Identification of Drivers from Cancer Genome Diversity in Hepatocellular Carcinoma

Abstract
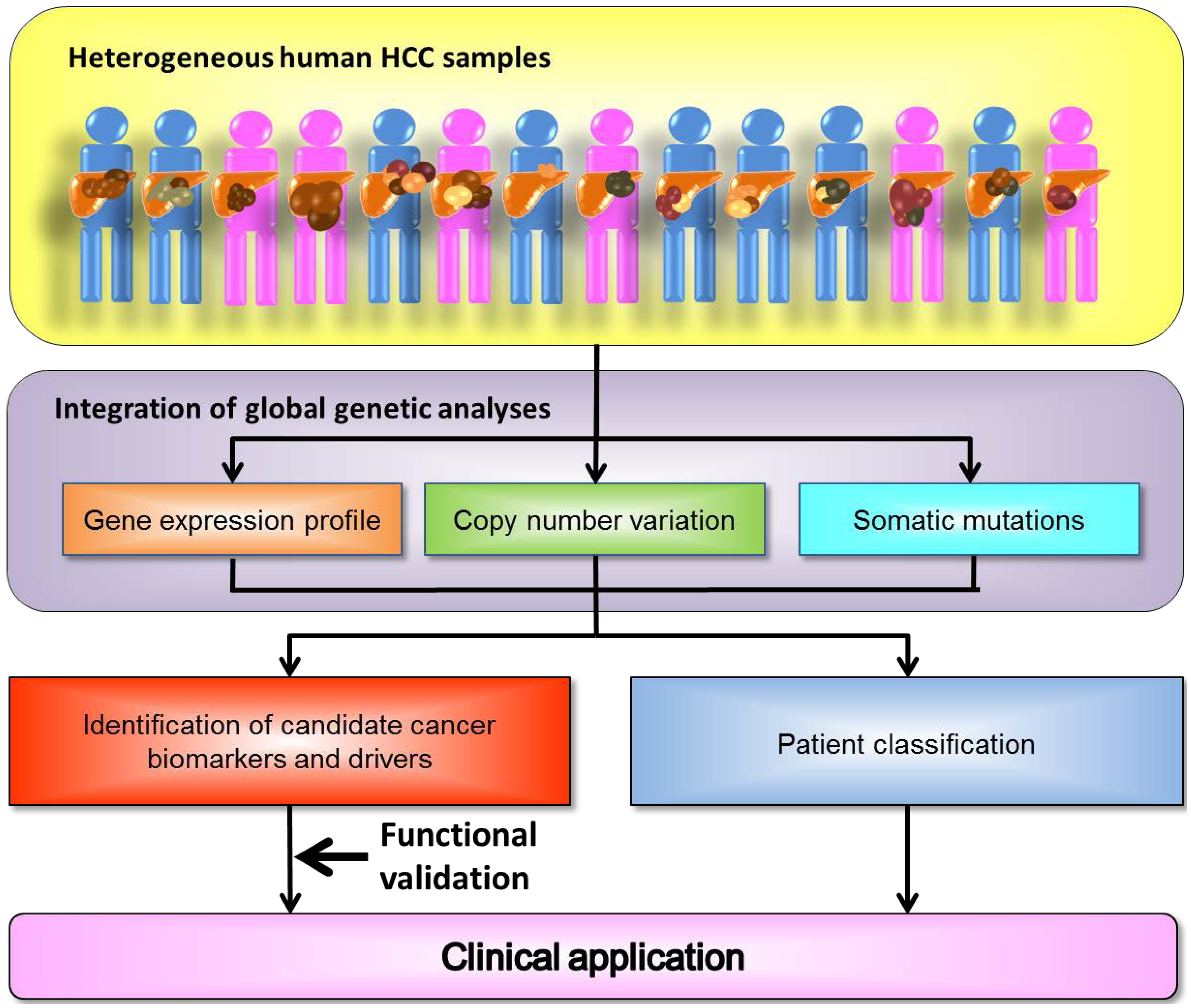
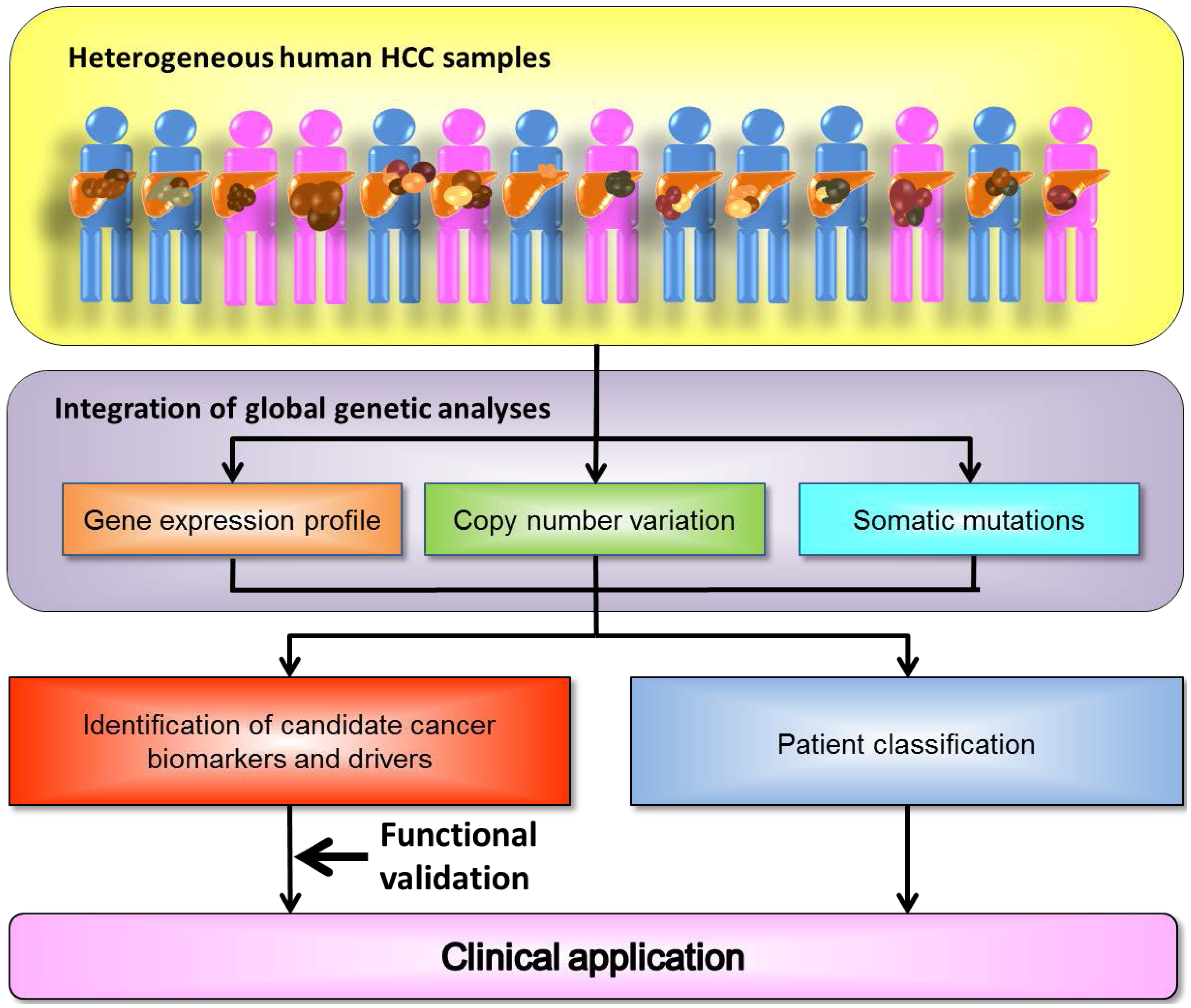
:1. The Landscape of the Cancer Genome
2. Heterogeneity of Genetic Alterations in Hepatocellular Carcinoma (HCC)
3. Gene Expression Profiles in HCC
4. Copy Number Alterations in HCC
5. Somatic Mutations in HCC

{kind=link}
| Cell Function | Gene Name | Frequency of Mutations | Gene Expression Dysregulation | Chromosomal Alterations the Gene Is Located | |
|---|---|---|---|---|---|
| Deep Sequencing * | COSMIC Database | ||||
| Cell cycle | TP53 | 48%, 27%, 21% | 31% | Decreased ** | 17p loss † |
| IRF2 | 0%, 0%, 5% | 1% | Unreported | 4q loss †† | |
| CDKN2A | 0%, 0%, 7%, | 10% | Decreased ‡ | 9p loss ‡‡ | |
| Cell proliferation | CTNNB1 | 11%, 0%, 33% | 19% | Increased ¶ | Unreported |
| AXIN1 | 0%, 0%, 15% | 13% | Decreased ¶¶ | 16p loss § | |
| KRAS | 0%, 0%, 1.6% | 2% | Unreported | Unreported | |
| PIK3CA | 7%, 0%, 1.6% | 6% | Unreported | Unreported | |
| ERRFI1 | 7%, 0%, 0% | 2% | Unreported | 1p loss §§ | |
| Chromatin remodeling | ARID1A | 11%, 13%, 17% | 14% | Unreported | Unreported |
| ARID2 | 7%, 4%, 6% | 9% | Unreported | Unreported | |
6. The Screening of Cancer Drivers
7. Future Directions
Acknowledgments
Conflicts of Interest
References
- Nowell, P.C. The clonal evolution of tumor cell populations. Science 1976, 194, 23–28. [Google Scholar]
- Hanahan, D.; Weinberg, R.A. The hallmarks of cancer. Cell 2000, 100, 57–70. [Google Scholar] [CrossRef]
- Vogelstein, B.; Kinzler, K.W. The multistep nature of cancer. Trends Genet. 1993, 9, 138–141. [Google Scholar] [CrossRef]
- Perreard, L.; Fan, C.; Quackenbush, J.F.; Mullins, M.; Gauthier, N.P.; Nelson, E.; Mone, M.; Hansen, H.; Buys, S.S.; Rasmussen, K.; et al. Classification and risk stratification of invasive breast carcinomas using a real-time quantitative RT-PCR assay. Breast Cancer Res. 2006, 8, R23. [Google Scholar]
- Greenman, C.; Stephens, P.; Smith, R.; Dalgliesh, G.L.; Hunter, C.; Bignell, G.; Davies, H.; Teague, J.; Butler, A.; Stevens, C.; et al. Patterns of somatic mutation in human cancer genomes. Nature 2007, 446, 153–158. [Google Scholar] [CrossRef]
- Wood, L.D.; Parsons, D.W.; Jones, S.; Lin, J.; Sjoblom, T.; Leary, R.J.; Shen, D.; Boca, S.M.; Barber, T.; Ptak, J.; et al. The genomic landscapes of human breast and colorectal cancers. Science 2007, 318, 1108–1113. [Google Scholar]
- Jones, S.; Zhang, X.; Parsons, D.W.; Lin, J.C.; Leary, R.J.; Angenendt, P.; Mankoo, P.; Carter, H.; Kamiyama, H.; Jimeno, A.; et al. Core signaling pathways in human pancreatic cancers revealed by global genomic analyses. Science 2008, 321, 1801–1806. [Google Scholar] [CrossRef]
- Cancer Genome Atlas Research Network. Integrated genomic analyses of ovarian carcinoma. Nature 2011, 474, 609–615. [Google Scholar] [CrossRef] [Green Version]
- Stransky, N.; Egloff, A.M.; Tward, A.D.; Kostic, A.D.; Cibulskis, K.; Sivachenko, A.; Kryukov, G.V.; Lawrence, M.S.; Sougnez, C.; McKenna, A.; et al. The mutational landscape of head and neck squamous cell carcinoma. Science 2011, 333, 1157–1160. [Google Scholar] [CrossRef] [Green Version]
- Barbieri, C.E.; Baca, S.C.; Lawrence, M.S.; Demichelis, F.; Blattner, M.; Theurillat, J.P.; White, T.A.; Stojanov, P.; van Allen, E.; Stransky, N.; et al. Exome sequencing identifies recurrent SPOP, FOXA1 and MED12 mutations in prostate cancer. Nat. Genet. 2012, 44, 685–689. [Google Scholar] [CrossRef]
- Cancer Genome Atlas Network. Comprehensive molecular characterization of human colon and rectal cancer. Nature 2012, 487, 330–337. [Google Scholar] [CrossRef]
- Cancer Genome Atlas Network. Comprehensive molecular portraits of human breast tumours. Nature 2012, 490, 61–70. [Google Scholar] [CrossRef]
- Imielinski, M.; Berger, A.H.; Hammerman, P.S.; Hernandez, B.; Pugh, T.J.; Hodis, E.; Cho, J.; Suh, J.; Capelletti, M.; Sivachenko, A.; et al. Mapping the hallmarks of lung adenocarcinoma with massively parallel sequencing. Cell 2012, 150, 1107–1120. [Google Scholar] [CrossRef]
- Lawrence, M.S.; Stojanov, P.; Polak, P.; Kryukov, G.V.; Cibulskis, K.; Sivachenko, A.; Carter, S.L.; Stewart, C.; Mermel, C.H.; Roberts, S.A.; et al. Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature 2013, 499, 214–218. [Google Scholar]
