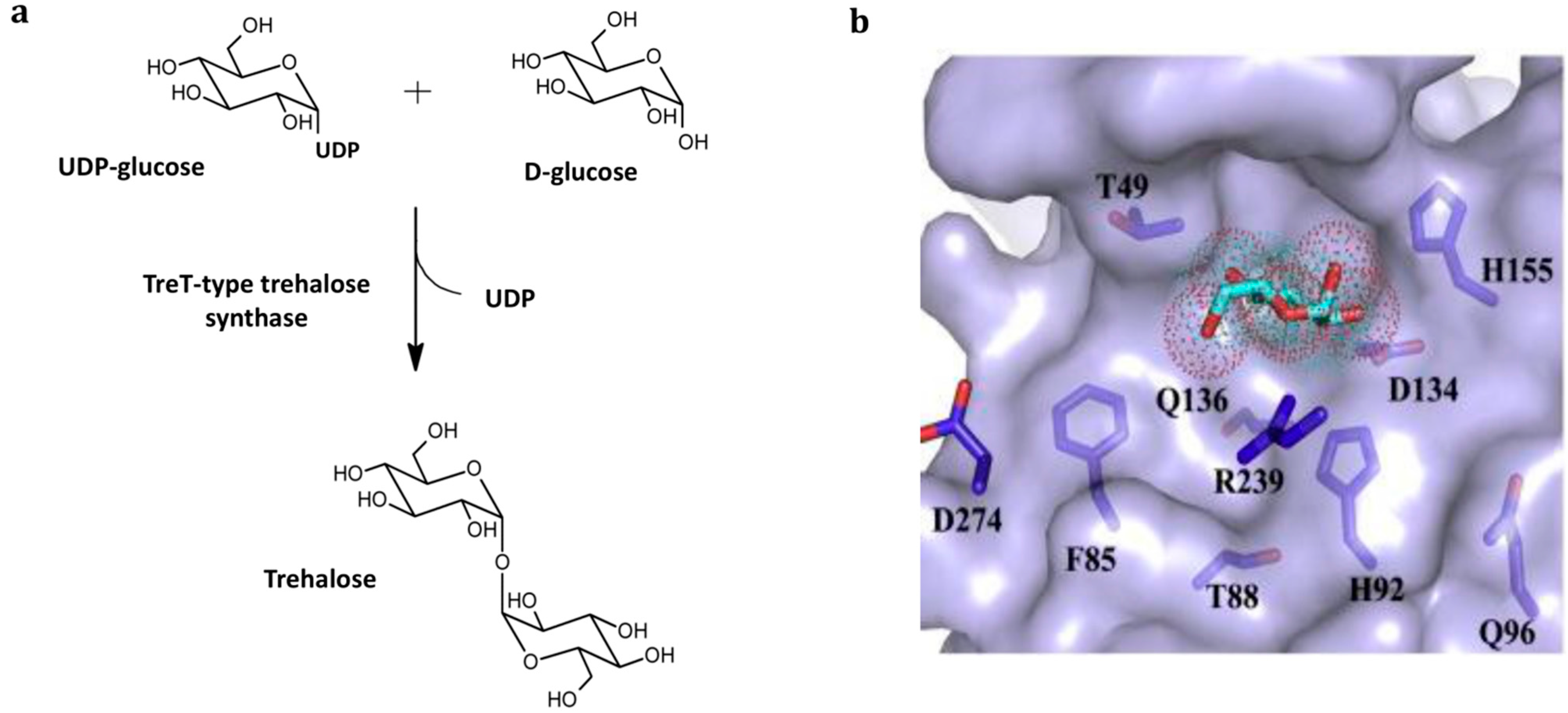
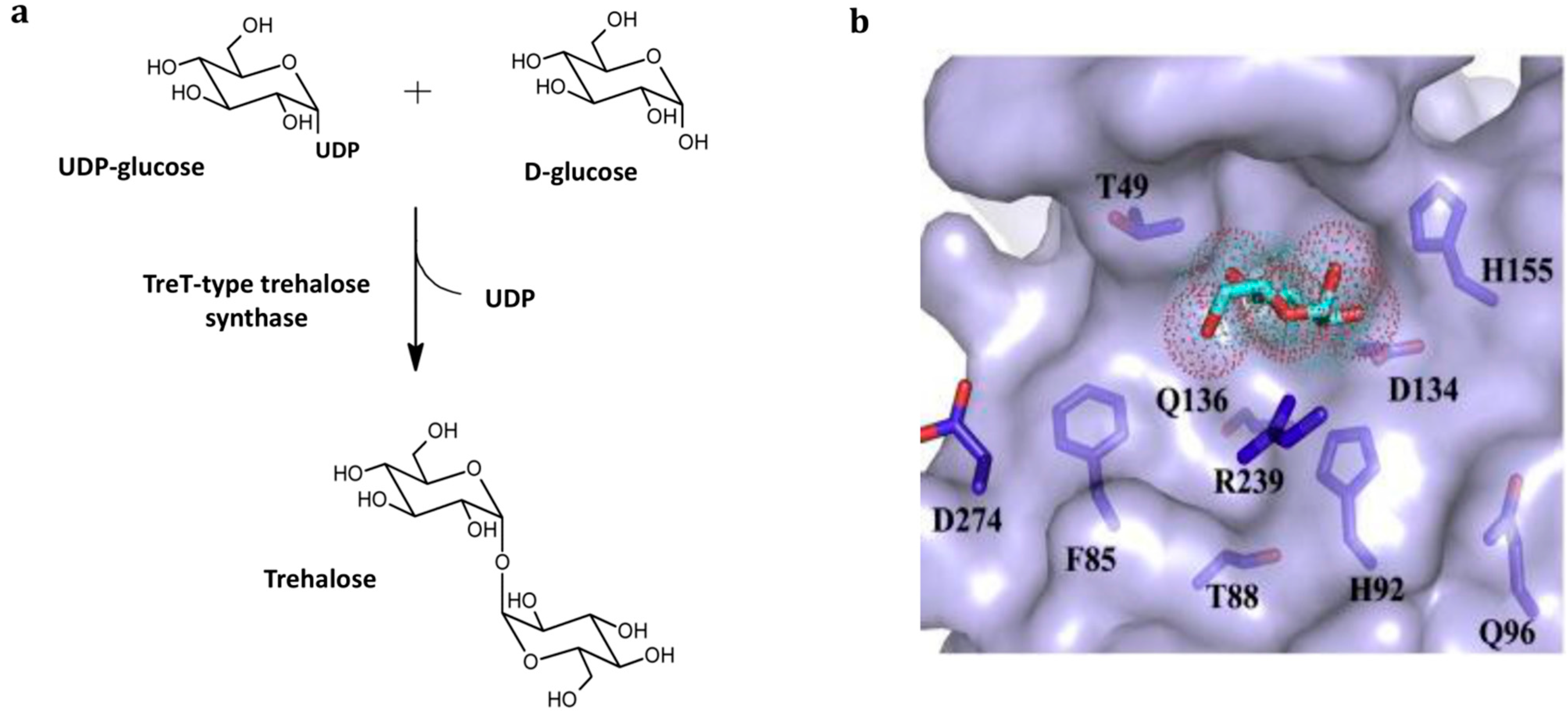
3.2. TreT-Type Trehalose Synthase-Catalyzed Production
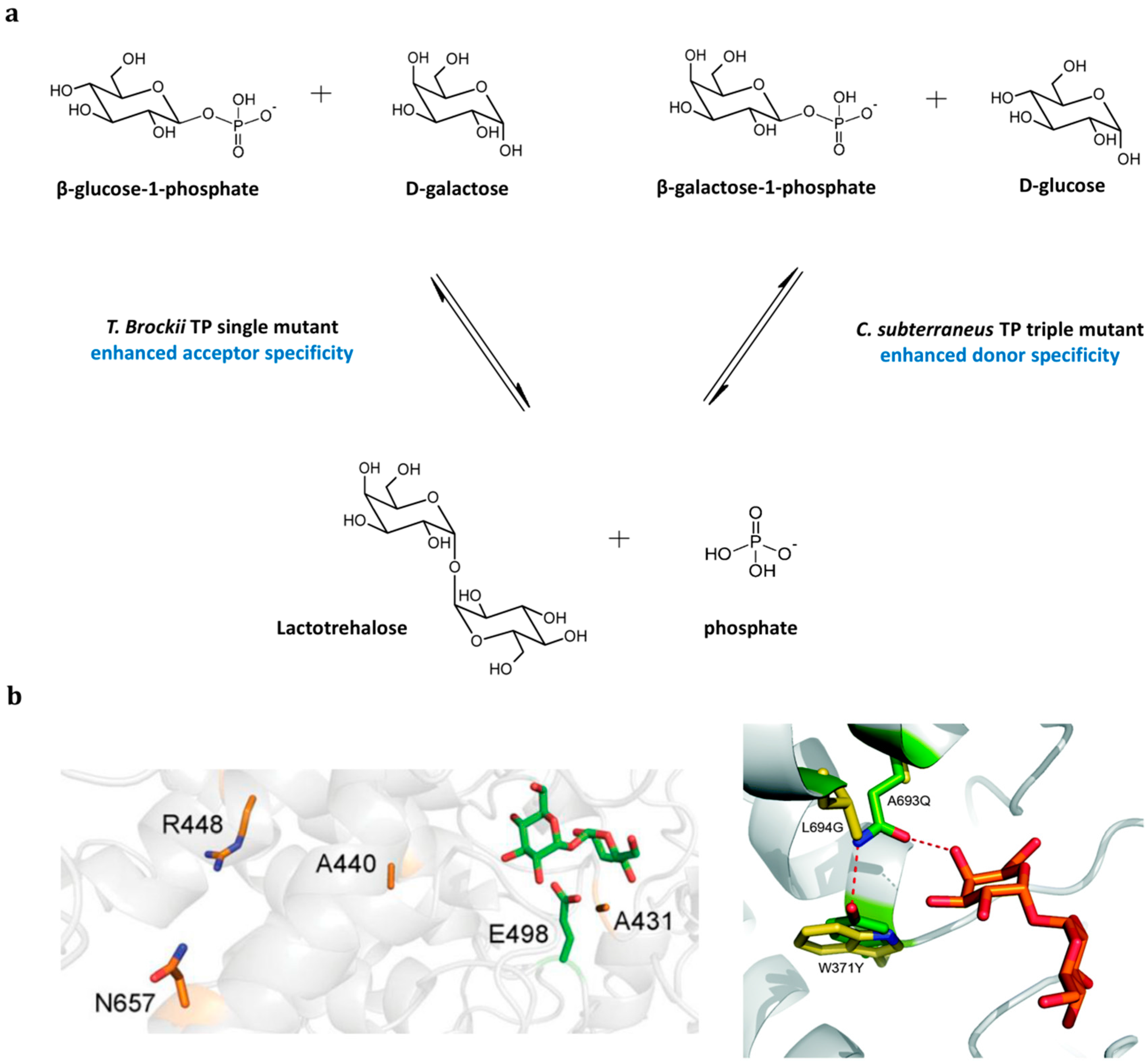
3.3. Inverting Trehalose Phosphorylase-Catalyzed Production
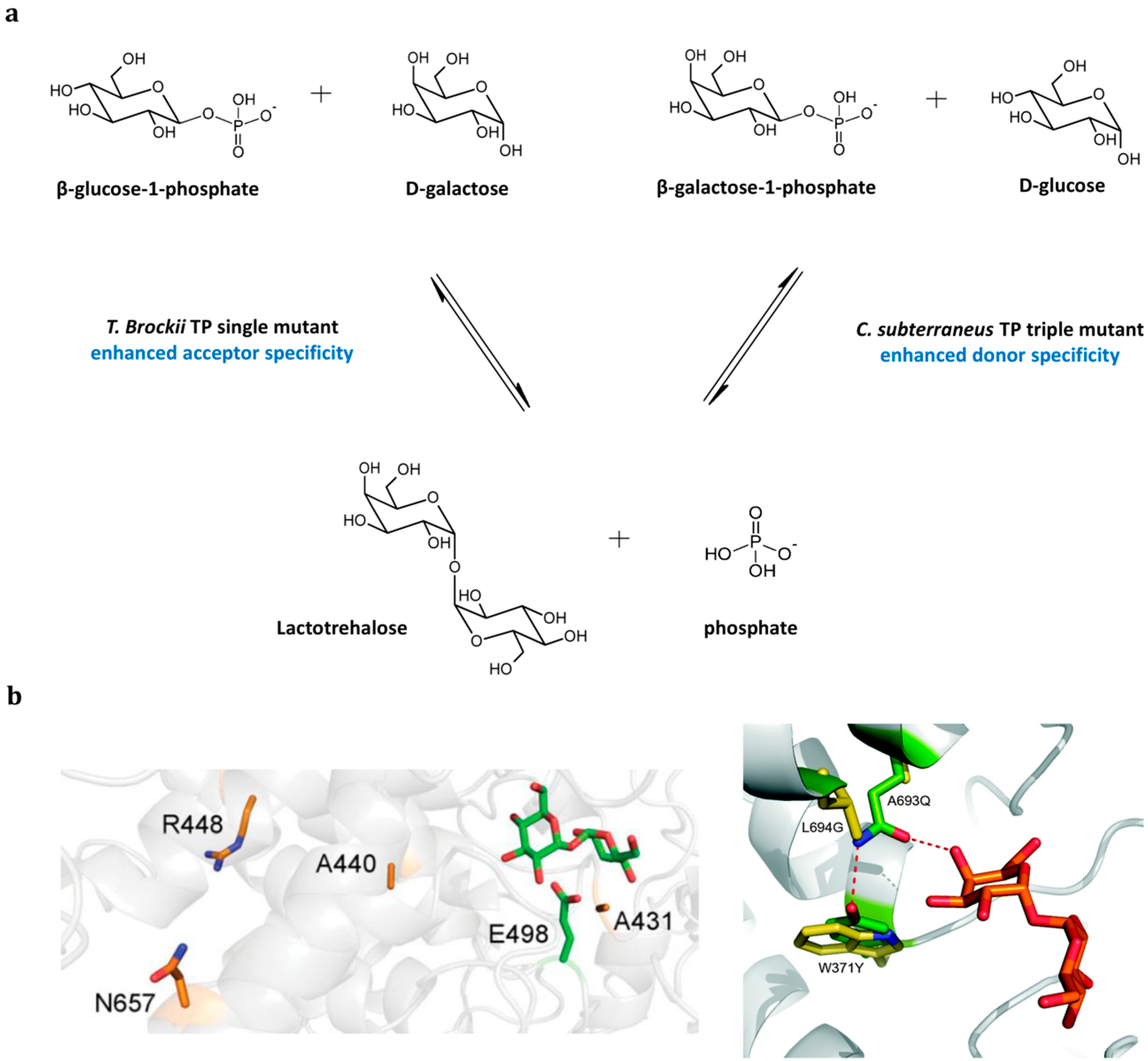
3.4. Retaining Trehalose Phosphorylase-Catalyzed Production
4. Conclusions, Outlook and Future Perspectives
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Higashiyama, T. Novel functions and applications of trehalose. Pure Appl. Chem. 2002, 74, 1263–1269. [Google Scholar]
- Nakakuki, T. Present status and future prospects of functional oligosaccharide development in Japan. J. Appl. Glycosci. 2005, 52, 267–271. [Google Scholar] [CrossRef]
- Teramoto, N.; Sachinvala, N.D.; Shibata, M. Trehalose and trehalose-based polymers for environmentally benign, biocompatible and bioactive materials. Molecules 2008, 13, 1773–1816. [Google Scholar] [CrossRef] [PubMed]
- LinGoerke, J.L.; Robbins, D.J.; Burczak, J.D. PCR-based random mutagenesis using manganese and reduced dNTP concentration. Biotechniques 1997, 23, 409–412. [Google Scholar]
- Albertorio, F.; Chapa, V.A.; Chen, X.; Diaz, A.J.; Cremer, P.S. The α,α-(1→1) linkage of trehalose is key to anhydrobiotic preservation. J. Am. Chem. Soc. 2007, 129, 10567–10574. [Google Scholar] [CrossRef] [PubMed]
- Urbanek, B.L.; Wing, D.C.; Haislop, K.S.; Hamel, C.J.; Kalscheuer, R.; Woodruff, P.J.; Swarts, B.M. Chemoenzymatic synthesis of trehalose analogues: Rapid access to chemical probes for investigating mycobacteria. ChemBioChem 2014, 15, 2066–2070. [Google Scholar] [CrossRef] [PubMed]
- Maruta, K.; Nakada, T.; Kubota, M.; Chaen, H.; Sugimoto, T.; Kurimoto, M.; Tsujisaka, Y. Formation of trehalose from maltooligosaccharides by a novel enzymatic system. Biosci. Biotechnol. Biochem. 1995, 59, 1829–1834. [Google Scholar] [CrossRef] [PubMed]
- Wyatt, T.T.; Gerwig, G.J.; Kamerling, J.P.; Wösten, H.A.; Dijksterhuis, J. Structural analysis of novel trehalose-based oligosaccharides from extremely stress-tolerant ascospores of Neosartorya fischeri (Aspergillus fischeri). Carbohydr. Res. 2015, 411, 49–55. [Google Scholar] [CrossRef] [PubMed]
- Babu, R.S.; O’Doherty, G.A. Palladium-catalyzed glycosylation reaction: De-novo synthesis of trehalose analogues. J. Carbohydr. Chem. 2005, 24, 169–177. [Google Scholar] [CrossRef]
