
Autophagy in DNA Damage Response

Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction

2. DNA Damage and Its Cellular Response
2.1. DNA Damage and Repair
2.2. DNA Damage Signaling
2.3. Programmed Cell Death and DNA Damage
3. Autophagy
4. DNA Damage Response and Autophagy
4.1. Autophagy and DNA Damage Response in the Nucleus
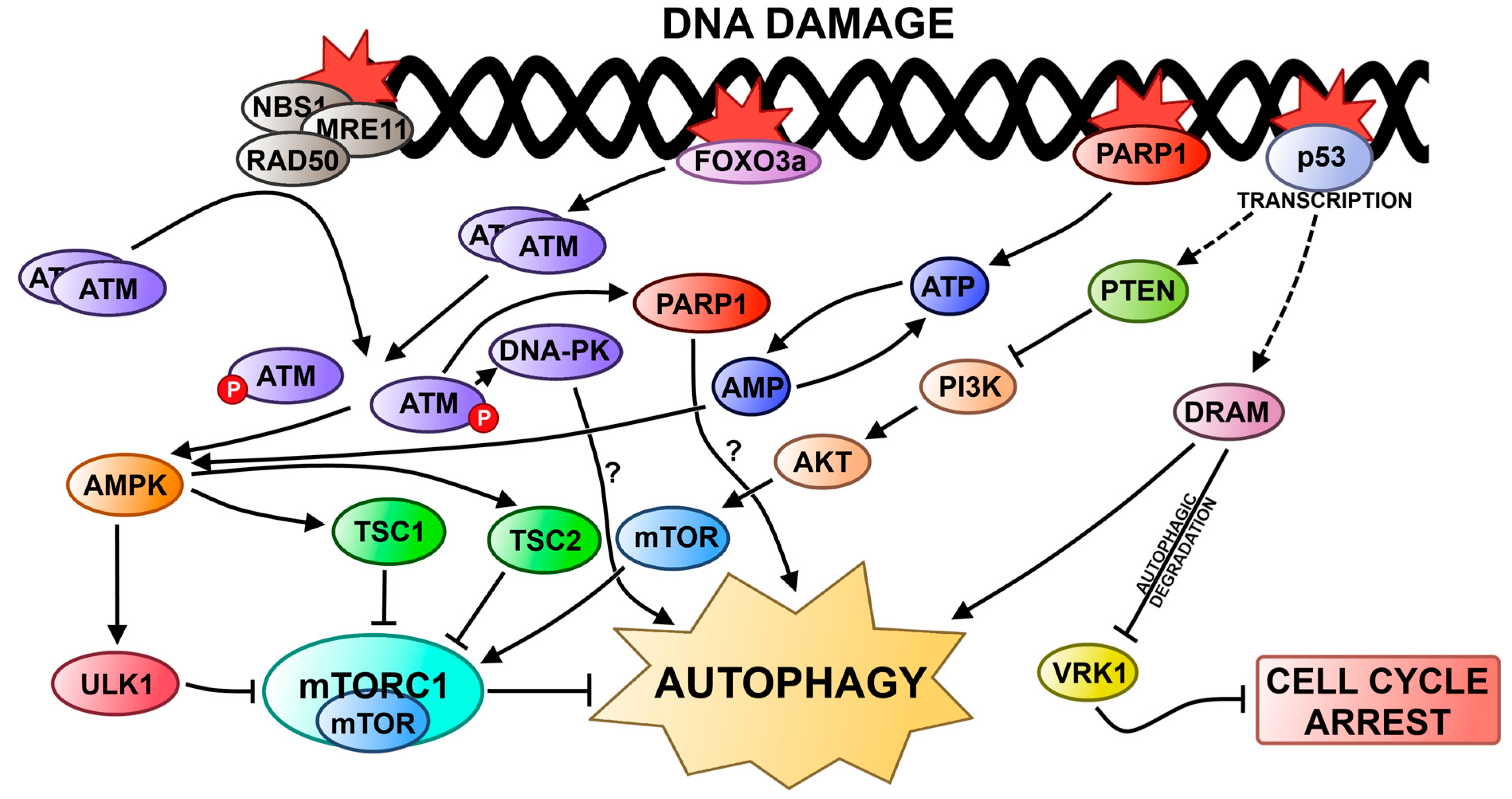
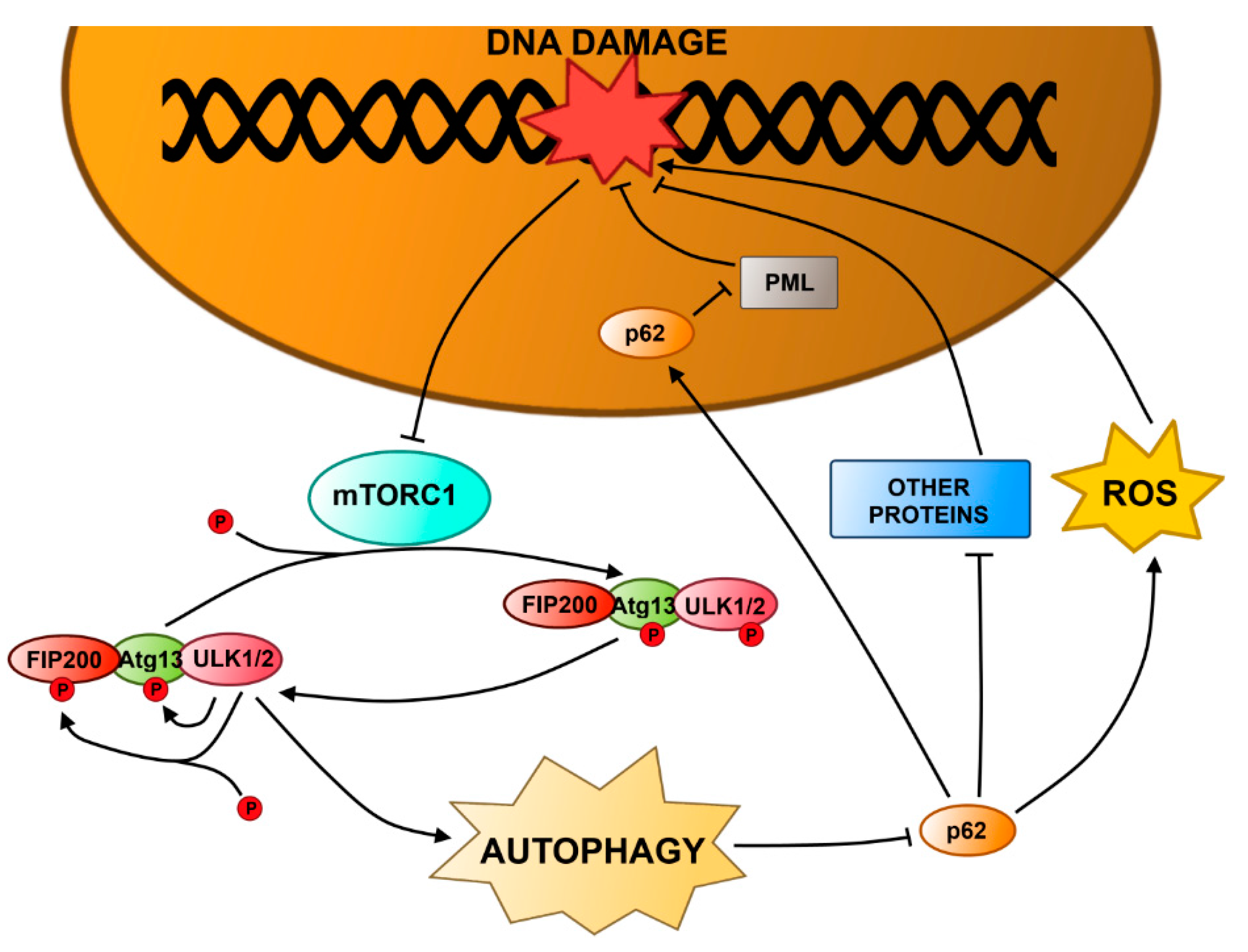
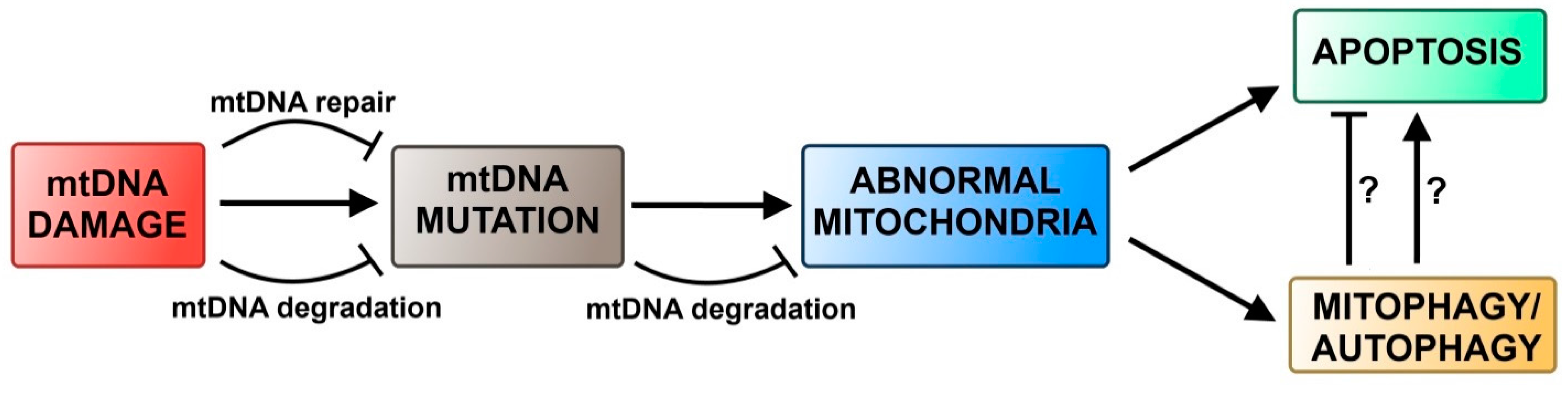
4.2. Autophagy and DNA Damage Response in Mitochondria
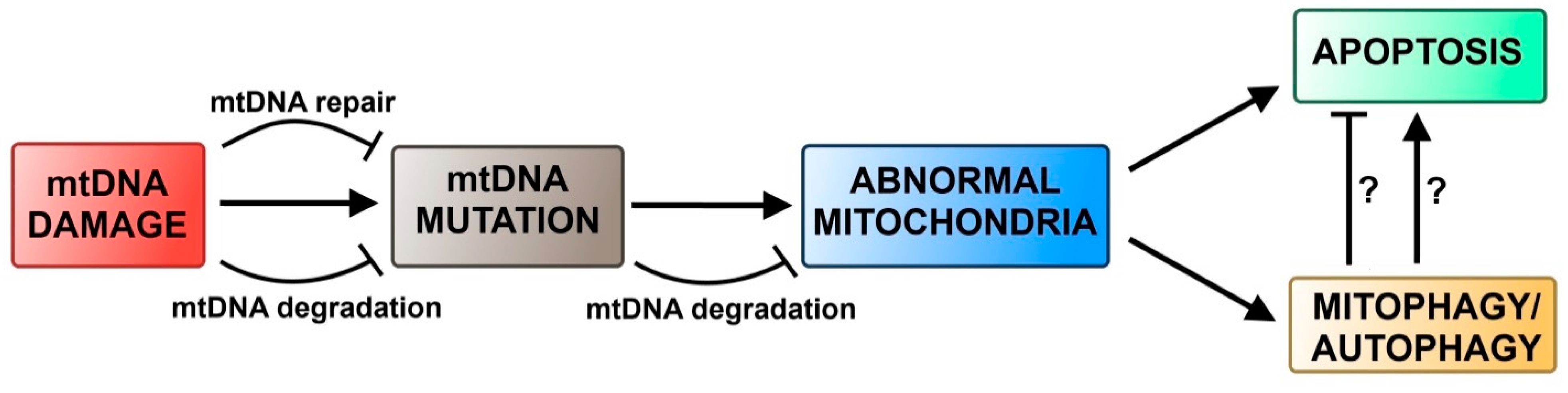
5. Conclusions and Perspectives
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Ciccia, A.; Elledge, S.J. The DNA damage response: Making it safe to play with knives. Mol. Cell 2010, 40, 179–204. [Google Scholar]
- Ferguson, L.R. Chronic inflammation and mutagenesis. Mutat. Res. 2010, 690, 3–11. [Google Scholar]
- Jackson, S.P.; Bartek, J. The DNA-damage response in human biology and disease. Nature 2009, 461, 1071–1078. [Google Scholar]
- Kaarniranta, K.; Sinha, D.; Blasiak, J.; Kauppinen, A.; Veréb, Z.; Salminen, A.; Boulton, M.E.; Petrovski, G. Autophagy and heterophagy dysregulation leads to retinal pigment epithelium dysfunction and development of age-related macular degeneration. Autophagy 2013, 9, 973–984. [Google Scholar]
- Simpson, P.T.; Vargas, A.C.; Al-Ejeh, F.; Khanna, K.K.; Chenevix-Trench, G.; Lakhani, S.R. Application of molecular findings to the diagnosis and management of breast disease: Recent advances and challenges. Hum. Pathol. 2011, 42, 153–165. [Google Scholar]
- Eker, A.P.; Quayle, C.; Chaves, I.; van der Horst, G.T. DNA repair in mammalian cells. Cell. Mol. Life Sci. 2009, 66, 968–980. [Google Scholar]
- Lindahl, T.; Barnes, D.E. Repair of endogenous DNA damage. Cold Spring Harb. Symp. Quant. Biol. 2000, 65, 127–133. [Google Scholar]
- Hoeijmakers, J.H. DNA damage, aging, and cancer. N. Engl. J. Med. 2009, 361, 1475–1485. [Google Scholar]
- Jiricny, J. The multifaceted mismatch-repair system. Nat. Rev. Mol. Cell Biol. 2006, 7, 335–346. [Google Scholar]
- Chapman, J.R.; Taylor., M.R.; Boulton, S.J. Playing the end game: DNA double-strand break repair pathway choice. Mol. Cell 2012, 47, 497–510. [Google Scholar]
- Yakes, F.M.; van Houten, B. Mitochondrial DNA damage is more extensive and persists longer than nuclear DNA damage in human cells following oxidative stress. Proc. Natl. Acad. Sci. USA 1997, 94, 514–519. [Google Scholar]
- Reddy, V.N.; Kasahara, E.; Hiraoka, M.; Lin, L.R.; Ho, Y.S. Effects of variation in superoxide dismutases (SOD) on oxidative stress and apoptosis in lens epithelium. Exp. Eye Res. 2004, 79, 859–868. [Google Scholar]
- Banmeyer, I.C.; Clippe, A.; Knoops, B. Human mitochondrial peroxiredoxin 5 protects from mitochondrial DNA damages induced by hydrogen peroxide. FEBS Lett. 2005, 579, 2327–2333. [Google Scholar]
- Pascucci, B.; Versteegh, A.; van Hoffen, A.; van Zeeland, A.A.; Mullenders, L.H.; Dogliotti, E. DNA repair of UV photoproducts and mutagenesis in human mitochondrial DNA. J. Mol. Biol. 1997, 273, 417–427. [Google Scholar]
- Boesch, P.; Weber-Lotfi, F.; Ibrahim, N.; Tarasenko, V.; Cosset, A.; Paulus, F.; Lightowlers, R.N.; Dietrich, A. DNA repair in organelles: Pathways, organization, regulation, relevance in disease and aging. Biochim. Biophys. Acta 2011, 1813, 186–200. [Google Scholar]
- Le Doux, S.P.; Wilson, G.L. Base excision repair of mitochondrial DNA damage in mammalian cells. Prog. Nucleic Acid Res. Mol. Biol. 2001, 68, 273–284. [Google Scholar]
- De Souza-Pinto, N.C.; Mason, P.A.; Hashiguchi, K.; Weissman, L.; Tian, J.; Guay, D.; Lebel, M.; Stevnsner, T.V.; Rasmussen, L.J.; Bohr, V.A.; et al. Novel DNA mismatch-repair activity involving YB-1 in human mitochondria. DNA Repair 2009, 8, 704–719. [Google Scholar]
- Kraytsberg, Y.; Schwartz, M.; Brown, T.A.; Ebralidse, K.; Kunz, W.S.; Clayton, D.A.; Vissing, J.; Khrapko, K. Recombination of human mitochondrial DNA. Science 2004, 304, 981. [Google Scholar]
- Sage, J.M.; Gildemeister, O.S.; Knight, K.L. Discovery of a novel function for human Rad51: Maintenance of the mitochondrial genome. J. Biol. Chem. 2010, 285, 18984–18990. [Google Scholar]
- Lakshmipathy, U.; Campbell, C. Double strand break rejoining by mammalian mitochondrial extracts. Nucleic Acids Res. 1999, 27, 11198–11204. [Google Scholar]
- Cui, R.; Widlund, H.R.; Feige, E.; Lin, J.Y.; Wilensky, D.L.; Igras, V.E.; D’Orazio, J.; Fung, C.Y.; Schanbacher, C.F.; Granter, S.R.; et al. Central role of p53 in the suntan response and pathologic hyperpigmentation. Cell 2007, 128, 853–864. [Google Scholar]
