A Cautionary Note on the Use of Split-YFP/BiFC in Plant Protein-Protein Interaction Studies

Abstract
:1. Introduction
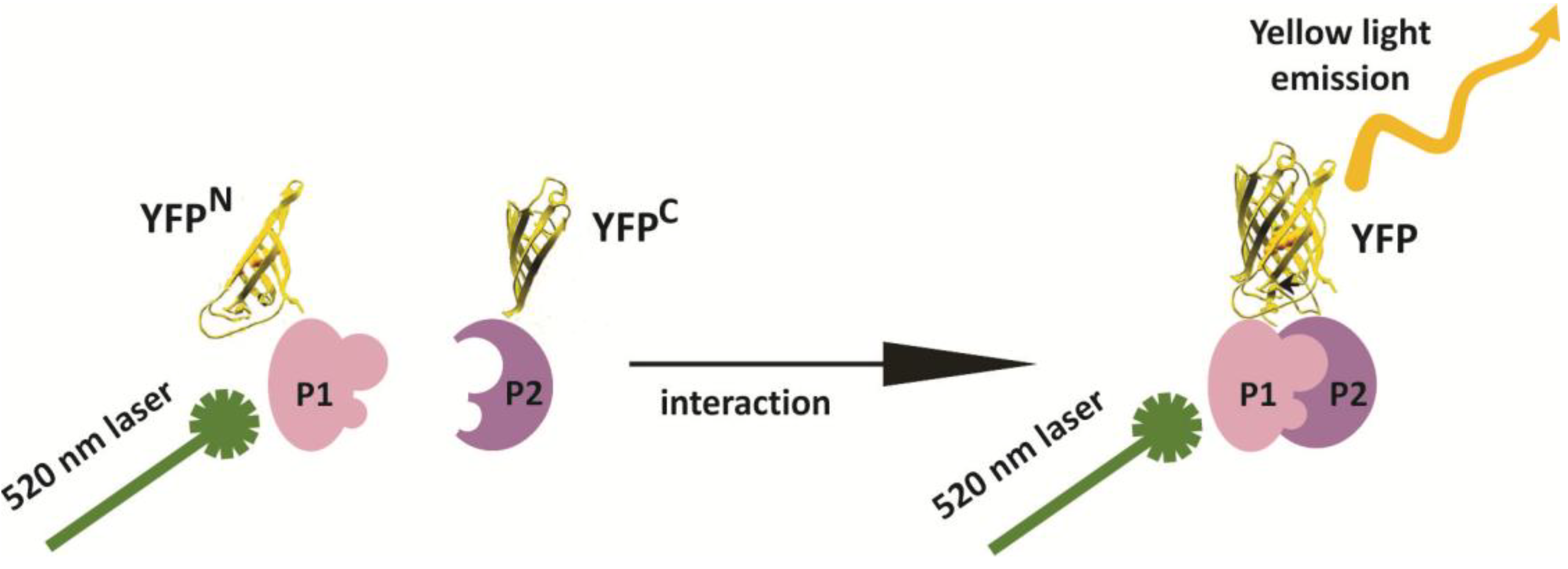
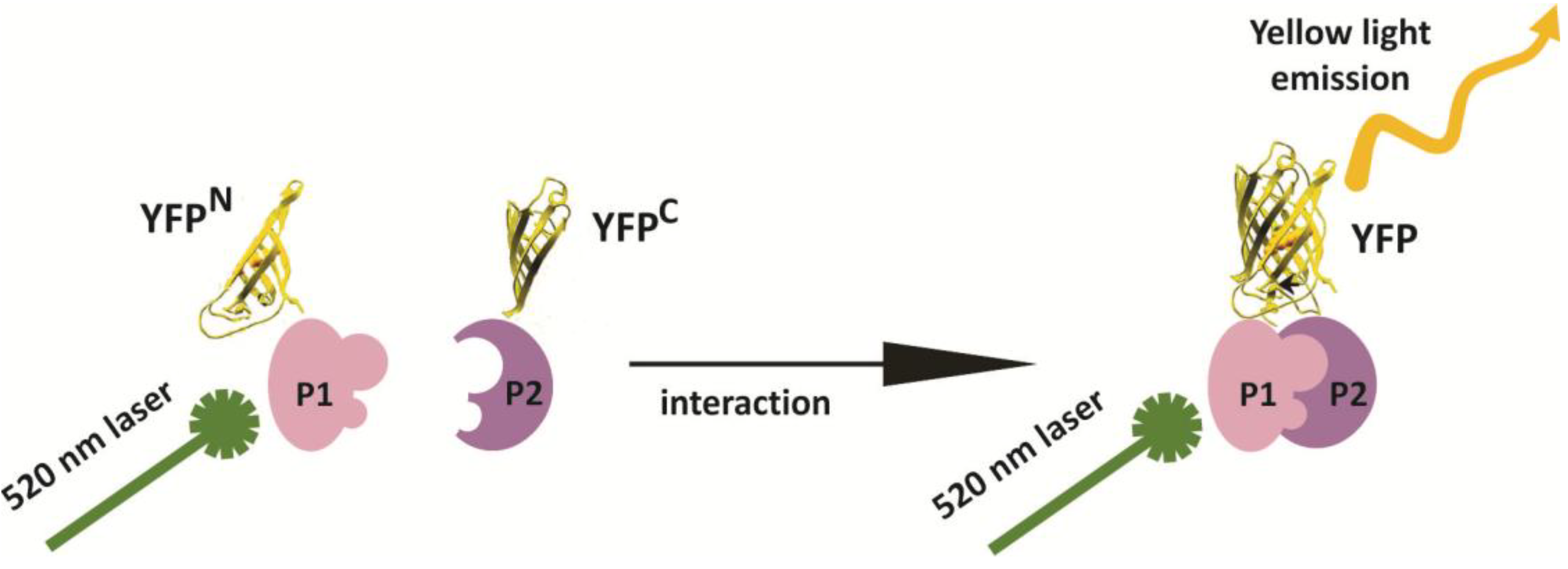
2. Results and Discussion
2.1. Inventory of BiFC (Bimolecular Fluorescence Complementation) Use in Plant Studies
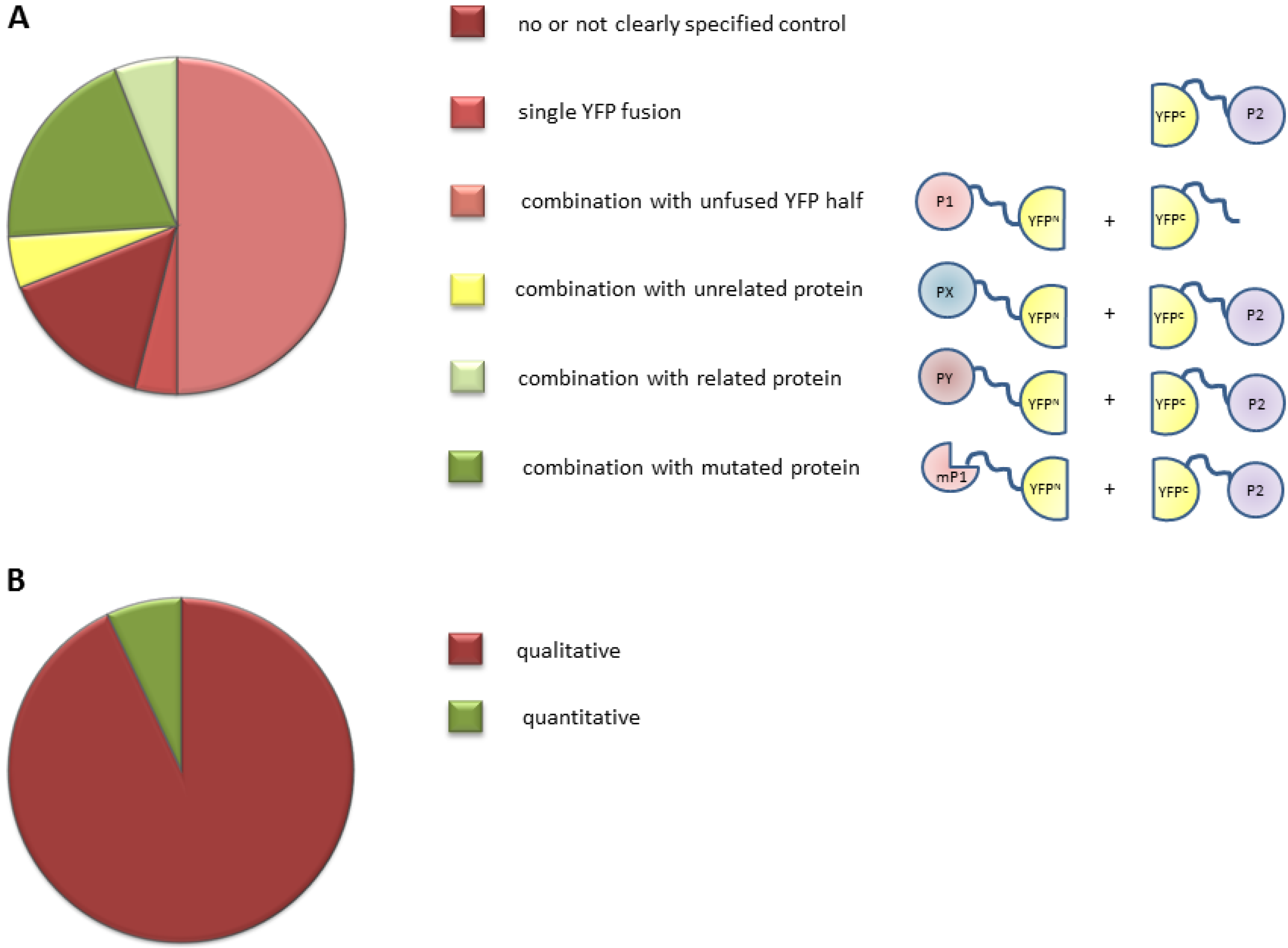
2.2. Overview of the BiFC Method
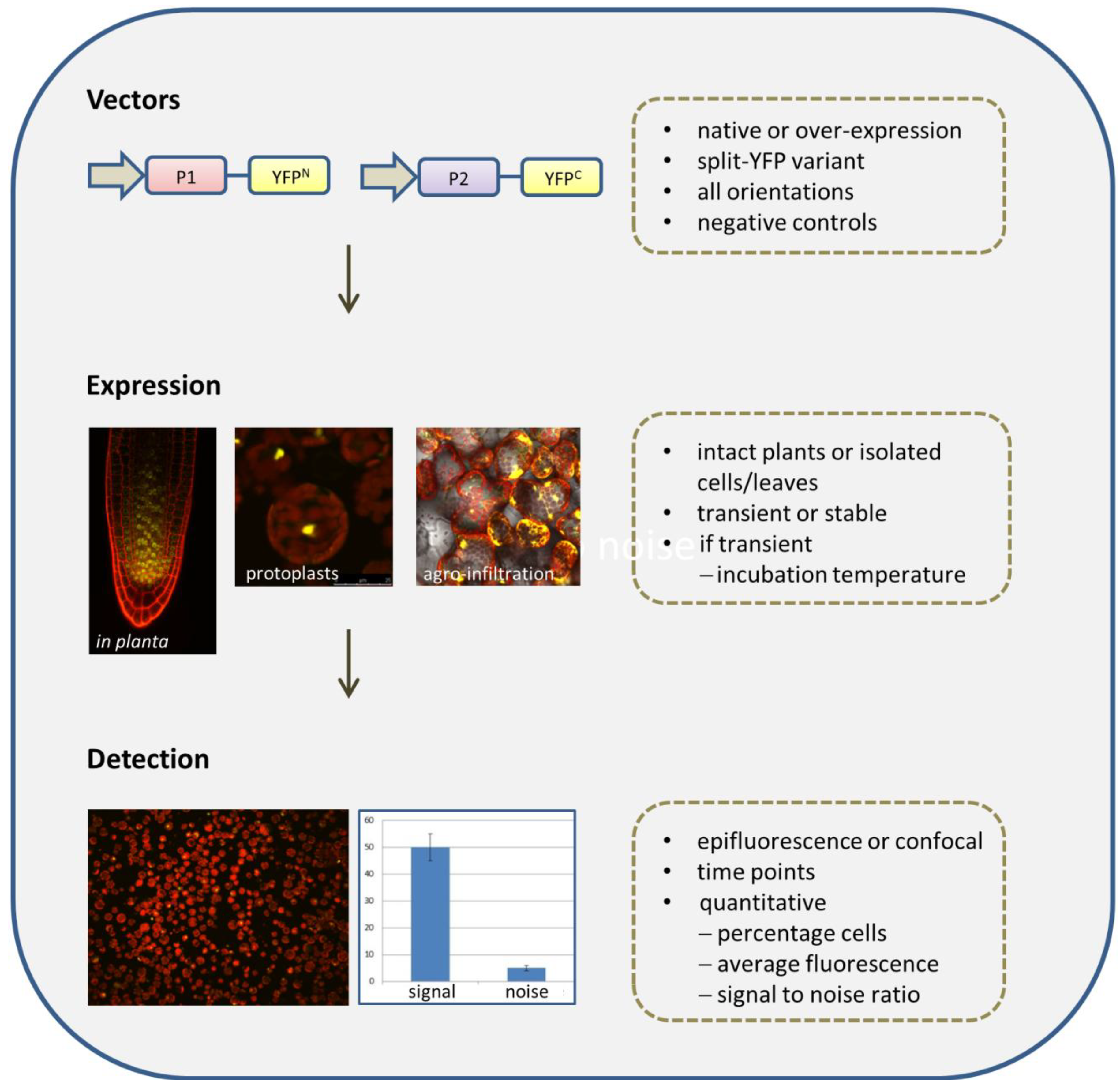
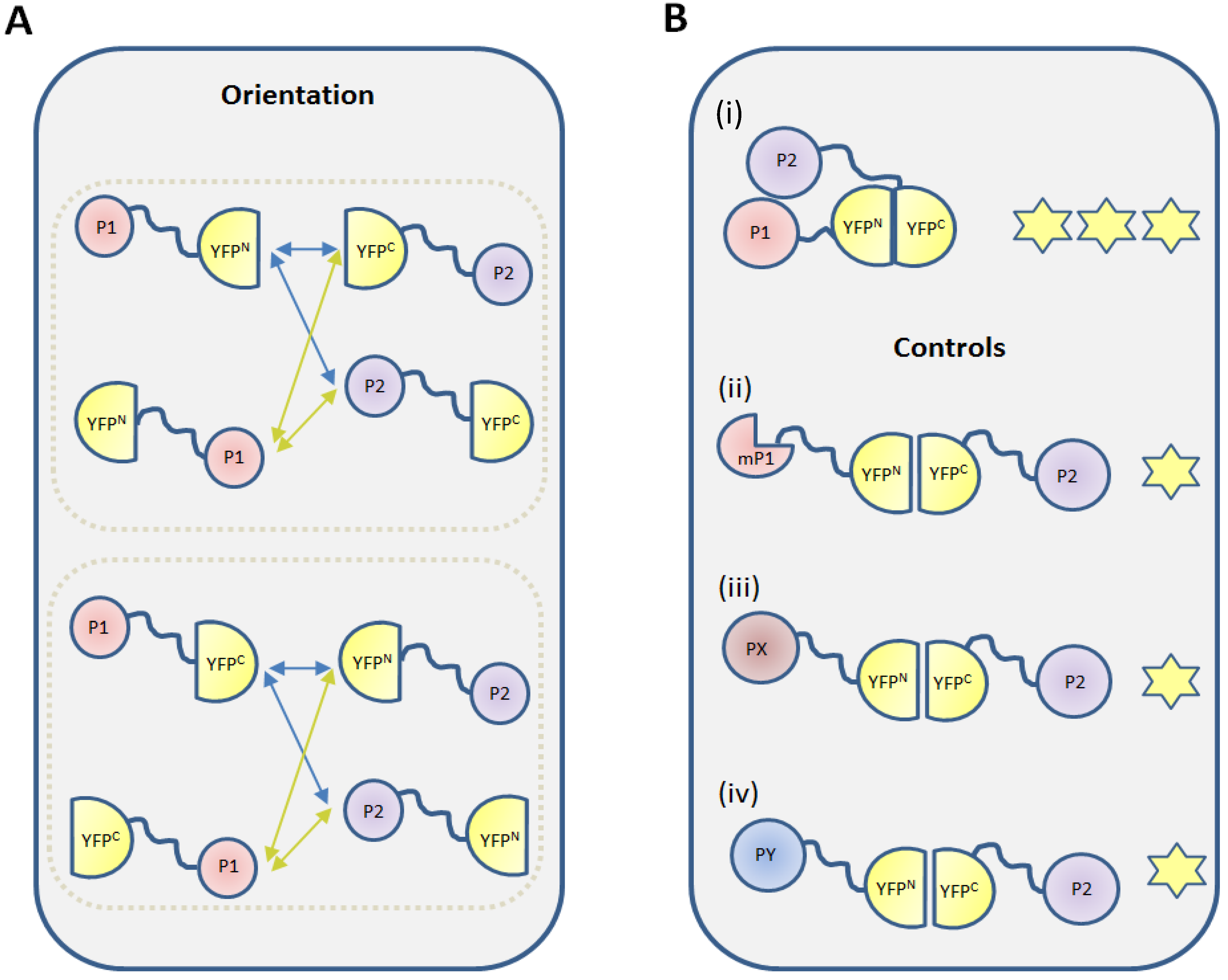
2.2.1. Selection of Vectors

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| BiFC Vector Set | Promoter | Protein of Interest | Peptide Linker | Length (AA) | YFP Part | Terminator |
|---|---|---|---|---|---|---|
| 1 [24] | CaMV35S | FBP2 | Gateway | 17 | YFPN | NOS |
| FBP11 | Gateway | 18 | YFPC | |||
| 2 | FBP2 | RSIAT | 15 | YFPN | ||
| FBP11 | KQKVMNH | 17 | YFPC | |||
| 3 [7] | FBP2 | Myc-c tag | 26 | YFPN | ||