- Umezawa, S.; Tatsuta, K.; Muto, R. Synthesis of trehalosamine. J. Antibiot. 1967, 20, 388. [Google Scholar] [PubMed]
- Uramoto, M.; Otake, N.; Yonehara, H. Mannosyl glucosaminide: A new antibiotic. J. Antibiot. 1967, 20, 236. [Google Scholar] [PubMed]
- Elbein, A.D.; Pan, Y.T.; Pastuszak, I.; Carroll, D. New insights on trehalose: A multifunctional molecule. Glycobiology 2003, 13, 17–27. [Google Scholar] [CrossRef] [PubMed]
- Singer, M.A.; Lindquist, S. Multiple effects of trehalose on protein folding in vitro and in vivo. Mol. Cell 1998, 1, 639–648. [Google Scholar] [CrossRef]
- Roser, B. Trehalose drying—A novel replacement for freeze-drying. Biopharm. Technol. Bus. Biopharm. 1991, 4, 47–52. [Google Scholar]
- Portmann, M.O.; Birch, G. Sweet taste and solution properties of α,α-trehalose. J. Sci. Food Agric. 1995, 69, 275–281. [Google Scholar] [CrossRef]
- Ohtake, S.; Wang, Y.J. Trehalose: Current use and future applications. J. Pharm. Sci. 2011, 100, 2020–2053. [Google Scholar] [CrossRef] [PubMed]
- Roser, B. Trehalose, a new approach to premium dried foods. Trends Food Sci. Technol. 1991, 2, 166–169. [Google Scholar] [CrossRef]
- Tanaka, K. Development of Treha(R) and its properties. Food Ind. 2009, 52, 45–51. [Google Scholar]
- Minami, Y.; Yazawa, K.; Nakamura, K.; Tamura, Z. Selectivity and efficiency of utilization of galactosyl-oligosaccharides by bifidobacteria. Chem. Pharm. Bull. 1985, 33, 710–714. [Google Scholar] [CrossRef] [PubMed]
- Miyachi, A.; Dohi, H.; Neri, P.; Mori, H.; Uzawa, H.; Seto, Y.; Nishida, Y. Multivalent galacto-trehaloses: Design, synthesis and biological evaluation under the concept of carbohydrate modules. Biomacromolecules 2009, 10, 1846–1853. [Google Scholar] [CrossRef] [PubMed]
- Dohi, H.; Nishida, Y.; Furuta, Y.; Uzawa, H.; Yokoyama, S.; Ito, S.; Mori, H; Kobayashi, K. Molecular design and biological potential of galacto-type trehalose as a nonnatural ligand of Shiga toxins. Org. Lett. 2002, 4, 355–357. [Google Scholar] [CrossRef] [PubMed]
- Nataro, J.P.; Kaper, J.B. Diarrheagenic Escherichia coli. Clin. Microbiol. Rev. 1998, 11, 142–201. [Google Scholar] [PubMed]