- Schreiber, V.; Dantzer, F.; Ame, J.C.; de Murcia, G. Poly(ADP-ribose): Novel functions for an old molecule. Nat. Rev. Mol. Cell Biol. 2006, 7, 517–528. [Google Scholar]
- Sancar, A.; Lindsey-Boltz, L.A.; Unsal-Kacmaz, K.; Linn, S. Molecular mechanisms of mammalian DNA repair and the DNA damage checkpoints. Annu. Rev. Biochem. 2004, 73, 39–85. [Google Scholar]
- Meek, K.; Dang, V.; Lees-Miller, S.P. DNA-PK: The means to justify the ends? Adv. Immunol. 2008, 99, 33–58. [Google Scholar]
- Harpe, J.W.; Elledge, S.J. The DNA damage response: Ten years after. Mol. Cell 2007, 28, 739–745. [Google Scholar]
- Matsuoka, S.; Ballif, B.A.; Smogorzewska, A.; McDonald, E.R., 3rd; Hurov, K.E.; Luo, J.; Bakalarski, C.E.; Zhao, Z.; Solimini, N.; Lerenthal, Y.; et al. ATM and ATR substrate analysis reveals extensive protein networks responsive to DNA damage. Science 2007, 316, 1160–1166. [Google Scholar]
- Paulsen, R.D.; Soni, D.V.; Wollman, R.; Hahn, A.T.; Yee, M.C.; Guan, A.; Hesley, J.A.; Miller, S.C.; Cromwell, E.F.; Solow-Cordero, D.E.; et al. A genome-wide siRNA screen reveals diverse cellular processes and pathways that mediate genome stability. Mol. Cell 2009, 35, 228–239. [Google Scholar]
- Cimprich, K.A.; Cortez, D. ATR: An essential regulator of genome integrity. Nat. Rev. Mol. Cell Biol. 2008, 9, 616–627. [Google Scholar]
- Paulovich, A.G.; Margulies, R.U.; Garvik, B.M.; Hartwell, L.H. RAD9, RAD17, and RAD24 are required for S phase regulation in Saccharomyces cerevisiae in response to DNA damage. Genetics 1997, 145, 45–62. [Google Scholar]
- Volkmer, E.; Karnitz, L.M. Human homologs of Schizosaccharomyces pombe Rad1, Hus1, and Rad9 form a DNA damage-responsive protein complex. J. Biol. Chem. 1999, 274, 567–570. [Google Scholar]
- Thelen, M.P.; Venclovas, C.; Fidelis, K. A sliding clamp model for the Rad1 family of cell cycle checkpoint proteins. Cell 1999, 96, 769–770. [Google Scholar]
- Griffiths, D.J.; Barbet, N.C.; McCready, S.; Lehmann, A.R.; Carr, A.M. Fission yeast rad17: A homologue of budding yeast RAD24 that shares regions of sequence similarity with DNA polymerase accessory proteins. EMBO J. 1995, 14, 5812–5823. [Google Scholar]
- Green, C.M.; Erdjument-Bromage, H.; Tempst, P.; Lowndes, N.F. A novel Rad24 checkpoint protein complex closely related to replication factor C. Curr. Biol. 2000, 10, 39–42. [Google Scholar]
- Naiki, T.; Shimomura, T.; Kondo, T.; Matsumoto, K.; Sugimoto, K. Rfc5, in cooperation with Rad24, controls DNA damage checkpoints throughout the cell cycle in Saccharomyces cerevisiae. Mol. Cell. Biol. 2000, 20, 5888–5896. [Google Scholar]
- Lindsey-Boltz, L.A.; Bermudez, V.P.; Hurwitz, J; Sancar, A. Purification and characterization of human DNA damage checkpoint Rad complexes. Proc. Natl. Acad. Sci. USA 2001, 98, 11236–11241. [Google Scholar]
- Golia, B.; Singh, H.R.; Timinszki, G. Poly-ADP-ribosylation signaling during DNA damage repair. Front. Biosci. 2015, 20, 440–457. [Google Scholar]
- Bakkenist, C.J.; Kastan, M.B. DNA damage activates ATM through intermolecular autophosphorylation and dimer dissociation. Nature 2003, 421, 499–506. [Google Scholar]
- Wang, Y.; Cortez, D.; Yazdi, P.; Neff, N.; Elledge, S.J.; Qin, J. BASC, a super complex “of BRCA1-associated proteins involved in the recognition and repair of aberrant DNA structures. Genes Dev. 2000, 14, 927–939. [Google Scholar]
- Kim, G.D.; Choi, Y.H.; Dimtchev, A.; Jeong, S.J.; Dritschilo, A.; Jung, M. Sensing of ionizing radiation-induced DNA damage by ATM through interaction with histone deacetylase. J. Biol. Chem. 1999, 274, 31127–33130. [Google Scholar]
- Schmidt, D.R.; Schreiber, S.L. Molecular association between ATR and two components of the nucleosome remodeling and deacetylating complex, HDAC2 and CHD4. Biochemistry 1999, 38, 14711–14717. [Google Scholar]
- Tran, H.; Brunet, A.; Grenier, J.M.; Datta, S.R.; Fornace, A.J., Jr.; DiStefano, P.S.; Chiang, L.W.; Greenberg, M.E. DNA repair pathway stimulated by the forkhead transcription factor FOXO3a through the Gadd45 protein. Science 2002, 296, 530–534. [Google Scholar]
- Thurn, K.T.; Thomas, S.; Raha, P.; Qureshi, I.; Munster, P.N. Histone deacetylase regulation of ATM-mediated DNA damage signaling. Mol. Cancer Ther. 2013, 12, 2078–2087. [Google Scholar]
- Rodriguez-Rochaa, H.; Garcia-Garciaa, A.; Panayiotidisb, M.I.; Francoa, R. DNA damage and autophagy. Mutat. Res. 2011, 711, 158–166. [Google Scholar]
- Festjens, N.; VandenBerghe, T.; Vandenabeele, P. Necrosis, a well-orchestrated form of cell demise: Signaling cascades, important mediators and concomitant immune response. Biochim. Biophys. Acta 2006, 1757, 1371–1387. [Google Scholar]