- Gerlinger, M.; Horswell, S.; Larkin, J.; Rowan, A.J.; Salm, M.P.; Varela, I.; Fisher, R.; McGranahan, N.; Matthews, N.; Santos, C.R.; et al. Genomic architecture and evolution of clear cell renal cell carcinomas defined by multiregion sequencing. Nat. Genet. 2014, 46, 225–233. [Google Scholar] [CrossRef]
- Futreal, P.A.; Coin, L.; Marshall, M.; Down, T.; Hubbard, T.; Wooster, R.; Rahman, N.; Stratton, M.R. A census of human cancer genes. Nat. Rev. Cancer 2004, 4, 177–183. [Google Scholar] [CrossRef]
- Lavanchy, D. Hepatitis B virus epidemiology, disease burden, treatment, and current and emerging prevention and control measures. J. Viral Hepat. 2004, 11, 97–107. [Google Scholar] [CrossRef]
- Chen, C.J.; Yang, H.I.; Su, J.; Jen, C.L.; You, S.L.; Lu, S.N.; Huang, G.T.; Iloeje, U.H. Risk of hepatocellular carcinoma across a biological gradient of serum hepatitis B virus DNA level. JAMA 2006, 295, 65–73. [Google Scholar]
- Chisari, F.V. Unscrambling hepatitis C virus-host interactions. Nature 2005, 436, 930–932. [Google Scholar] [CrossRef]
- Bowen, D.G.; Walker, C.M. Adaptive immune responses in acute and chronic hepatitis C virus infection. Nature 2005, 436, 946–952. [Google Scholar] [CrossRef]
- Hoek, J.B.; Pastorino, J.G. Ethanol, oxidative stress, and cytokine-induced liver cell injury. Alcohol 2002, 27, 63–68. [Google Scholar]
- McClain, C.J.; Hill, D.B.; Song, Z.; Deaciuc, I.; Barve, S. Monocyte activation in alcoholic liver disease. Alcohol 2002, 27, 53–61. [Google Scholar]
- Osna, N.A.; Clemens, D.L.; Donohue, T.M., Jr. Ethanol metabolism alters interferon γ signaling in recombinant HepG2 cells. Hepatology 2005, 42, 1109–1117. [Google Scholar] [CrossRef]
- Feitelson, M.A.; Sun, B.; Satiroglu Tufan, N.L.; Liu, J.; Pan, J.; Lian, Z. Genetic mechanisms of hepatocarcinogenesis. Oncogene 2002, 21, 2593–2604. [Google Scholar] [CrossRef]
- Aguilar, F.; Harris, C.C.; Sun, T.; Hollstein, M.; Cerutti, P. Geographic variation of p53 mutational profile in nonmalignant human liver. Science 1994, 264, 1317–1319. [Google Scholar]
- Bressac, B.; Kew, M.; Wands, J.; Ozturk, M. Selective G to T mutations of p53 gene in hepatocellular carcinoma from Southern Africa. Nature 1991, 350, 429–431. [Google Scholar] [CrossRef]
- Hsu, I.C.; Metcalf, R.A.; Sun, T.; Welsh, J.A.; Wang, N.J.; Harris, C.C. Mutational hotspot in the p53 gene in human hepatocellular carcinomas. Nature 1991, 350, 427–428. [Google Scholar] [CrossRef]
- Ozturk, M. p53 mutation in hepatocellular carcinoma after aflatoxin exposure. Lancet 1991, 338, 1356–1359. [Google Scholar] [CrossRef]
- Calle, E.E.; Rodriguez, C.; Walker-Thurmond, K.; Thun, M.J. Overweight, obesity, and mortality from cancer in a prospectively studied cohort of U.S. adults. N. Engl. J. Med. 2003, 348, 1625–1638. [Google Scholar]
- Thorgeirsson, S.S.; Grisham, J.W. Molecular pathogenesis of human hepatocellular carcinoma. Nat. Genet. 2002, 31, 339–346. [Google Scholar] [CrossRef]
- Badvie, S. Hepatocellular carcinoma. Postgrad. Med. J. 2000, 76, 4–11. [Google Scholar] [CrossRef]
- Limdi, J.K.; Crampton, J.R. Hereditary haemochromatosis. QJM 2004, 97, 315–324. [Google Scholar] [CrossRef]
- Farrell, G.C.; Larter, C.Z. Nonalcoholic fatty liver disease: From steatosis to cirrhosis. Hepatology 2006, 43, S99–S112. [Google Scholar] [CrossRef]
- Adams, L.A.; Angulo, P. Recent concepts in non-alcoholic fatty liver disease. Diabet. Med. 2005, 22, 1129–1133. [Google Scholar] [CrossRef]
- Kondo, Y.; Kanai, Y.; Sakamoto, M.; Mizokami, M.; Ueda, R.; Hirohashi, S. Genetic instability and aberrant DNA methylation in chronic hepatitis and cirrhosis—A comprehensive study of loss of heterozygosity and microsatellite instability at 39 loci and DNA hypermethylation on 8 CpG islands in microdissected specimens from patients with hepatocellular carcinoma. Hepatology 2000, 32, 970–979. [Google Scholar] [CrossRef]
- Kawai, H.; Suda, T.; Aoyagi, Y.; Isokawa, O.; Mita, Y.; Waguri, N.; Kuroiwa, T.; Igarashi, M.; Tsukada, K.; Mori, S.; et al. Quantitative evaluation of genomic instability as a possible predictor for development of hepatocellular carcinoma: Comparison of loss of heterozygosity and replication error. Hepatology 2000, 31, 1246–1250. [Google Scholar] [CrossRef]