- Gibson, R.P.; Gloster, T.M.; Roberts, S.; Warren, R.A.; Storch de Gracia, I.; García, A.; Chiara, J.L.; Davies, G.J. Molecular basis for trehalase inhibition revealed by the structure of trehalase in complex with potent inhibitors. Angew. Chem. Int. Ed. 2007, 46, 4115–4119. [Google Scholar] [CrossRef] [PubMed]
- Swarts, B.M.; Holsclaw, C.M.; Jewett, J.C.; Alber, M.; Fox, D.M.; Siegrist, M.S.; Leary, J.A.; Kalscheuer, R.; Bertozzi, C.R. Probing the mycobacterial trehalome with bioorthogonal chemistry. J. Am. Chem. Soc. 2012, 134, 16123–16126. [Google Scholar] [CrossRef] [PubMed]
- Tournu, H.; Fiori, A.; van Dijck, P. Relevance of trehalose in pathogenicity: Some general rules, yet many exceptions. PLoS Pathog. 2013, 9, e1003447. [Google Scholar] [CrossRef] [PubMed]
- Backus, K.M.; Boshoff, H.I.; Barry, C.S.; Boutureira, O.; Patel, M.K.; D’Hooge, F.; Lee, S.S.; Via, L.E.; Tahlan, K.; Barry, C.E., 3rd; et al. Uptake of unnatural trehalose analogs as a reporter for Mycobacterium tuberculosis. Nat. Chem. Biol. 2011, 7, 228–235. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Elchert, B.; Takemoto, J.Y.; Bensaci, M.; Wennergren, J.; Chang, H.; Rai, R.; Chang, C.W. Synthesis of trehalose-based compounds and their inhibitory activities against Mycobacterium smegmatis. Bioorg. Med. Chem. 2004, 12, 6397–6413. [Google Scholar] [CrossRef] [PubMed]
- Gobec, S.; Plantan, I.; Mravljak, J.; Svajger, U.; Wilson, R.A.; Besra, G.S.; Soares, S.L.; Appelberg, R.; Kikelj, D. Design, synthesis, biochemical evaluation and antimycobacterial action of phosphonate inhibitors of antigen 85C, a crucial enzyme involved in biosynthesis of the mycobacterial cell wall. Eur. J. Med. Chem. 2007, 42, 54–63. [Google Scholar] [CrossRef] [PubMed]
- Belisle, J.T.; Viss, V.D.; Sievert, T.; Takayama, K.; Brennan, P.J.; Besra, G.S. Role of the major antigen of Mycobacterium tuberculosis in cell wall biogenesis. Science 1997, 276, 1420–1422. [Google Scholar] [CrossRef] [PubMed]
- Stocker, B.L.; Khan, A.A.; Chee, S.H.; Kamena, F.; Timmer, M.S. On one leg: Trehalose monoesters activate macrophages in a mincle-dependant manner. ChemBioChem 2014, 15, 382–388. [Google Scholar] [CrossRef] [PubMed]