| FBP11 | HA-tag | 25 | YFPC |
2.2.2. Fusion Orientations

2.2.3. Negative Controls

2.2.4. The BiFC Assay: Expression Systems
2.2.5. Detection Methods
3. Experimental Section
3.1. Test of Different Peptide Linker Sequences
3.2. Self-Assembly Capacity of YFP (Yellow Fluorescent Protein) Halves
4. Conclusions
Supplementary Files
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Braun, P.; Aubourg, S.; van Leene, J.; de Jaeger, G.; Lurin, C. Plant Protein Interactomes. Annu. Rev. Plant Biol. 2013, 64, 161–187. [Google Scholar]
- Immink, R.G.H.; Angenent, G.C. Transcription factors do it together: the hows and whys of studying protein-protein interactions. Trends Plant Sci. 2002, 7, 531–534. [Google Scholar]
- Kodama, Y.; Hu, C.-D. Bimolecular fluorescence complementation (BiFC): A 5-year update and future perspectives. BioTechniques 2012, 53, 285–298. [Google Scholar]
- Ghosh, I.; Hamilton, A.D.; Regan, L. Antiparallel leucine zipper-directed protein reassembly: Application to the green fluorescent protein. J. Am. Chem. Soc. 2000, 122, 5658–5659. [Google Scholar]
- Hu, C.-D.; Chinenov, Y.; Kerppola, T.K. Visualization of interactions among bZIP and Rel family proteins in living cells using bimolecular fluorescence complementation. Mol. Cell 2002, 9, 789–798. [Google Scholar]
- Bracha-Drori, K.; Shichrur, K.; Katz, A.; Oliva, M.; Angelovici, R.; Yalovsky, S.; Ohad, N. Detection of protein-protein interactions in plants using bimolecular fluorescence complementation. Plant J. 2004, 40, 419–427. [Google Scholar]
- Walter, M.; Chaban, C.; Schütze, K.; Batistic, O.; Weckermann, K.; Näke, C.; Blazevic, D.; Grefen, C.; Schumacher, K.; Oecking, C.; et al. Visualization of protein interactions in living plant cells using bimolecular fluorescence complementation. Plant J. 2004, 40, 428–438. [Google Scholar]