- Maggioni, M.; Coggi, G.; Cassani, B.; Bianchi, P.; Romagnoli, S.; Mandelli, A.; Borzio, M.; Colombo, P.; Roncalli, M. Molecular changes in hepatocellular dysplastic nodules on microdissected liver biopsies. Hepatology 2000, 32, 942–946. [Google Scholar] [CrossRef]
- Sun, M.; Eshleman, J.R.; Ferrell, L.D.; Jacobs, G.; Sudilovsky, E.C.; Tuthill, R.; Hussein, M.R.; Sudilovsky, O. An early lesion in hepatic carcinogenesis: Loss of heterozygosity in human cirrhotic livers and dysplastic nodules at the 1p36–p34 region. Hepatology 2001, 33, 1415–1424. [Google Scholar] [CrossRef]
- Farazi, P.A.; DePinho, R.A. Hepatocellular carcinoma pathogenesis: From genes to environment. Nat. Rev. Cancer 2006, 6, 674–687. [Google Scholar] [CrossRef]
- Heller, M.J. DNA microarray technology: Devices, systems, and applications. Annu. Rev. Biomed. Eng. 2002, 4, 129–153. [Google Scholar] [CrossRef]
- Chen, X.; Cheung, S.T.; So, S.; Fan, S.T.; Barry, C.; Higgins, J.; Lai, K.M.; Ji, J.; Dudoit, S.; Ng, I.O.; et al. Gene expression patterns in human liver cancers. Mol. Biol. Cell 2002, 13, 1929–1939. [Google Scholar] [CrossRef] [Green Version]
- Villanueva, A.; Newell, P.; Chiang, D.Y.; Friedman, S.L.; Llovet, J.M. Genomics and signaling pathways in hepatocellular carcinoma. Semin. Liver Dis. 2007, 27, 55–76. [Google Scholar] [CrossRef]
- Okabe, H.; Satoh, S.; Kato, T.; Kitahara, O.; Yanagawa, R.; Yamaoka, Y.; Tsunoda, T.; Furukawa, Y.; Nakamura, Y. Genome-wide analysis of gene expression in human hepatocellular carcinomas using cDNA microarray: Identification of genes involved in viral carcinogenesis and tumor progression. Cancer Res. 2001, 61, 2129–2137. [Google Scholar]
- Delpuech, O.; Trabut, J.B.; Carnot, F.; Feuillard, J.; Brechot, C.; Kremsdorf, D. Identification, using cDNA macroarray analysis, of distinct gene expression profiles associated with pathological and virological features of hepatocellular carcinoma. Oncogene 2002, 21, 2926–2937. [Google Scholar] [CrossRef]
- Iizuka, N.; Oka, M.; Yamada-Okabe, H.; Mori, N.; Tamesa, T.; Okada, T.; Takemoto, N.; Tangoku, A.; Hamada, K.; Nakayama, H.; et al. Comparison of gene expression profiles between hepatitis B virus- and hepatitis C virus-infected hepatocellular carcinoma by oligonucleotide microarray data on the basis of a supervised learning method. Cancer Res. 2002, 62, 3939–3944. [Google Scholar]
- Smith, M.W.; Yue, Z.N.; Geiss, G.K.; Sadovnikova, N.Y.; Carter, V.S.; Boix, L.; Lazaro, C.A.; Rosenberg, G.B.; Bumgarner, R.E.; Fausto, N.; et al. Identification of novel tumor markers in hepatitis C virus-associated hepatocellular carcinoma. Cancer Res. 2003, 63, 859–864. [Google Scholar]
- Ye, Q.H.; Qin, L.X.; Forgues, M.; He, P.; Kim, J.W.; Peng, A.C.; Simon, R.; Li, Y.; Robles, A.I.; Chen, Y.; et al. Predicting hepatitis B virus-positive metastatic hepatocellular carcinomas using gene expression profiling and supervised machine learning. Nat. Med. 2003, 9, 416–423. [Google Scholar] [CrossRef]
- Iizuka, N.; Oka, M.; Yamada-Okabe, H.; Nishida, M.; Maeda, Y.; Mori, N.; Takao, T.; Tamesa, T.; Tangoku, A.; Tabuchi, H.; et al. Oligonucleotide microarray for prediction of early intrahepatic recurrence of hepatocellular carcinoma after curative resection. Lancet 2003, 361, 923–929. [Google Scholar] [CrossRef]
- Lee, J.S.; Chu, I.S.; Heo, J.; Calvisi, D.F.; Sun, Z.; Roskams, T.; Durnez, A.; Demetris, A.J.; Thorgeirsson, S.S. Classification and prediction of survival in hepatocellular carcinoma by gene expression profiling. Hepatology 2004, 40, 667–676. [Google Scholar] [CrossRef]
- Boyault, S.; Rickman, D.S.; de Reynies, A.; Balabaud, C.; Rebouissou, S.; Jeannot, E.; Herault, A.; Saric, J.; Belghiti, J.; Franco, D.; et al. Transcriptome classification of HCC is related to gene alterations and to new therapeutic targets. Hepatology 2007, 45, 42–52. [Google Scholar] [CrossRef] [Green Version]
- Hoshida, Y.; Nijman, S.M.; Kobayashi, M.; Chan, J.A.; Brunet, J.P.; Chiang, D.Y.; Villanueva, A.; Newell, P.; Ikeda, K.; Hashimoto, M.; et al. Integrative transcriptome analysis reveals common molecular subclasses of human hepatocellular carcinoma. Cancer Res. 2009, 69, 7385–7392. [Google Scholar] [CrossRef]