- Schoenen, H.; Bodendorfer, B.; Hitchens, K.; Manzanero, S.; Werninghaus, K.; Nimmerjahn, F.; Agger, E.M.; Stenger, S.; Andersen, P.; Ruland, J. Cutting edge: Mincle is essential for recognition and adjuvanticity of the mycobacterial cord factor and its synthetic analog trehalose-dibehenate. J. Immunol. 2010, 184, 2756–2760. [Google Scholar] [CrossRef] [PubMed]
- Birch, G.; Richardson, A.C. Chemical modification of trehalose. Part II. Synthesis of the galacto-analogue of trehalose. J. Chem. Soc. 1970, 6, 749–752. [Google Scholar] [CrossRef]
- Belocopitow, E.; Maréchal, L.R.; Bar-Guilloux, E.; Robic, D. Enzymatic synthesis of 6-deoxy-α-d-glucopyranosyl-α-d-glucopyranoside and α-d-xylopyranosyl-α-d-glucopyranoside. Carbohydr. Res. 1971, 19, 268–271. [Google Scholar] [CrossRef]
- Ryu, S.I.; Kim, J.E.; Huong, N.T. Molecular cloning and characterization of trehalose synthase from Thermotoga maritima DSM3109: Syntheses of trehalose disaccharide analogues and NDP-glucoses. Enzym. Microb. Technol. 2010, 47, 249–256. [Google Scholar] [CrossRef]
- Lemieux, R.U.; Bauer, H.F. A chemical synthesis of d-trehalose. Can. J. Chem. 1953, 2, 340–343. [Google Scholar]
- Thompson, A.; Anno, K.; Wolfrom, M.L.; Inatome, M. Acid reversion products from d-glucose. J. Am. Chem. Soc. 1954, 76, 1309–1311. [Google Scholar] [CrossRef]
- Cabib, E.; Leloir, L.F. The biosynthesis of trehalose phosphate. J. Am. Chem. Soc. 1957, 75, 259–275. [Google Scholar]
- Murao, S.; Nagano, H.; Ogura, S.; Nishino, T. Enzymatic synthesis of trehalose from maltose. Agric. Biol. Chem. 1985, 49, 2113–2118. [Google Scholar] [CrossRef]
- Kubota, M.; Sawatani, I.; Oku, K.; Takeuchi, K.; Murai, S. The development of α,α-trehalose production and its applications. J. Appl. Glycosci. 2004, 51, 63–70. [Google Scholar] [CrossRef]
- Nishimoto, T.; Nakano, M.; Ikegami, S.; Chaen, H.; Fukuda, S.; Sugimoto, T.; Kurimoto, M.; Tsujisaka, Y. Existence of a novel enzyme converting maltose into trehalose. Biosci. Biotechnol. Biochem. 1995, 59, 2189–2190. [Google Scholar] [CrossRef]
- Nishimoto, T.; Nakada, T.; Chaen, H.; Fukuda, S.; Sugimoto, T.; Kurimoto, M. Purification and characterization of a thermostable trehalose synthase from Thermus aquaticus. Biosci. Biotechnol. Biochem. 1996, 60, 835–839. [Google Scholar] [CrossRef]
- Chaen, H.N.; Mukai, T.N.; Chaen, H.; Fukud, S.; Sugimoto, T.; Kurimoto, M.; Tsujisaka, Y. Efficient enzymatic synthesis of disaccharide α-d-galactosyl α-d-glucoside by trehalose phosphorylase from Thermoanaerobacter brockii. J. Appl. Glycosci. 2001, 48, 135–137. [Google Scholar] [CrossRef]