- Huang, C.; Freter, C. Lipid metabolism, apoptosis and cancer therapy. Int. J. Mol. Sci. 2015, 16, 924–949. [Google Scholar]
- Hsu, H.; Xiong, J.; Goeddel, D.V. The TNF receptor 1-associated protein TRADD signals cell death and NFκB activation. Cell 1995, 81, 495–504. [Google Scholar]
- Wajant, H. The Fas signaling pathway: More than a paradigm. Science 2002, 29, 1635–1636. [Google Scholar]
- Kischkel, F.C.; Hellbardt, S.; Behrmann, I.; Germer, M.; Pawlita, M.; Krammer, P.H.; Peter, M.E. Cytotoxicity dependent APO-1 (Fas/CD95)-associated proteins form a death-inducing signaling complex (DISC) with the receptor. EMBO J. 1995, 14, 5579–5588. [Google Scholar]
- Saelens, X.; Festjens, N.; vande Walle, L.; van Gurp, M.; van Loo, G.; Vandenabeele, P. Toxic proteins released from mitochondria in cell death. Oncogene 2004, 23, 2861–2874. [Google Scholar]
- Parsons, M.J.; Green, D.R. Mitochondria in cell death. Essays Biochem. 2010, 47, 99–114. [Google Scholar]
- Elmore, S. Apoptosis: A review of programmed cell death. Toxicol. Pathol. 2007, 35, 495–516. [Google Scholar]
- Schuler, M.; Green, D.R. Mechanisms of p53-dependent apoptosis. Biochem. Soc. Trans. 2001, 29, 684–688. [Google Scholar]
- Roos, W.P.; Kaina, B. DNA damage-induced cell death by apoptosis. Trends Mol. Med. 2006, 12, 440–450. [Google Scholar]
- Chipuk, J.E.; Green, D.R. Dissecting p53-dependent apoptosis. Cell Death Differ. 2006, 13, 994–1002. [Google Scholar]
- Kung, C.P.; Budina, A.; Balaburski, G.; Bergenstock, M.K.; Murphy, M. Autophagy in tumor suppression and cancer therapy. Crit. Rev. Eukaryot. Gene Expr. 2011, 21, 71–100. [Google Scholar]
- Levine, B.; Klionsky, D.J. Development by self-digestion: Molecular mechanisms and biological functions of autophagy. Cell 2004, 6, 463–477. [Google Scholar]
- Cuervo, A.M. Autophagy: Many paths to the same end. Mol. Cell. Biochem. 2004, 263, 55–72. [Google Scholar]
- Marzella, L.; Ahlberg, J.; Glaumann, H. Autophagy, heterophagy, microautophagy and crinophagy as the means for intracellular degradation. Virchows Arch. B 1981, 36, 219–234. [Google Scholar]
- Shao, N.; Chai, Y.L.; Shyam, E.; Reddy, P.; Rao, V.N. Induction of apoptosis by the tumor suppressor protein BRCA1. Oncogene 1996, 13, 1–7. [Google Scholar]
- Martin, S.A.; Ouchi, T. BRCA1 phosphorylation regulates caspase-3 activation in UV-induced apoptosis. Cancer Res. 2005, 65, 10657–10662. [Google Scholar]
- Burma, S.; Chen, D.J. Role of DNA-PK in the cellular response to DNA double-strand breaks. DNA Repair 2004, 3, 909–918. [Google Scholar]
- Espejel, S.; Franco, S.; Sgura, A.; Gae, D.; Bailey, S.M.; Taccioli, G.E.; Blasco, M.A. Functional interaction between DNA-PKcs and telomerase in telomere length maintenance. EMBO J. 2002, 21, 6275–6287. [Google Scholar]
- Espejel, S.; Martín, M.; Klatt, P.; Martín-Caballero, J.; Flores, J.M.; Blasco, M.A. Shorter telomeres, accelerated ageing and increased lymphoma in DNA-PKcs deficient mice. EMBO Rep. 2004, 5, 503–509. [Google Scholar]
- Luo, X.; Kraus, W.L. On PAR with PARP: Cellular stress signaling through poly (ADP-ribose) and PARP-1. Genes Dev. 2012, 26, 417–432. [Google Scholar]
- Nowsheen, S.; Bonner, J.A.; Lo Buglio, A.F.; Trummell, H.; Whitley, A.C.; Dobelbower, M.C.; Yang, E.S. Cetuximab augments cytotoxicity with poly (ADP-ribose) polymerase inhibition in head and neck cancer. PLoS One 2011, 6, e24148. [Google Scholar]
- Al-Ejeh, F.; Shi, W.; Miranda, M.; Simpson, P.T.; Vargas, A.C.; Song, S.; Wiegmans, A.P.; Swarbrick, A.; Welm, A.L.; Brown, M.P.; et al. Treatment of triple-negative breast cancer using anti-EGFR-directed radioimmunotherapy combined with radiosensitizing chemotherapy and PARP inhibitor. J. Nucl. Med. 2013, 54, 913–921. [Google Scholar]
- Yu, S.W.; Wang, H.; Poitras, M.F.; Coombs, C.; Bowers, W.J.; Federoff, H.J.; Poirier, G.G.; Dawson, T.M.; Dawson, V.L. Mediation of poly(ADP-ribose) polymerase-1-dependent cell death by apoptosis-inducing factor. Science 2002, 297, 259–263. [Google Scholar]
- Cregan, S.P.; Dawson, V.L.; Slack, R.S. Role of AIF in caspase-dependent and caspase-independent cell death. Oncogene 2004, 23, 2785–2796. [Google Scholar]
- Al-Ejeh, F.; Kumar, R.; Wiegmans, A.; Lakhani, S.R.; Brown, M.P.; Khanna, K.K. Harnessing the complexity of DNA-damage response pathways to improve cancer treatment outcomes. Oncogene 2010, 29, 6085–6098. [Google Scholar]