- Budhu, A.; Forgues, M.; Ye, Q.H.; Jia, L.H.; He, P.; Zanetti, K.A.; Kammula, U.S.; Chen, Y.; Qin, L.X.; Tang, Z.Y.; et al. Prediction of venous metastases, recurrence and prognosis in hepatocellular carcinoma based on a unique immune response signature of the liver microenvironment. Cancer Cell 2006, 10, 99–111. [Google Scholar] [CrossRef]
- Roskams, T.A.; Libbrecht, L.; Desmet, V.J. Progenitor cells in diseased human liver. Semin. Liver Dis. 2003, 23, 385–396. [Google Scholar] [CrossRef]
- Lee, J.S.; Heo, J.; Libbrecht, L.; Chu, I.S.; Kaposi-Novak, P.; Calvisi, D.F.; Mikaelyan, A.; Roberts, L.R.; Demetris, A.J.; Sun, Z.; et al. A novel prognostic subtype of human hepatocellular carcinoma derived from hepatic progenitor cells. Nat. Med. 2006, 12, 410–416. [Google Scholar] [CrossRef]
- Jallepalli, P.V.; Lengauer, C. Chromosome segregation and cancer: Cutting through the mystery. Nat. Rev. Cancer 2001, 1, 109–117. [Google Scholar] [CrossRef]
- Gollin, S.M. Chromosomal instability. Curr. Opin. Oncol. 2004, 16, 25–31. [Google Scholar] [CrossRef]
- Moinzadeh, P.; Breuhahn, K.; Stutzer, H.; Schirmacher, P. Chromosome alterations in human hepatocellular carcinomas correlate with aetiology and histological grade—Results of an explorative CGH meta-analysis. Br. J. Cancer 2005, 92, 935–941. [Google Scholar] [CrossRef]
- Hermsen, M.A.; Meijer, G.A.; Baak, J.P.; Joenje, H.; Walboomers, J.J. Comparative genomic hybridization: A new tool in cancer pathology. Hum. Pathol. 1996, 27, 342–349. [Google Scholar] [CrossRef]
- Bentz, M.; Plesch, A.; Stilgenbauer, S.; Dohner, H.; Lichter, P. Minimal sizes of deletions detected by comparative genomic hybridization. Genes Chromosomes Cancer 1998, 21, 172–175. [Google Scholar] [CrossRef]
- Pollack, J.R.; Perou, C.M.; Alizadeh, A.A.; Eisen, M.B.; Pergamenschikov, A.; Williams, C.F.; Jeffrey, S.S.; Botstein, D.; Brown, P.O. Genome-wide analysis of DNA copy-number changes using cDNA microarrays. Nat. Genet. 1999, 23, 41–46. [Google Scholar]
- Chochi, Y.; Kawauchi, S.; Nakao, M.; Furuya, T.; Hashimoto, K.; Oga, A.; Oka, M.; Sasaki, K. A copy number gain of the 6p arm is linked with advanced hepatocellular carcinoma: An array-based comparative genomic hybridization study. J. Pathol. 2009, 217, 677–684. [Google Scholar] [CrossRef]
- Patil, M.A.; Gutgemann, I.; Zhang, J.; Ho, C.; Cheung, S.T.; Ginzinger, D.; Li, R.; Dykema, K.J.; So, S.; Fan, S.T.; et al. Array-based comparative genomic hybridization reveals recurrent chromosomal aberrations and Jab1 as a potential target for 8q gain in hepatocellular carcinoma. Carcinogenesis 2005, 26, 2050–2057. [Google Scholar] [CrossRef]
- Kakar, S.; Chen, X.; Ho, C.; Burgart, L.J.; Adeyi, O.; Jain, D.; Sahai, V.; Ferrell, L.D. Chromosomal abnormalities determined by comparative genomic hybridization are helpful in the diagnosis of atypical hepatocellular neoplasms. Histopathology 2009, 55, 197–205. [Google Scholar] [CrossRef]
- Schlaeger, C.; Longerich, T.; Schiller, C.; Bewerunge, P.; Mehrabi, A.; Toedt, G.; Kleeff, J.; Ehemann, V.; Eils, R.; Lichter, P.; et al. Etiology-dependent molecular mechanisms in human hepatocarcinogenesis. Hepatology 2008, 47, 511–520. [Google Scholar]
- Guo, X.; Yanna; Ma, X.; An, J.; Shang, Y.; Huang, Q.; Yang, H.; Chen, Z.; Xing, J. A meta-analysis of array-CGH studies implicates antiviral immunity pathways in the development of hepatocellular carcinoma. PLoS One 2011, 6, e28404. [Google Scholar]
- Midorikawa, Y.; Tsutsumi, S.; Nishimura, K.; Kamimura, N.; Kano, M.; Sakamoto, H.; Makuuchi, M.; Aburatani, H. Distinct chromosomal bias of gene expression signatures in the progression of hepatocellular carcinoma. Cancer Res. 2004, 64, 7263–7270. [Google Scholar] [CrossRef]
- Midorikawa, Y.; Yamamoto, S.; Ishikawa, S.; Kamimura, N.; Igarashi, H.; Sugimura, H.; Makuuchi, M.; Aburatani, H. Molecular karyotyping of human hepatocellular carcinoma using single-nucleotide polymorphism arrays. Oncogene 2006, 25, 5581–5590. [Google Scholar] [CrossRef]
- Furge, K.A.; Dykema, K.J.; Ho, C.; Chen, X. Comparison of array-based comparative genomic hybridization with gene expression-based regional expression biases to identify genetic abnormalities in hepatocellular carcinoma. BMC Genomics 2005, 6, 67. [Google Scholar]