- Kim, H.M.; Chang, Y.K.; Ryu, S.-I.; Moon, S.-G.; Lee, S.-B. Enzymatic synthesis of a galactose-containing trehalose analogue disaccharide by Pyrococcus horikoshii trehalose-synthesizing glycosyltransferase: Inhibitory effects on several disaccharidase activities. J. Mol. Catal. 2007, 49, 98–103. [Google Scholar] [CrossRef]
- Hashimoto, H.; Katayama, C.; Goto, M.; Okinaga, T.; Kitahata, S. Enzymatic synthesis of α-linked galactooligosaccharides using the reverse reaction of a cell-bound α-galactosidase from Candida guilliermondii H-404. Biosci. Biotechnol. Biochem. 1995, 59, 179–183. [Google Scholar] [CrossRef]
- Kim, B.G.; Lee, K.J.; Han, N.-S.; Park, K.-H.; Lee, S.-B. Enzymatic synthesis and characterization of galactosyl trehalose trisaccharides. Food Sci. Biotechnol. 2007, 16, 127–132. [Google Scholar]
- Desmet, T.; Soetaert, W. Enzymatic glycosylation of small molecules: Challenging substrates require tailored catalysts. Chem. Eur. J. 2012, 18, 10786–10801. [Google Scholar] [CrossRef] [PubMed]
- Desmet, T.; Soetaert, W. Enzymatic glycosyl transfer: Mechanisms and applications. Biocatal. Biotransform. 2011, 29, 1–18. [Google Scholar] [CrossRef]
- Seibel, J.; Joerdening, H.J.; Buchholza, K. Glycosylation with activated sugars using glycosyltransferases and transglycosidases. Biocatal. Biotransform. 2006, 24, 311–342. [Google Scholar] [CrossRef]
- Ryu, S.I.; Park, C.S.; Cha, J.; Wood, E.J.; Lee, S.-B. A novel trehalose-synthesizing glycosyltransferase from Pyrococcus horikoshii: Molecular cloning and characterization. Biochem. Biophys. Res. Commun. 2005, 329, 429–436. [Google Scholar] [CrossRef] [PubMed]
- Qu, Q.; Lee, S.J.; Boos, W. TreT, a novel trehalose glycosyltransferring synthase of the hyperthermophilic archaeon Thermococcus litoralis. J. Biol. Chem. 2004, 279, 47890–47897. [Google Scholar] [CrossRef] [PubMed]
- Kouril, T.; Zaparty, M.; Marrero, J.; Brinkmann, H.; Siebers, B. A novel trehalose synthesizing pathway in the hyperthermophilic Crenarchaeon Thermoproteus tenax: The unidirectional TreT pathway. Arch. Microbiol. 2011, 190, 355–369. [Google Scholar] [CrossRef] [PubMed]