- Kerppola, T.K. Multicolor Bimolecular Fluorescence Complementation (BiFC) Analysis of Protein Interactions with Alternative Partners. Cold Spring Harb. Protoc. 2013, 2013. [Google Scholar] [CrossRef]
- Lee, L.-Y.; Fang, M.-J.; Kuang, L.-Y.; Gelvin, S. Vectors for multi-color bimolecular fluorescence complementation to investigate protein-protein interactions in living plant cells. Plant Methods 2008, 4. [Google Scholar] [CrossRef]
- Waadt, R.; Schmidt, L.K.; Lohse, M.; Hashimoto, K.; Bock, R.; Kudla, J. Multicolor bimolecular fluorescence complementation reveals simultaneous formation of alternative CBL/CIPK complexes in planta. Plant J. 2008, 56, 505–516. [Google Scholar]
- Shyu, Y.J.; Suarez, C.D.; Hu, C.-D. Visualization of ternary complexes in living cells by using a BiFC-based FRET assay. Nat. Protoc. 2008, 3, 1693–1702. [Google Scholar]
- Shyu, Y.J.; Suarez, C.D.; Hu, C.-D. Visualization of AP-1-NF-κB ternary complexes in living cells by using a BiFC-based FRET. Proc. Natl. Acad. Sci.USA 2008, 105, 151–156. [Google Scholar]
- Smaczniak, C.; Immink, R.G.H.; Muiño, J.M.; Blanvillain, R.; Busscher, M.; Busscher-Lange, J.; Dinh, Q.D.; Liu, S.; Westphal, A.H.; Boeren, S.; et al. Characterization of MADS-domain transcription factor complexes in Arabidopsis flower development. Proc. Natl. Acad Sci. USA 2012, 109, 1560–1565. [Google Scholar]
- Ohashi, K.; Kiuchi, T.; Shoji, K.; Sampei, K.; Mizuno, K. Visualization of cofilin-actin and Ras-Raf interactions by bimolecular fluorescence complementation assays using a new pair of split Venus fragments. BioTechniques 2012, 52, 45–50. [Google Scholar]
- Kodama, Y.; Hu, C.-D. An improved bimolecular fluorescence complementation assay with a high signal-to-noise ratio. BioTechniques 2010, 49, 793–805. [Google Scholar]
- Nakagawa, C.; Inahata, K.; Nishimura, S.; Sugimoto, K. Improvement of a venus-based bimolecular fluorescence complementation assay to visualize bFos-bJun interaction in living cells. Biosci. Biotechnol. Biochem. 2011, 75, 1399–1401. [Google Scholar]
- Kerppola, T.K. Design and implementation of bimolecular fluorescence complementation (BiFC) assays for the visualization of protein interactions in living cells. Nat. Protoc. 2006, 1, 1278–1286. [Google Scholar]
- Fang, Y.; Spector, D.L. BiFC imaging assay for plant protein-protein interactions. Cold Spring Harb. Protoc. 2010, 2010. [Google Scholar] [CrossRef]
- Saka, Y.; Hagemann, A.I.; Smith, J.C. Visualizing protein interactions by bimolecular fluorescence complementation in Xenopus. Methods 2008, 45, 192–195. [Google Scholar]
- PubMed-NCBI: US National Library of Medicine National Institutes of Health. Available online: http://www.ncbi.nlm.nih.gov/pubmed (accessed on 1 February 2014).