- Pollack, J.R.; Sorlie, T.; Perou, C.M.; Rees, C.A.; Jeffrey, S.S.; Lonning, P.E.; Tibshirani, R.; Botstein, D.; Borresen-Dale, A.L.; Brown, P.O. Microarray analysis reveals a major direct role of DNA copy number alteration in the transcriptional program of human breast tumors. Proc. Natl. Acad. Sci. USA 2002, 99, 12963–12968. [Google Scholar] [CrossRef]
- Adler, A.S.; Lin, M.; Horlings, H.; Nuyten, D.S.; van de Vijver, M.J.; Chang, H.Y. Genetic regulators of large-scale transcriptional signatures in cancer. Nat. Genet. 2006, 38, 421–430. [Google Scholar] [CrossRef]
- Woo, H.G.; Park, E.S.; Lee, J.S.; Lee, Y.H.; Ishikawa, T.; Kim, Y.J.; Thorgeirsson, S.S. Identification of potential driver genes in human liver carcinoma by genomewide screening. Cancer Res. 2009, 69, 4059–4066. [Google Scholar] [CrossRef]
- Roessler, S.; Long, E.L.; Budhu, A.; Chen, Y.; Zhao, X.; Ji, J.; Walker, R.; Jia, H.L.; Ye, Q.H.; Qin, L.X.; et al. Integrative genomic identification of genes on 8p associated with hepatocellular carcinoma progression and patient survival. Gastroenterology 2012, 142, 957–966. [Google Scholar] [CrossRef]
- Laurent-Puig, P.; Legoix, P.; Bluteau, O.; Belghiti, J.; Franco, D.; Binot, F.; Monges, G.; Thomas, G.; Bioulac-Sage, P.; Zucman-Rossi, J. Genetic alterations associated with hepatocellular carcinomas define distinct pathways of hepatocarcinogenesis. Gastroenterology 2001, 120, 1763–1773. [Google Scholar] [CrossRef]
- Anzola, M.; Saiz, A.; Cuevas, N.; Lopez-Martinez, M.; Martinez de Pancorbo, M.A.; Burgos, J.J. High levels of p53 protein expression do not correlate with p53 mutations in hepatocellular carcinoma. J. Viral Hepat. 2004, 11, 502–510. [Google Scholar] [CrossRef]
- Boix-Ferrero, J.; Pellin, A.; Blesa, R.; Adrados, M.; Llombart-Bosch, A. Absence of p53 gene mutations in hepatocarcinomas from a Mediterranean area of Spain. A study of 129 archival tumour samples. Virchows Arch. 1999, 434, 497–501. [Google Scholar]
- Buetow, K.H.; Sheffield, V.C.; Zhu, M.; Zhou, T.; Shen, F.M.; Hino, O.; Smith, M.; McMahon, B.J.; Lanier, A.P.; London, W.T.; et al. Low frequency of p53 mutations observed in a diverse collection of primary hepatocellular carcinomas. Proc. Natl. Acad. Sci. USA 1992, 89, 9622–9626. [Google Scholar]
- Hussain, S.P.; Schwank, J.; Staib, F.; Wang, X.W.; Harris, C.C. TP53 mutations and hepatocellular carcinoma: Insights into the etiology and pathogenesis of liver cancer. Oncogene 2007, 26, 2166–2176. [Google Scholar] [CrossRef]
- Vautier, G.; Bomford, A.B.; Portmann, B.C.; Metivier, E.; Williams, R.; Ryder, S.D. p53 mutations in british patients with hepatocellular carcinoma: Clustering in genetic hemochromatosis. Gastroenterology 1999, 117, 154–160. [Google Scholar]
- Oda, T.; Tsuda, H.; Scarpa, A.; Sakamoto, M.; Hirohashi, S. p53 gene mutation spectrum in hepatocellular carcinoma. Cancer Res. 1992, 52, 6358–6364. [Google Scholar]
- Murakami, Y.; Hayashi, K.; Hirohashi, S.; Sekiya, T. Aberrations of the tumor suppressor p53 and retinoblastoma genes in human hepatocellular carcinomas. Cancer Res. 1991, 51, 5520–5525. [Google Scholar]
- Tanaka, S.; Toh, Y.; Adachi, E.; Matsumata, T.; Mori, R.; Sugimachi, K. Tumor progression in hepatocellular carcinoma may be mediated by p53 mutation. Cancer Res. 1993, 53, 2884–2887. [Google Scholar]
- Hayashi, H.; Sugio, K.; Matsumata, T.; Adachi, E.; Takenaka, K.; Sugimachi, K. The clinical significance of p53 gene mutation in hepatocellular carcinomas from Japan. Hepatology 1995, 22, 1702–1707. [Google Scholar]
- Hsu, H.C.; Jeng, Y.M.; Mao, T.L.; Chu, J.S.; Lai, P.L.; Peng, S.Y. β-Catenin mutations are associated with a subset of low-stage hepatocellular carcinoma negative for hepatitis B virus and with favorable prognosis. Am. J. Pathol. 2000, 157, 763–770. [Google Scholar]
- Park, J.Y.; Park, W.S.; Nam, S.W.; Kim, S.Y.; Lee, S.H.; Yoo, N.J.; Lee, J.Y.; Park, C.K. Mutations of β-catenin and AXIN I genes are a late event in human hepatocellular carcinogenesis. Liver Int. 2005, 25, 70–76. [Google Scholar] [CrossRef]