- Hokke, C.H.; Zervosen, A.; Elling, L.; Joziasse, D.H.; van den Eijnden, D.H. One-pot enzymatic synthesis of the Gal-α-1,3-β-1,4-GlcNAc sequence with in situ UDP-Gal regeneration. Glycoconj. J. 1996, 13, 687–692. [Google Scholar] [CrossRef] [PubMed]
- Ichikawa, Y.; Wang, R.; Wong, C.H. Regeneration of sugar nucleotide for enzymatic oligosaccharide synthesis. Methods Enzymol. 1994, 247, 107–127. [Google Scholar] [PubMed]
- Masada, S.; Kawase, Y.; Nagatoshi, M.; Oguchi, Y.; Terasaka, K.; Mizukami, H. An efficient chemoenzymatic production of small molecule glucosides with in situ UDP-glucose recycling. FEBS Lett. 2007, 581, 2562–2566. [Google Scholar] [CrossRef] [PubMed]
- Bungaruang, L.; Gutmann, A.; Nidetzkya, B. Leloir glycosyltransferases and natural product glycosylation: Biocatalytic synthesis of the C-glucoside nothofagin, a major antioxidant of redbush herbal tea. Adv. Synth. Catal. 2013, 355, 2757–2763. [Google Scholar] [CrossRef] [PubMed]
- Luley-Goedl, C.; Nidetzky, B. Carbohydrate synthesis by disaccharide phosphorylases: Reactions, catalytic mechanisms and application in the glycosciences. Biotechnol. J. 2010, 5, 1324–1338. [Google Scholar] [CrossRef] [PubMed]
- Belocopitow, E.; Maréchal, L.R. Trehalose phosphorylase from Euglena gracilis. Biochim. Biophys. Acta 1970, 198, 151–154. [Google Scholar] [CrossRef]
- Kizawa, H.; Miyagawa, K.; Sugiyama, Y. Purification and characterization of trehalose phosphorylase from Micrococcus varians. Biosci. Biotechnol. Biochem. 1995, 59, 1908–1912. [Google Scholar] [CrossRef]
- Maruta, K.; Watanabe, H.; Nishimoto, T.; Kubota, M.; Chaen, H.; Fukuda, S.; Kurimoto, M.; Tsujisaka, Y. Acceptor specificity of trehalose phosphorylase from Thermoanaerobacter brockii: Production of novel nonreducing trisaccharide, 6-O-α-d-galactopyranosyl trehalose. J. Biosci. Bioeng. 2006, 101, 385–390. [Google Scholar] [CrossRef] [PubMed]
- Inoue, Y.; Ishii, K.; Tomita, T.; Yatake, T.; Fukui, F. Characterization of trehalose phosphorylase from Bacillus stearothermophilus SK-1 and nucleotide sequence of the corresponding gene. Biosci. Biotechnol. Biochem. 2002, 66, 1835–1843. [Google Scholar] [CrossRef] [PubMed]
- Yuj, M.H. Novel Microorganism, Maltose Phosphorylase, Trehalose Phosphorylase, and Process for Producing These. Jpn. Patent WO2005003343, 13 January 2005. [Google Scholar]