- Mortimore, G.E.; Lardeux, B.R.; Adams, C.E. Regulation of microautophagy and basal protein turnover in rat liver. Effects of short-term starvation. J. Biol. Chem. 1988, 263, 2506–2512. [Google Scholar]
- Agarraberes, F.; Dice, J.F. A molecular chaperone complex at the lysosomal membrane is required for protein translocation. J. Cell Sci. 2001, 114, 2491–2499. [Google Scholar]
- Majeski, A.E.; Dice, J.F. Mechanisms of chaperone-mediated autophagy. Int. J. Biochem. Cell Biol. 2004, 36, 2435–2444. [Google Scholar]
- Massey, A.C.; Zhang, C.; Cuervo, A.M. Chaperone-mediated autophagy in aging and disease. Curr. Top. Dev. Biol. 2006, 73, 205–235. [Google Scholar]
- Klionsky, D.J.; Emr, S.D. Autophagy as a regulated pathway of cellular degradation. Science 2000, 290, 1717–1721. [Google Scholar]
- Baehrecke, E.H. Autophagy: Dual roles in life and death? Nat. Rev. Mol. Cell Biol 2005, 6, 505–510. [Google Scholar]
- Klionsky, D.J.; Abdalla, F.C.; Abeliovich, H.; Abraham, R.T.; Acevedo-Arozena, A.; Adeli, K.; Agholme, L.; Agnello, M.; Agostinis, P.; Aguirre-Ghiso, J.A.; et al. Guidelines for the use and interpretation of assays for monitoring autophagy. Autophagy 2012, 8, 445–544. [Google Scholar]
- Filomeni, G.; de Zio, D.; Cecconi, F. Oxidative stress and autophagy: the clash between damage and metabolic needs. Cell Death Differ. 2014. [Google Scholar] [CrossRef]
- Kroemer, G.; Mariño, G.; Levine, B. Autophagy and the integrated stress response. Mol. Cell 2010, 40, 280–293. [Google Scholar]
- Gozuacik, D.; Kimchi, A. Autophagy and cell death. Curr. Top. Dev. Biol. 2007, 78, 217–245. [Google Scholar]
- Wei, H.; Wang, C.; Croce, C.M.; Guan, J.L. p62/SQSTM1 synergizes with autophagy for tumor growth in vivo. Genes Dev. 2014, 28, 1204–1216. [Google Scholar]
- Kamada, Y.; Funakoshi, T.; Shintani, T.; Nagano, K.; Ohsumi, M.; Ohsumi, Y. Tor-mediated induction of autophagy via an Apg1 protein kinase complex. J. Cell Biol. 2000, 150, 1507–1513. [Google Scholar]
- Mizushima, N. The role of the Atg1/ULK1 complex in autophagy regulation. Curr. Opin. Cell Biol. 2010, 22, 132–139. [Google Scholar]
- Wei, H.; Guan, J.L. Pro-tumorigenic function of autophagy in mammalian oncogenesis. Autophagy 2012, 8, 129–131. [Google Scholar]
- Dunlop, E.A.; Tee, A.R. mTOR and autophagy: A dynamic relationship governed by nutrients and energy. Semin. Cell Dev. Biol. 2014, 36, 121–129. [Google Scholar]
- Roberts, D.J.; Miyamoto, S. Hexokinase II integrates energy metabolism and cellular protection: Akting on mitochondria and TORcing to autophagy. Cell Death Differ. 2015, 22, 248–257. [Google Scholar]
- Mizushima, N.; Noda, T.; Ohsumi, Y. Apg16p is required for the function of the Apg12p–Apg5p conjugate in the yeast autophagy pathway. EMBO J. 1999, 18, 3888–3896. [Google Scholar]
- Kuma, A.; Mizuchima, N.; Ishihara, N.; Ohsumi, Y. Formation of the approximately 350 kDa Apg12–Apg5–Apg16 multimeric complex, mediated by Apg16 oligomerization, is essential for autophagy in yeast. J. Biol. Chem. 2002, 277, 18619–18625. [Google Scholar]
- Scott, S.V.; Nice, D.C., 3rd; Nau, J.J.; Weisman, L.S.; Kamada, Y.; Keizer-Gunnink, I.; Funakoshi, T.; Veenhuis, M.; Ohsumi, Y.; Klionsky, D.J.; et al. Apg13p and Vac8p are part of a complex of phosphoproteins that are required for cytoplasm to vacuole targeting. J. Biol. Chem. 2000, 275, 25840–25849. [Google Scholar]
- Itakura, E.; Kishi, C.; Inoue, K.; Mizushima, N. Beclin-1 forms two distinct phosphatidylinositol 3-kinase complexes with mammalian Atg14 and UVRAG. Mol. Biol. Cell 2008, 19, 5360–5372. [Google Scholar]
- Liang, C.; Feng, P.; Ku, B.; Dotan, I.; Canaani, D.; Oh, B.H.; Jung, J.U. Autophagic and tumour suppressor activity of a novel Beclin1-binding protein UVRAG. Nat. Cell Biol. 2006, 8, 688–699. [Google Scholar]
- Sun, Q.; Fan, W.; Chen, K.; Ding, X.; Chen, S.; Zhong, Q. Identification of Barkor as a mammalian autophagy-specific factor for Beclin 1 and class III phosphatidylinositol 3-kinase. Proc. Natl. Acad. Sci. USA 2008, 105, 19211–19216. [Google Scholar]
- Takahashi, Y.; Coppola, D.; Matsushita, N.; Cualing, H.D.; Sun, M.; Sato, Y.; Liang, C.; Jung, J.U.; Cheng, J.Q.; Mulé, J.J.; et al. Bif-1 interacts with Beclin 1 through UVRAG and regulates autophagy and tumorigenesis. Nat. Cell Biol. 2007, 9, 1142–1151. [Google Scholar]
- Kihara, A.; Noda, T.; Ishihara, N.; Ohsumi, Y. Two distinct Vps34 phosphatidylinositol 3-kinase complexes function in autophagy and carboxypeptidase Y sorting in Saccharomyces cerevisiae. J. Cell Biol. 2001, 152, 519–530. [Google Scholar]
- Ichimura, Y.; Kirisako, T.; Takao, T.; Satomi, Y.; Shimonishi, Y.; Ishihara, N.; Mizushima, N.; Tanida, I.; Kominami, E.; Ohsumi, M.; et al. An ubiquitin-like system mediates protein lipidation. Nature 2000, 408, 488–492. [Google Scholar]