- Legoix, P.; Bluteau, O.; Bayer, J.; Perret, C.; Balabaud, C.; Belghiti, J.; Franco, D.; Thomas, G.; Laurent-Puig, P.; Zucman-Rossi, J. β-Catenin mutations in hepatocellular carcinoma correlate with a low rate of loss of heterozygosity. Oncogene 1999, 18, 4044–4046. [Google Scholar] [CrossRef]
- Cui, J.; Zhou, X.; Liu, Y.; Tang, Z.; Romeih, M. Wnt signaling in hepatocellular carcinoma: Analysis of mutation and expression of β-catenin, T-cell factor-4 and glycogen synthase kinase 3-β genes. J. Gastroenterol. Hepatol. 2003, 18, 280–287. [Google Scholar] [CrossRef]
- Ishizaki, Y.; Ikeda, S.; Fujimori, M.; Shimizu, Y.; Kurihara, T.; Itamoto, T.; Kikuchi, A.; Okajima, M.; Asahara, T. Immunohistochemical analysis and mutational analyses of β-catenin, AXIN family and APC genes in hepatocellular carcinomas. Int. J. Oncol. 2004, 24, 1077–1083. [Google Scholar]
- Satoh, S.; Daigo, Y.; Furukawa, Y.; Kato, T.; Miwa, N.; Nishiwaki, T.; Kawasoe, T.; Ishiguro, H.; Fujita, M.; Tokino, T.; et al. AXIN1 mutations in hepatocellular carcinomas, and growth suppression in cancer cells by virus-mediated transfer of AXIN1. Nat. Genet. 2000, 24, 245–250. [Google Scholar] [CrossRef]
- Nault, J.C.; Mallet, M.; Pilati, C.; Calderaro, J.; Bioulac-Sage, P.; Laurent, C.; Laurent, A.; Cherqui, D.; Balabaud, C.; Zucman-Rossi, J. High frequency of telomerase reverse-transcriptase promoter somatic mutations in hepatocellular carcinoma and preneoplastic lesions. Nat. Commun. 2013, 4, 2218. [Google Scholar]
- Wei, X.; Walia, V.; Lin, J.C.; Teer, J.K.; Prickett, T.D.; Gartner, J.; Davis, S.; Stemke-Hale, K.; Davies, M.A.; Gershenwald, J.E.; et al. Exome sequencing identifies GRIN2A as frequently mutated in melanoma. Nat. Genet. 2011, 43, 442–446. [Google Scholar]
- Wang, L.; Tsutsumi, S.; Kawaguchi, T.; Nagasaki, K.; Tatsuno, K.; Yamamoto, S.; Sang, F.; Sonoda, K.; Sugawara, M.; Saiura, A.; et al. Whole-exome sequencing of human pancreatic cancers and characterization of genomic instability caused by MLH1 haploinsufficiency and complete deficiency. Genome Res. 2012, 22, 208–219. [Google Scholar]
- Fujimoto, A.; Totoki, Y.; Abe, T.; Boroevich, K.A.; Hosoda, F.; Nguyen, H.H.; Aoki, M.; Hosono, N.; Kubo, M.; Miya, F.; et al. Whole-genome sequencing of liver cancers identifies etiological influences on mutation patterns and recurrent mutations in chromatin regulators. Nat. Genet. 2012, 44, 760–764. [Google Scholar] [CrossRef]
- Huang, J.; Deng, Q.; Wang, Q.; Li, K.Y.; Dai, J.H.; Li, N.; Zhu, Z.D.; Zhou, B.; Liu, X.Y.; Liu, R.F.; et al. Exome sequencing of hepatitis B virus-associated hepatocellular carcinoma. Nat. Genet. 2012, 44, 1117–1121. [Google Scholar] [CrossRef]
- Guichard, C.; Amaddeo, G.; Imbeaud, S.; Ladeiro, Y.; Pelletier, L.; Maad, I.B.; Calderaro, J.; Bioulac-Sage, P.; Letexier, M.; Degos, F.; et al. Integrated analysis of somatic mutations and focal copy-number changes identifies key genes and pathways in hepatocellular carcinoma. Nat. Genet. 2012, 44, 694–698. [Google Scholar] [CrossRef] [Green Version]
- Guan, B.; Wang, T.L.; Shih, I. ARID1A, a factor that promotes formation of SWI/SNF-mediated chromatin remodeling, is a tumor suppressor in gynecologic cancers. Cancer Res. 2011, 71, 6718–6727. [Google Scholar] [CrossRef]
- Wilson, B.G.; Roberts, C.W. SWI/SNF nucleosome remodellers and cancer. Nat. Rev. Cancer 2011, 11, 481–492. [Google Scholar] [CrossRef]
- Jones, S.; Wang, T.L.; Shih, I.; Mao, T.L.; Nakayama, K.; Roden, R.; Glas, R.; Slamon, D.; Diaz, L.A., Jr.; Vogelstein, B.; et al. Frequent mutations of chromatin remodeling gene ARID1A in ovarian clear cell carcinoma. Science 2010, 330, 228–231. [Google Scholar] [CrossRef]
- Wiegand, K.C.; Shah, S.P.; Al-Agha, O.M.; Zhao, Y.; Tse, K.; Zeng, T.; Senz, J.; McConechy, M.K.; Anglesio, M.S.; Kalloger, S.E.; et al. ARID1A mutations in endometriosis-associated ovarian carcinomas. N. Engl. J. Med. 2010, 363, 1532–1543. [Google Scholar] [CrossRef]
- Gui, Y.; Guo, G.; Huang, Y.; Hu, X.; Tang, A.; Gao, S.; Wu, R.; Chen, C.; Li, X.; Zhou, L.; et al. Frequent mutations of chromatin remodeling genes in transitional cell carcinoma of the bladder. Nat. Genet. 2011, 43, 875–878. [Google Scholar]