- Van der Borght, J.; Chen, C.; Hoflack, L.; van Renterghem, L.; Desmet, T.; Soetaert, W. Enzymatic properties and substrate specificity of the trehalose phosphorylase from Caldanaerobacter subterraneus. Appl. Environ. Microbiol. 2011, 77, 6939–6944. [Google Scholar] [CrossRef] [PubMed]
- Avonce, N.; Mendoza-Vargas, A.; Morett, E.; Iturriaga, G. Insights on the evolution of trehalose biosynthesis. BMC Evol. Biol. 2006, 6, 109–123. [Google Scholar] [CrossRef] [PubMed]
- Chaen, H.; Nakada, T.; Nishimoto, T.; Kuroda, N.; Fukuda, S.; Sugimoto, T.; Kurimoto, M.; Tsujisaka, Y. Purification and characterization of thermostable trehalose phosphorylase from Thermoanaerobium brockii. J. Appl. Glycosci. 1999, 46, 399–405. [Google Scholar] [CrossRef]
- Maréchal, L.R.; Belocopitow, E. Metabolism of trehalose in Euglena gracilis. J. Biol. Chem. 1972, 47, 3223–3228. [Google Scholar]
- Van der Borght, J.; Soetaert, W.; Desmet, T. Engineering the acceptor specificity of trehalose phosphorylase for the production of trehalose analogs. Biotechnol. Prog. 2012, 28, 1257–1262. [Google Scholar]
- Chen, C.; van der Borght, J.; de Vreese, R.; D’hooghe, M.; Soetaert, W.; Desmet, T. Engineering the specificity of trehalose phosphorylase as a general strategy for the production of glycosyl phosphates. Chem. Commun. 2014, 50, 7834–7836. [Google Scholar] [CrossRef] [PubMed]
- Van der Borght, J.; Desmet, T.; Soetaert, W. Enzymatic production of β-d-glucose-1-phosphate from trehalose. Biotechnol. J. 2010, 5, 986–993. [Google Scholar] [CrossRef] [PubMed]
- Goedl, C.; Schwarz, A.; Mueller, M.; Brecker, L.; Nidetzky, B. Mechanistic differences among retaining disaccharide phosphorylases: Insights from kinetic analysis of active site mutants of sucrose phosphorylase and α,α-trehalose phosphorylase. Carbohydr. Res. 2008, 343, 2032–2040. [Google Scholar] [CrossRef] [PubMed]
- Wannet, W.J.; Op den Camp, H.J.; Wisselink, H.W.; van der Drift, C.; van Griensven, L.J.; Vogels, G.D. Purification and characterization of trehalose phosphorylase from the commercial mushroom Agaricus bisporus. Biochim. Biophys. Acta 1998, 1425, 177–188. [Google Scholar] [CrossRef]