- Lee, L.Y.; Wu, F.H.; Hsu, C.T.; Shen, S.C.; Yeh, H.Y.; Liao, D.C.; Fang, M.J.; Liu, N.T.; Yen, Y.C.; Dokladal, L.; et al. Screening a cDNA library for protein-protein interactions directly in planta. Plant Cell 2012, 24, 1746–1759. [Google Scholar]
- Arai, R.; Ueda, H.; Kitayama, A.; Kamiya, N.; Nagamune, T. Design of the linkers which effectively separate domains of a bifunctional fusion protein. Protein Eng. 2001, 14, 529–532. [Google Scholar]
- Welch, D.; Hassan, H.; Blilou, I.; Immink, R.; Heidstra, R.; Scheres, B. Arabidopsis JACKDAW and MAGPIE zinc finger proteins delimit asymmetric cell division and stabilize tissue boundaries by restricting SHORT-ROOT action. Genes Dev. 2007, 21, 2196–2204. [Google Scholar]
- Hartley, J.L.; Temple, G.F.; Brasch, M.A. DNA cloning using in vitro site-specific recombination. Genome Res. 2000, 10, 1788–1795. [Google Scholar]
- Busso, D.; Dleagoutte-Busso, B.; Moras, D. Construction of a set gateway-based destination vectors for high-throughput cloning and expression screening in Eschericia coli. Anal. Biochem. 2005, 343, 313–321. [Google Scholar]
- Jang, C.; Seo, E.Y.; Nam, J.; Bae, H.; Gim, Y.G.; Kim, H.G.; Cho, I.S.; Lee, Z.W.; Bauchan, G.R.; Hammond, J.; et al. Insights into alternanthera mosaic virus TGB3 functions: Interactions with nicotiana benthamiana PsbO correlate with chloroplast vesiculation and veinal necrosis caused by TGB3 over-expression. Front. Plant Sci. 2013, 4. [Google Scholar] [CrossRef]
- Park, M.-R.; Kim, K.-H. Molecular characterization of the interaction between the N-terminal region of Potato virus X (PVX) coat protein (CP) and Nicotiana benthamiana PVX CP-interacting protein, NbPCIP1. Virus Genes 2013, 46, 517–523. [Google Scholar]
- Dong, C.H.; Jang, M.; Scharein, B.; Malach, A.; Rivarola, M.; Liesch, J.; Groth, G.; Hwang, I.; Chang, C. Molecular association of the Arabidopsis ETR1 ethylene receptor and a regulator of ethylene signaling, RTE1. J. Biol. Chem. 2010, 285, 40706–40713. [Google Scholar]
- Ren, X.L.; Qi, G.N.; Feng, H.Q.; Zhao, S.; Zhao, S.S.; Wang, Y.; Wu, W.H. Calcineurin B-like protein CBL10 directly interacts with AKT1 and modulates K+ homeostasis in Arabidopsis. Plant J. Cell Mol. Biol. 2013, 74, 258–266. [Google Scholar]
- Merzlyak, E.M.; Goedhart, J.; Shcherbo, D.; Bulina, M.E.; Shcheglov, A.S.; Fradkov, A.F.; Gaintzeva, A.; Lukyanov, K.A.; Lukyanov, S.; Gadella, T.W.J.; et al. Bright monomeric red fluorescent protein with an extended fluorescence lifetime. Nat. Methods 2007, 4, 555–557. [Google Scholar]
- Boutilier, K.; Offringa, R.; Sharma, V.K.; Kieft, H.; Ouellet, T.; Zhang, L.; Hattori, J.; Liu, C.-M.; van Lammeren, A.A.M.; Miki, B.L.A.; et al. Ectopic Expression of BABY BOOM Triggers a Conversion from Vegetative to Embryonic Growth. Plant Cell 2002, 14, 1737–1749. [Google Scholar]
- Boevink, P.; McLellan, H.; Bukharova, T.; Engelhardt, S.; Birch, P. In vivo protein-protein interaction studies with BiFC: Conditions, cautions, and caveats. Methods Mol. Biol. 2014, 1127, 81–90. [Google Scholar]