- Wang, K.; Kan, J.; Yuen, S.T.; Shi, S.T.; Chu, K.M.; Law, S.; Chan, T.L.; Kan, Z.; Chan, A.S.; Tsui, W.Y.; et al. Exome sequencing identifies frequent mutation of ARID1A in molecular subtypes of gastric cancer. Nat. Genet. 2011, 43, 1219–1223. [Google Scholar] [CrossRef]
- Jones, S.; Li, M.; Parsons, D.W.; Zhang, X.; Wesseling, J.; Kristel, P.; Schmidt, M.K.; Markowitz, S.; Yan, H.; Bigner, D.; et al. Somatic mutations in the chromatin remodeling gene ARID1A occur in several tumor types. Hum. Mutat. 2012, 33, 100–103. [Google Scholar] [CrossRef] [Green Version]
- Li, M.; Zhao, H.; Zhang, X.; Wood, L.D.; Anders, R.A.; Choti, M.A.; Pawlik, T.M.; Daniel, H.D.; Kannangai, R.; Offerhaus, G.J.; et al. Inactivating mutations of the chromatin remodeling gene ARID2 in hepatocellular carcinoma. Nat. Genet. 2011, 43, 828–829. [Google Scholar] [CrossRef]
- Xu, X.R.; Huang, J.; Xu, Z.G.; Qian, B.Z.; Zhu, Z.D.; Yan, Q.; Cai, T.; Zhang, X.; Xiao, H.S.; Qu, J.; et al. Insight into hepatocellular carcinogenesis at transcriptome level by comparing gene expression profiles of hepatocellular carcinoma with those of corresponding noncancerous liver. Proc. Natl. Acad. Sci. USA 2001, 98, 15089–15094. [Google Scholar] [CrossRef]
- Marchio, A.; Meddeb, M.; Pineau, P.; Danglot, G.; Tiollais, P.; Bernheim, A.; Dejean, A. Recurrent chromosomal abnormalities in hepatocellular carcinoma detected by comparative genomic hybridization. Genes Chromosomes Cancer 1997, 18, 59–65. [Google Scholar] [CrossRef]
- Guan, X.Y.; Fang, Y.; Sham, J.S.; Kwong, D.L.; Zhang, Y.; Liang, Q.; Li, H.; Zhou, H.; Trent, J.M. Recurrent chromosome alterations in hepatocellular carcinoma detected by comparative genomic hybridization. Genes Chromosomes Cancer 2000, 29, 110–116. [Google Scholar] [CrossRef]
- Katoh, H.; Shibata, T.; Kokubu, A.; Ojima, H.; Loukopoulos, P.; Kanai, Y.; Kosuge, T.; Fukayama, M.; Kondo, T.; Sakamoto, M.; et al. Genetic profile of hepatocellular carcinoma revealed by array-based comparative genomic hybridization: Identification of genetic indicators to predict patient outcome. J. Hepatol. 2005, 43, 863–874. [Google Scholar] [CrossRef]
- Kusano, N.; Okita, K.; Shirahashi, H.; Harada, T.; Shiraishi, K.; Oga, A.; Kawauchi, S.; Furuya, T.; Sasaki, K. Chromosomal imbalances detected by comparative genomic hybridization are associated with outcome of patients with hepatocellular carcinoma. Cancer 2002, 94, 746–751. [Google Scholar]
- Wong, N.; Lai, P.; Pang, E.; Fung, L.F.; Sheng, Z.; Wong, V.; Wang, W.; Hayashi, Y.; Perlman, E.; Yuna, S.; et al. Genomic aberrations in human hepatocellular carcinomas of differing etiologies. Clin. Cancer Res. 2000, 6, 4000–4009. [Google Scholar]
- Sy, S.M.; Wong, N.; Lai, P.B.; To, K.F.; Johnson, P.J. Regional over-representations on chromosomes 1q, 3q and 7q in the progression of hepatitis B virus-related hepatocellular carcinoma. Mod. Pathol. 2005, 18, 686–692. [Google Scholar] [CrossRef]
- Wong, N.; Lai, P.; Lee, S.W.; Fan, S.; Pang, E.; Liew, C.T.; Sheng, Z.; Lau, J.W.; Johnson, P.J. Assessment of genetic changes in hepatocellular carcinoma by comparative genomic hybridization analysis: Relationship to disease stage, tumor size, and cirrhosis. Am. J. Pathol. 1999, 154, 37–43. [Google Scholar]
- Tornillo, L.; Carafa, V.; Richter, J.; Sauter, G.; Moch, H.; Minola, E.; Gambacorta, M.; Bianchi, L.; Vecchione, R.; Terracciano, L.M. Marked genetic similarities between hepatitis B virus-positive and hepatitis C virus-positive hepatocellular carcinomas. J. Pathol. 2000, 192, 307–312. [Google Scholar] [CrossRef]
- Pang, A.; Ng, I.O.; Fan, S.T.; Kwong, Y.L. Clinicopathologic significance of genetic alterations in hepatocellular carcinoma. Cancer Genet. Cytogenet. 2003, 146, 8–15. [Google Scholar] [CrossRef]
- Matsuda, Y.; Ichida, T.; Matsuzawa, J.; Sugimura, K.; Asakura, H. p16(INK4) is inactivated by extensive CpG methylation in human hepatocellular carcinoma. Gastroenterology 1999, 116, 394–400. [Google Scholar]
- De La Coste, A.; Romagnolo, B.; Billuart, P.; Renard, C.A.; Buendia, M.A.; Soubrane, O.; Fabre, M.; Chelly, J.; Beldjord, C.; Kahn, A.; et al. Somatic mutations of the β-catenin gene are frequent in mouse and human hepatocellular carcinomas. Proc. Natl. Acad. Sci. USA 1998, 95, 8847–8851. [Google Scholar] [CrossRef]