- Kawai, A.; Uchiyama, H.; Takano, S.; Nakamura, N.; Ohkuma, S. Autophagosome-lysosome fusion depends on the pH in acidic compartments in CHO cells. Autophagy 2007, 3, 154–157. [Google Scholar]
- Rieber, M.; Rieber, M.S. Sensitization to radiation-induced DNA damage accelerates loss of Bcl-2 and increases apoptosis and autophagy. Cancer Biol. Ther. 2008, 7, 1561–1566. [Google Scholar]
- Katayama, M.; Kawaguchi, T.; Berger, M.S.; Pieper, R.O. DNA damaging agent induced autophagy produces a cytoprotective adenosine triphosphate surge in malignant glioma cells. Cell Death Differ. 2007, 14, 548–558. [Google Scholar]
- Abedin, M.J.; Wang, D.; McDonnell, M.A.; Lehmann, U.; Kelekar, A. Autophagy delays apoptotic death in breast cancer cells following DNA damage. Cell Death Differ. 2007, 14, 500–510. [Google Scholar]
- Elliott, A.; Reiners, J.J., Jr. Suppression of autophagy enhances the cytotoxicity of the DNA-damaging aromatic amine p-anilinoaniline. Toxicol. Appl. Pharmacol. 2008, 232, 169–179. [Google Scholar]
- Apel, A.; Herr, I.; Schwarz, H.; Rodemann, H.P.; Mayer, A. Blocked autophagy sensitizes resistant carcinoma cells to radiation therapy. Cancer Res. 2008, 68, 1485–1494. [Google Scholar]
- Qadir, M.A.; Kwok, B.; Dragowska, W.H.; To, K.H.; Le, D.; Bally, M.B.; Gorski, S.M. Macroautophagy inhibition sensitizes tamoxifen-resistant breast cancer cells and enhances mitochondrial depolarization. Breast Cancer Res. Treat. 2008, 112, 389–403. [Google Scholar]
- Amaravadi, R.K.; Yu, D.; Lum, J.J.; Bui, T.; Christophorou, M.A.; Evan, G.I.; Thomas-Tikhonenko, A.; Thompson, C.B. Autophagy inhibition enhances therapy-induced apoptosis in a Myc-induced model of lymphoma. J. Clin. Investig. 2007, 117, 326–336. [Google Scholar]
- Zhao, Z.; Ni, D.; Ghozalli, I.; Piroz, S.D.; Ma, B.; Liang, C. UVARG at the crossroad of autophagy and genomic stability. Autophagy 2012, 8, 1392–1393. [Google Scholar]
- Karantza-Wadsworth, V.; Patel, S.; Kravchuk, O.; Chen, G.; Mathew, R.; Jin, S.; White, E. Autophagy mitigates metabolic stress and genome damage in mammary tumorigenesis. Genes Dev. 2007, 21, 1621–1635. [Google Scholar]
- Alexander, A.; Kim, J.; Walker, C.L. ATM engages the TSC2/mTORC1 signaling node to regulate autophagy. Autophagy 2010, 6, 672–673. [Google Scholar]
- Alexander, A.; Cai, S.L.; Kim, J.; Nanez, A.; Sahin, M.; MacLean, K.H.; Inoki, K.; Guan, K.L.; Shen, J.; Person, M.D.; et al. ATM signals to TSC2 in the cytoplasm to regulate mTORC1 in response to ROS. Proc. Natl. Acad. Sci. USA 2010, 107, 4153–4158. [Google Scholar]
- Inoki, K.; Zhu, T.; Guan, K.L. TSC2 mediates cellular energy response to control cell growth and survival. Cell 2003, 115, 577–590. [Google Scholar]
- Zhao, M.; Klionsky, D.J. AMPK-dependent phosphorylation of ULK1 induces autophagy. Cell Metab. 2011, 13, 119–120. [Google Scholar]
- Tsai, W.B.; Chung, Y.M.; Takahashi, Y.; Xu, Z.; Hu, M.C. Functional interaction between FOXO3a and ATM regulates DNA damage response. Nat. Cell Biol. 2008, 10, 460–467. [Google Scholar]
- Mammucari, C.; Milan, G.; Romanello, V.; Masiero, E.; Rudolf, R.; del Piccolo, P.; Burden, S.J.; di Lisi, R.; Sandri, C.; Zhao, J.; et al. FoxO3 controls autophagy in skeletal muscle in vivo. Cell Metab. 2007, 6, 458–471. [Google Scholar]
- Salminen, A.; Kaarniranta, K. Regulation of the aging process by autophagy. Trends Mol. Med. 2009, 15, 217–224. [Google Scholar]
- Chiacchiera, F.; Simone, C. The AMPK-FoxO3A axis as a target for cancer treatment. Cell Cycle 2010, 9, 1091–1096. [Google Scholar]
- Tasdemir, E.; Chiara Maiuri, M.; Morselli, E.; Criollo, A.; D’Amelio, M.; Djavaheri-Mergny, M.; Cecconi, F.; Tavernarakis, N.; Kroemer, G. A dual role of p53 in the control of autophagy. Autophagy 2008, 4, 810–814. [Google Scholar]
- Zong, W.X.; Moll, U. p53 in autophagy control. Cell Cycle 2008, 7, 2947–2948. [Google Scholar]
- Kang, K.B.; Zhu, C.; Yong, S.K.; Gao, Q.; Wong, M.C. Enhanced sensitivity of celecoxib in human glioblastoma cells: Induction of DNA damage leading to p53-dependent G1 cell cycle arrest and autophagy. Mol. Cancer 2009, 8, 66. [Google Scholar]
- Feng, Z.; Zhang, H.; Levine, A.J.; Jin, S. The coordinate regulation of the p53 and mTOR pathways in cells. Proc. Natl. Acad. Sci. USA 2005, 102, 8204–8209. [Google Scholar]
- Fortini, P.; Dogliotti, E. Mechanisms of dealing with DNA damage in terminally differentiated cells. Mutat. Res. 2010, 685, 38–44. [Google Scholar]