- Schutze, K.; Harter, K.; Chaban, C. Bimolecular fluorescence complementation (BiFC) to study protein-protein interactions in living plant cells. Methods Mol. Biol. 2009, 479, 189–202. [Google Scholar]
- Waadt, R.; Kudla, J. In planta visualization of protein interactions using bimolecular fluorescence complementation (BiFC). CSH Protoc. 2008, 2008. [Google Scholar] [CrossRef]
- Denecke, J.; Gossele, V.; Botterman, J.; Cornelissen, M. Quantitative analysis of transiently expressed genes in plant cells. Methods Mol. Cell. Biol. 1989, 1, 19–27. [Google Scholar]
- Immink, R.G.H.; Gadella, T.W.J.; Ferrario, S.; Busscher, M.; Angenent, G.C. Analysis of MADS box protein–protein interactions in living plant cells. Proc. Natl. Acad. Sci.USA 2002, 99, 2416–2421. [Google Scholar]
- Tonaco, I.A.N.; Borst, J.W.; de Vries, S.C.; Angenent, G.C.; Immink, R.G.H. In vivo imaging of MADS-box transcription factor interactions. J. Exp. Bot. 2006, 57, 33–42. [Google Scholar]
- Shyu, Y.J.; Liu, H.; Deng, X.; Hu, C.-D. Identification of new fluorescent protein fragments for bimolecular fluorescence complementation analysis under physiological conditions. BioTechniques 2006, 40, 61–66. [Google Scholar]
- Berendzen, K.W.; Bohmer, M.; Wallmeroth, N.; Peter, S.; Vesic, M.; Zhou, Y.; Tiesler, F.K.E.; Schleifenbaum, F.; Harter, K. Screening for in planta protein-protein interactions combining bimolecular fluorescence complementation with flow cytometry. Plant Methods 2012, 8. [Google Scholar] [CrossRef]
- Denecke, J.; Vitale, A. The use of protoplasts to study protein synthesis and transport by the plant endomembrane system. Methods Cell Biol. 1995, 50, 335–348. [Google Scholar]
- Bücherl, C.; Aker, J.; Vries, S.; Borst, J.W. Probing protein-protein interactions with FRET-FLIM. Methods Mol. Biol. 2010, 655, 389–399. [Google Scholar]
- Morsy, M.; Gouthu, S.; Orchard, S.; Thorneycroft, D.; Harper, J.F.; Mittler, R.; Cushman, J.C. Charting plant interactomes: possibilities and challenges. Trends Plant Sci. 2008, 13, 183–191. [Google Scholar]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Horstman, A.; Tonaco, I.A.N.; Boutilier, K.; Immink, R.G.H. A Cautionary Note on the Use of Split-YFP/BiFC in Plant Protein-Protein Interaction Studies. Int. J. Mol. Sci. 2014, 15, 9628-9643. https://doi.org/10.3390/ijms15069628
Horstman A, Tonaco IAN, Boutilier K, Immink RGH. A Cautionary Note on the Use of Split-YFP/BiFC in Plant Protein-Protein Interaction Studies. International Journal of Molecular Sciences. 2014; 15(6):9628-9643. https://doi.org/10.3390/ijms15069628
Chicago/Turabian StyleHorstman, Anneke, Isabella Antonia Nougalli Tonaco, Kim Boutilier, and Richard G. H. Immink. 2014. "A Cautionary Note on the Use of Split-YFP/BiFC in Plant Protein-Protein Interaction Studies" International Journal of Molecular Sciences 15, no. 6: 9628-9643. https://doi.org/10.3390/ijms15069628
APA StyleHorstman, A., Tonaco, I. A. N., Boutilier, K., & Immink, R. G. H. (2014). A Cautionary Note on the Use of Split-YFP/BiFC in Plant Protein-Protein Interaction Studies. International Journal of Molecular Sciences, 15(6), 9628-9643. https://doi.org/10.3390/ijms15069628