- Goedl, C.; Griessler, R.; Schwarz, A.; Nidetzky, B. Structure and function relationships for Schizophyllum commune trehalose phosphorylase and their implications for the catalytic mechanism of family GT-4 glycosyltransferases. Biochem. J. 2006, 397, 491–500. [Google Scholar] [PubMed]
- Saito, K.; Yamazaki, H. Production of trehalose synthase from a basidiomycete, Grifola frondosa, in Escherichia coli. Appl. Microbiol. Biotechnol. 1998, 50, 193–198. [Google Scholar] [CrossRef] [PubMed]
- Zhao, R.; Ghent University, Ghent, Belgium. 2015; Unpublished work.
- Saito, K.; Kase, T.; Takahashi, E.; Horinouchi, S. Purification and characterization of a trehalose synthase from the basidiomycete Grifola frondosa. Appl. Environ. Microbiol. 1998, 64, 4340–4345. [Google Scholar] [PubMed]
- Eis, C.; Nidetzky, B. Characterization of trehalose phosphorylase from Schizophyllum commune. Biochem. J. 1999, 15, 385–393. [Google Scholar] [CrossRef]
- Eis, C.; Watkins, M.; Prohaska, T.; Nidetzky, B. Fungal trehalose phosphorylase: Kinetic mechanism, pH-dependence of the reaction and some structural properties of the enzyme from Schizophyllum commune. Biochem. J. 2001, 15, 757–767. [Google Scholar] [CrossRef]
- Chen, C.; Desmet, T.; van der Borghta, J.; Ki Carol Linb, S.; Soetaerta, W. Adsorption-desorption of trehalose analogues from a bioconversion mixture using activated carbon. Sep. Purif. Technol. 2012, 96, 161–167. [Google Scholar] [CrossRef]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Walmagh, M.; Zhao, R.; Desmet, T. Trehalose Analogues: Latest Insights in Properties and Biocatalytic Production. Int. J. Mol. Sci. 2015, 16, 13729-13745. https://doi.org/10.3390/ijms160613729
Walmagh M, Zhao R, Desmet T. Trehalose Analogues: Latest Insights in Properties and Biocatalytic Production. International Journal of Molecular Sciences. 2015; 16(6):13729-13745. https://doi.org/10.3390/ijms160613729
Chicago/Turabian StyleWalmagh, Maarten, Renfei Zhao, and Tom Desmet. 2015. "Trehalose Analogues: Latest Insights in Properties and Biocatalytic Production" International Journal of Molecular Sciences 16, no. 6: 13729-13745. https://doi.org/10.3390/ijms160613729
APA StyleWalmagh, M., Zhao, R., & Desmet, T. (2015). Trehalose Analogues: Latest Insights in Properties and Biocatalytic Production. International Journal of Molecular Sciences, 16(6), 13729-13745. https://doi.org/10.3390/ijms160613729