- Jin, S. p53, Autophagy and tumor suppression. Autophagy 2005, 1, 171–173. [Google Scholar]
- Crighton, D.; Wilkinson, S.; O’Prey, J.; Syed, N.; Smith, P.; Harrison, P.R.M.; Garrone, O.; Crook, T.; Ryan, K.M. DRAM, a p53-induced modulator of autophagy, is critical for apoptosis. Cell 2006, 126, 121–134. [Google Scholar]
- Crighton, D.; Wilkinson, S.; Ryan, K.M. DRAM links autophagy to p53 and programmed cell death. Autophagy 2007, 3, 72–74. [Google Scholar]
- Valbuena, A.; Castro-Obregón, S.; Lazo, P.A. Down-regulation of VRK1 by p53 in response to DNA damage is mediated by the autophagic pathway. PLoS One 2011, 6, e17320. [Google Scholar]
- Klerkx, E.P.; Lazo, P.A.; Askjaer, P. Emerging biological functions of the vaccinia-related kinase (VRK) family. Histol. Histopathol. 2009, 24, 749–759. [Google Scholar]
- Sanz-Garcia, M.; Valbuena González, M.; López-Sánchez, A.; Blanco, I.; Fernández, S.; Vázquez Cedeira, I.F.; Lazo, M.; Pedro, A. Vaccinia-related kinase (VRK) signaling in cell and tumor biology. In Emerging Signaling Pathways in Tumor Biology; Lazo, P.A., Ed.; Transworld Research Networks: Kerala, India, 2010; pp. 135–156. [Google Scholar]
- Dyavaiah, M.; Rooney, J.P.; Chittur, S.V.; Lin, Q.; Begley, T.J. Autophagy-dependent regulation of the DNA damage response protein ribonucleotide reductase 1. Mol. Cancer Res. 2011, 9, 462–475. [Google Scholar]
- Ha, H.C.; Snyder, S.H. Poly(ADP-ribose) polymerase is a mediator of necrotic cell death by ATP depletion. Proc. Natl. Acad. Sci. USA 1999, 96, 13978–13982. [Google Scholar]
- Rodríguez-Vargas, J.M.; Ruiz-Magaña, M.J.; Ruiz-Ruiz, C.; Majuelos-Melguizo, J.; Peralta-Leal, A.; Rodríguez, M.I.; Muñoz-Gámez, J.A.; de Almodóvar, M.R.; Siles, E.; Rivas, A.L.; et al. ROS-induced DNA damage and PARP-1 are required for optimal induction of starvation-induced autophagy. Cell Res. 2012, 22, 1181–1198. [Google Scholar]
- Hoyer-Hansen, M.; Jaattela, M. AMP-activated protein kinase: A universal regulator of autophagy? Autophagy 2007, 3, 381–383. [Google Scholar]
- Munoz-Gamez, J.A.; Rodríguez-Vargas, J.M.; Quiles-Pérez, R.; Aguilar-Quesada, R.; Martín-Oliva, D.; de Murcia, G.; Menissier de Murcia, J.; Almendros, A.; Ruiz de Almodóvar, M.; Oliver, F.J.; et al. PARP-1 is involved in autophagy induced by DNA damage. Autophagy 2009, 5, 61–74. [Google Scholar]
- Huang, Q.; Shen, H.M. To die or to live: The dual role of poly(ADP-ribose) polymerase-1 in autophagy and necrosis under oxidative stress and DNA damage. Autophagy 2009, 5, 273–276. [Google Scholar]
- Yoon, J.H.; Ahn, S.G.; Lee, B.H.; Jung, S.H.; Oh, S.H. Role of autophagy in chemoresistance: Regulation of the ATM-mediated DNA-damage signaling pathway through activation of DNA-PKcs and PARP-1. Biochem. Pharmacol. 2012, 83, 747–757. [Google Scholar]
- Abbi, S.; Ueda, H.; Zheng, C.; Cooper, L.A.; Zhao, J.; Christopher, R.; Guan, J.L. Regulation of focal adhesion kinase by a novel protein inhibitor FIP200. Mol. Biol. Cell 2002, 13, 3178–3191. [Google Scholar]
- Rajendran, R.; Garva, R.; Krstic-Demonacos, M.; Demonacos, C. Sirtuins: Molecular traffic lights in the crossroad of oxidative stress, chromatin remodeling, and transcription. J. Biomed. Biotechnol. 2011, 2011, 368276. [Google Scholar]
- Kitada, M.; Kume, S.; Takeda-Watanabe, A.; Kanasaki, K.; Koya, D. Sirtuins and renal diseases: Relationship with aging and diabetic nephropathy. Clin. Sci. 2013, 124, 153–164. [Google Scholar]
- Hariharan, N.; Maejima, Y.; Nakae, J.; Paik, J.; Depinho, R.A.; Sadoshima, J. Deacetylation of FoxO by Sirt1 plays an essential role in mediating starvation-induced autophagy in cardiac myocytes. Circ. Res. 2010, 107, 1470–1482. [Google Scholar]
- Hands, S.L.; Proud, C.G.; Wyttenbach, A. mTOR’s role in ageing: Protein synthesis or autophagy? Aging 2009, 1, 586–597. [Google Scholar]
- Yi, J.; Luo, J. SIRT1 and p53, effect on cancer, senescence and beyond. Biochim. Biophys. Acta 2010, 1804, 1684–1689. [Google Scholar]
- Ueda, H.; Abbi, S.; Zheng, C.; Guan, J.L. Suppression of Pyk2 kinase and cellular activities by FIP200. J. Cell Biol. 2000, 149, 423–430. [Google Scholar]
- Gan, B.; Melkoumian, Z.K.; Wu, X.; Guan, K.L.; Guan, J.L. Identification of FIP200 interaction with the TSC1–TSC2 complex and its role in regulation of cell size control. J. Cell Biol. 2005, 170, 379–389. [Google Scholar]
- Melkoumian, Z.K.; Peng, X.; Gan, B.; Wu, X.; Guan, J.L. Mechanism of cell cycle regulation by FIP200 in human breast cancer cells. Cancer Res. 2005, 65, 6676–6684. [Google Scholar]
- Gan, B.; Peng, X.; Nagy, T.; Alcaraz, A.; Gu, H.; Guan, J.L. Role of FIP200 in cardiac and liver development and its regulation of TNFα and TSC–mTOR signaling pathways. J. Cell Biol. 2006, 175, 121–133. [Google Scholar]
- Ganley, I.G.; du Lam, H.; Wang, J.; Ding, X.; Chen, S.; Jiang, X. ULK1–ATG13–FIP200 complex mediates mTOR signaling and is essential for autophagy. J. Biol. Chem. 2009, 284, 12297–12305. [Google Scholar]
- Hosokawa, N.; Hara, T.; Kaizuka, T.; Kishi, C.; Takamura, A.; Miura, Y.; Iemura, S.; Natsume, T.; Takehana, K.; Yamada, N.; et al. Nutrient-dependent mTORC1 association with the ULK1–Atg13–FIP200 complex required for autophagy. Mol. Biol. Cell 2009, 20, 1981–1991. [Google Scholar]
- Jung, C.H.; Jun, C.B.; Ro, S.H.; Kim, Y.M.; Otto, N.M.; Cao, J.; Kundu, M.; Kim, D.H. ULK–Atg13–FIP200 complexes mediate mTOR signaling to the autophagy machinery. Mol. Biol. Cell 2009, 20, 1992–2003. [Google Scholar]
- Alers, S.; Löffler, A.S.; Wesselborg, S.; Stork, B. The incredible ULKs. Cell Commun. Signal. 2012, 10, 7. [Google Scholar]
- Bae, H.; Guan, J.L. Suppression of autophagy by FIP200 deletion impairs DNA damage repair and increases cell death upon treatments with anticancer agents. Mol. Cancer Res. 2011, 9, 1232–1241. [Google Scholar]
- Moscat, J.; Diaz-Meco, M.T.; Wooten, M.W. Signal integration and diversification through the p62 scaffold protein. Trends Biochem. Sci. 2007, 32, 95–100. [Google Scholar]
- Vadlamudi, R.K.; Joung, I.; Strominger, J.L.; Shin, J. p62, a phosphotyrosine-independent ligand of the SH2 domain of p56lck, belongs to a new class of ubiquitin-binding proteins. J. Biol. Chem. 1996, 271, 20235–20237. [Google Scholar]
- Pohl, C.; Jentsch, S. Midbody ring disposal by autophagy is a post-abscission event of cytokinesis. Nat. Cell Biol. 2009, 11, 65–70. [Google Scholar]
- Pankiv, S.; Clausen, T.H.; Lamark, T.; Brech, A.; Bruun, J.A.; Outzen, H.; Øvervatn, A.; Bjørkøy, G.; Johansen, T. p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J. Biol. Chem. 2007, 282, 24131–24145. [Google Scholar]
- Wooten, M.W.; Geetha, T.; Babu, J.R.; Seibenhener, M.L.; Peng, J.; Cox, N.; Diaz-Meco, M.T.; Moscat, J. Essential role of sequestosome 1/p62 in regulating accumulation of Lys63-ubiquitinated proteins. J. Biol. Chem. 2008, 283, 6783–6789. [Google Scholar]
- Pankiv, S.; Lamark, T.; Bruun, J.A.; Øvervatn, A.; Bjørkøy, G.; Johansen, T. Nucleocytoplasmic shuttling of p62/SQSTM1 and its role in recruitment of nuclear polyubiquitinated proteins to promyelocytic leukemia bodies. J. Biol. Chem. 2010, 285, 5941–5953. [Google Scholar]
- Lallemand-Breitenbach, V.; de The, H. PML nuclear bodies. Cold Spring Harb. Perspect. Biol. 2010, 2, a000661. [Google Scholar]
- Mathew, R.; Kongara, S.; Beaudoin, B.; Karp, C.M.; Bray, K.; Degenhardt, K.; Chen, G.; Jin, S.; White, E. Autophagy suppresses tumor progression by limiting chromosomal instability. Genes Dev. 2007, 21, 1367–1381. [Google Scholar]
- Scheffler, I.E. Mitochondria,, 2nd ed.; Wiley: Hoboken, NJ, USA, 2008. [Google Scholar]
- Greaves, L.C.; Reeve, A.K.; Taylor, R.W.; Turnbull, D.M. Mitochondrial DNA and disease. J. Pathol. 2012, 226, 274–286. [Google Scholar]
- Kim, I.; Rodriguez-Enriquez, S.; Lemasters, J.J. Selective degradation of mitochondria by mitophagy. Arch. Biochem. Biophys. 2007, 462, 245–253. [Google Scholar]
- Ashford, T.P.; Porter, K.R. Cytoplasmic components in hepatic cell lysosomes. J. Cell Biol. 1962, 12, 198–202. [Google Scholar]
- Novak, I.; Kirkin, V.; McEwan, D.G.; Zhang, J.; Wild, P.; Rozenknop, A.; Rogov, V.; Löhr, F.; Popovic, D.; Occhipinti, A.; et al. Nix is a selective autophagy receptor for mitochondrial clearance. EMBO Rep. 2010, 11, 45–51. [Google Scholar]
- Ding, W.X.; Ni, H.M.; Li, M.; Liao, Y.; Chen, X.; Stolz, D.B.; Dorn, G.W., 2nd.; Yin, X.M. Nix is critical to two distinct phases of mitophagy, reactive oxygen species-mediated autophagy induction and Parkin-ubiquitin-p62-mediated mitochondrial priming. J. Biol. Chem. 2010, 285, 27879–27890. [Google Scholar]
- Matsuda, S.; nakanishi, A.; Minami, A.; Wada, Y.; Kitagishi, Y. Functions and characteristics of PINK1 and Parkin in cancer. Front. Biosci. 2015, 20, 491–501. [Google Scholar]
- Geisler, S.; Holmström, K.M.; Treis, A.; Skujat, D.; Weber, S.S.; Fiesel., F.C.; Kahle, P.J.; Springer, W. The PINK1/Parkin-mediated mitophagy is compromised by PD-associated mutations. Autophagy 2010, 6, 871–878. [Google Scholar]
- Narendra, D.; Tanaka, A.; Suen, D.F.; Youle, R.J. Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J. Cell Biol. 2008, 183, 795–803. [Google Scholar]
- Vives-Bauza, C.; Zhou, C.; Huang, Y.; Cui, M.; de Vries, R.L.; Kim, J.; May, J.; Tocilescu, M.A.; Liu, W.; Ko, H.S.; et al. PINK1-dependent recruitment of Parkin to mitochondria in mitophagy. Proc. Natl. Acad. Sci. USA 2010, 107, 378–383. [Google Scholar]
- Vila, M.; Ramonet, D.; Perier, C. Mitochondrial alterations in Parkinson’s disease: New clues. J. Neurochem. 2008, 107, 317–328. [Google Scholar]
- Gil, J.M.; Rego, A.C. Mechanisms of neurodegeneration in Huntington’s disease. Eur. J. Neurosci. 2008, 27, 2803–2820. [Google Scholar]
- Bess, A.S.; Ryde, I.T.; Hinton, D.E.; Meyer, J.N. UVC-induced mitochondrial degradation via autophagy correlates with mtDNA damage removal in primary human fibroblasts. J. Biochem. Mol. Toxicol. 2013, 27, 28–41. [Google Scholar]
- Scheibye-Knudsen, M.; Ramamoorthy, M.; Sykora, P.; Maynard, S.; Lin, P.C.; Minor, R.K.; Wilson, D.M., 3rd.; Cooper, M.; Spencer, R.; de Cabo, R.; et al. Cockayne syndrome group B protein prevents the accumulation of damaged mitochondria by promoting mitochondrial autophagy. J. Exp. Med. 2012, 209, 855–869. [Google Scholar]
- Monick, M.M.; Powers, L.S.; Walters, K.; Lovan, N.; Zhang, M.; Gerke, A.; Hansdottir, S.; Hunninghake, G.W. Identification of an autophagy defect in smokers’ alveolar macrophages. J. Immunol. 2010, 185, 5425–5435. [Google Scholar]
- Chen, L.H.; Chu, P.M.; Lee, Y.J.; Tu, P.H.; Chi, C.W.; Lee, H.C.; Chiou, S.H. Targeting protective autophagy exacerbates UV-triggered apoptotic cell death. Int. J. Mol. Sci. 2012, 13, 1209–1224. [Google Scholar]
- Rodriguez-Hernandez, A.; Cordero, M.D.; Salviati, L.; Artuch, R.; Pineda, M.; Briones, P.; Gómez Izquierdo, L.; Cotán, D.; Navas, P.; Sánchez-Alcázar, J.A.; et al. Coenzyme Q deficiency triggers mitochondria degradation by mitophagy. Autophagy 2009, 5, 19–32. [Google Scholar] [Green Version]
- Cotan, D.; Cordero, M.D.; Garrido-Maraver, J.; Oropesa-Ávila, M.; Rodríguez-Hernández, A.; Gómez Izquierdo, L.; de la Mata, M.; de Miguel, M.; Lorite, J.B.; Infante, E.R.; et al. Secondary coenzyme Q10 dficiency triggers mitochondria degradation by mitophagy in MELAS fibroblasts. FASEB J. 2011, 25, 2669–2687. [Google Scholar]
- Priault, M.; Salin, B.; Schaeffer, J.; Vallette, F.M.; di Rago, J.P.; Martinou, J.C. Impairing the bioenergetic status and the biogenesis of mitochondria triggers mitophagy in yeast. Cell Death Differ. 2005, 12, 1613–1621. [Google Scholar]
- Elmore, S.P.; Qian, T.; Grissom, S.F.; Lemasters, J.J. The mitochondrial permeability transition initiates autophagy in rat hepatocytes. FASEB J. 2001, 15, 2286–2287. [Google Scholar]
- Gu, Y.; Wang, C.; Cohen, A. Effect of IGF-1 on the balance between autophagy of dysfunctional mitochondria and apoptosis. FEBS Lett. 2004, 577, 357–360. [Google Scholar]
- Amaravadi, R.K.; Lippincott-Schwartz, J.; Yin, X.M.; Weiss, W.A.; Takebe, N.; Timmer, W.; di Paola, R.S.; Lotze, M.T.; White, E. Principles and current strategies for targeting autophagy for cancer treatment. Clin. Cancer Res. 2011, 17, 654–666. [Google Scholar]
- Edinger, A.L.; Thompson, C.B. Death by design: Apoptosis, necrosis and autophagy. Curr. Opin. Cell Biol. 2004, 16, 663–669. [Google Scholar]
- Maycotte, P.; Thorburn, A. Autophagy and cancer therapy. Cancer Biol. Ther. 2011, 11, 127–137. [Google Scholar]
- Kenzelmann Broz, D.; Spano Mello, S.; Bieging, K.T.; Jiang, D.; Dusek, R.L.; Brady, C.A.; Sidow, A.; Attardi, L.D. Global genomic profiling reveals an extensive p53-regulated autophagy program contributing to key p53 responses. Genes Dev. 2013, 27, 1016–1031. [Google Scholar]
- Liu, H.; He, Z.; Simon, H.U. Targeting autophagy as a potential therapeutic approach for melanoma therapy. Semin. Cancer Biol. 2013, 23, 352–360. [Google Scholar]
- Cui, J.; Gong, Z.; Shen, H.M. The role of autophagy in liver cancer: Molecular mechanisms and potential therapeutic targets. Biochim. Biophys. Acta 2013, 1836, 15–26. [Google Scholar]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Czarny, P.; Pawlowska, E.; Bialkowska-Warzecha, J.; Kaarniranta, K.; Blasiak, J. Autophagy in DNA Damage Response. Int. J. Mol. Sci. 2015, 16, 2641-2662. https://doi.org/10.3390/ijms16022641
Czarny P, Pawlowska E, Bialkowska-Warzecha J, Kaarniranta K, Blasiak J. Autophagy in DNA Damage Response. International Journal of Molecular Sciences. 2015; 16(2):2641-2662. https://doi.org/10.3390/ijms16022641
Chicago/Turabian StyleCzarny, Piotr, Elzbieta Pawlowska, Jolanta Bialkowska-Warzecha, Kai Kaarniranta, and Janusz Blasiak. 2015. "Autophagy in DNA Damage Response" International Journal of Molecular Sciences 16, no. 2: 2641-2662. https://doi.org/10.3390/ijms16022641