- Wei, Y.; van Nhieu, J.T.; Prigent, S.; Srivatanakul, P.; Tiollais, P.; Buendia, M.A. Altered expression of E-cadherin in hepatocellular carcinoma: Correlations with genetic alterations, β-catenin expression, and clinical features. Hepatology 2002, 36, 692–701. [Google Scholar]
- Taniguchi, K.; Roberts, L.R.; Aderca, I.N.; Dong, X.; Qian, C.; Murphy, L.M.; Nagorney, D.M.; Burgart, L.J.; Roche, P.C.; Smith, D.I.; et al. Mutational spectrum of β-catenin, AXIN1, and AXIN2 in hepatocellular carcinomas and hepatoblastomas. Oncogene 2002, 21, 4863–4871. [Google Scholar] [CrossRef]
- Li, J.; Quan, H.; Liu, Q.; Si, Z.; He, Z.; Qi, H. Alterations of axis inhibition protein 1 (AXIN1) in hepatitis B virus-related hepatocellular carcinoma and overexpression of AXIN1 induces apoptosis in hepatocellular cancer cells. Oncol. Res. 2013, 20, 281–288. [Google Scholar]
- Huang, J.; Sheng, H.H.; Shen, T.; Hu, Y.J.; Xiao, H.S.; Zhang, Q.; Zhang, Q.H.; Han, Z.G. Correlation between genomic DNA copy number alterations and transcriptional expression in hepatitis B virus-associated hepatocellular carcinoma. FEBS Lett. 2006, 580, 3571–3581. [Google Scholar] [CrossRef]
- Sawey, E.T.; Chanrion, M.; Cai, C.; Wu, G.; Zhang, J.; Zender, L.; Zhao, A.; Busuttil, R.W.; Yee, H.; Stein, L.; et al. Identification of a therapeutic strategy targeting amplified FGF19 in liver cancer by oncogenomic screening. Cancer Cell 2011, 19, 347–358. [Google Scholar] [CrossRef]
- Deane, N.G.; Parker, M.A.; Aramandla, R.; Diehl, L.; Lee, W.J.; Washington, M.K.; Nanney, L.B.; Shyr, Y.; Beauchamp, R.D. Hepatocellular carcinoma results from chronic cyclin D1 overexpression in transgenic mice. Cancer Res. 2001, 61, 5389–5395. [Google Scholar]
- Wang, R.; Ferrell, L.D.; Faouzi, S.; Maher, J.J.; Bishop, J.M. Activation of the Met receptor by cell attachment induces and sustains hepatocellular carcinomas in transgenic mice. J. Cell Biol. 2001, 153, 1023–1034. [Google Scholar] [CrossRef]
- Zender, L.; Xue, W.; Zuber, J.; Semighini, C.P.; Krasnitz, A.; Ma, B.; Zender, P.; Kubicka, S.; Luk, J.M.; Schirmacher, P.; et al. An oncogenomics-based in vivo RNAi screen identifies tumor suppressors in liver cancer. Cell 2008, 135, 852–864. [Google Scholar] [CrossRef]
- Schramek, D.; Sendoel, A.; Segal, J.P.; Beronja, S.; Heller, E.; Oristian, D.; Reva, B.; Fuchs, E. Direct in vivo RNAi screen unveils myosin IIa as a tumor suppressor of squamous cell carcinomas. Science 2014, 343, 309–313. [Google Scholar]
- Bard-Chapeau, E.A.; Nguyen, A.T.; Rust, A.G.; Sayadi, A.; Lee, P.; Chua, B.Q.; New, L.S.; de Jong, J.; Ward, J.M.; Chin, C.K.; et al. Transposon mutagenesis identifies genes driving hepatocellular carcinoma in a chronic hepatitis B mouse model. Nat. Genet. 2014, 46, 24–32. [Google Scholar]
- Schlabach, M.R.; Luo, J.; Solimini, N.L.; Hu, G.; Xu, Q.; Li, M.Z.; Zhao, Z.; Smogorzewska, A.; Sowa, M.E.; Ang, X.L.; et al. Cancer proliferation gene discovery through functional genomics. Science 2008, 319, 620–624. [Google Scholar] [CrossRef]
- Silva, J.M.; Marran, K.; Parker, J.S.; Silva, J.; Golding, M.; Schlabach, M.R.; Elledge, S.J.; Hannon, G.J.; Chang, K. Profiling essential genes in human mammary cells by multiplex RNAi screening. Science 2008, 319, 617–620. [Google Scholar] [CrossRef]
- Luo, J.; Emanuele, M.J.; Li, D.; Creighton, C.J.; Schlabach, M.R.; Westbrook, T.F.; Wong, K.K.; Elledge, S.J. A genome-wide RNAi screen identifies multiple synthetic lethal interactions with the Ras oncogene. Cell 2009, 137, 835–848. [Google Scholar] [CrossRef]
- Faber, A.C.; Wong, K.K.; Engelman, J.A. Differences underlying EGFR and HER2 oncogene addiction. Cell Cycle 2010, 9, 851–852. [Google Scholar] [CrossRef]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Takai, A.; Dang, H.T.; Wang, X.W. Identification of Drivers from Cancer Genome Diversity in Hepatocellular Carcinoma. Int. J. Mol. Sci. 2014, 15, 11142-11160. https://doi.org/10.3390/ijms150611142
Takai A, Dang HT, Wang XW. Identification of Drivers from Cancer Genome Diversity in Hepatocellular Carcinoma. International Journal of Molecular Sciences. 2014; 15(6):11142-11160. https://doi.org/10.3390/ijms150611142
Chicago/Turabian StyleTakai, Atsushi, Hien T. Dang, and Xin W. Wang. 2014. "Identification of Drivers from Cancer Genome Diversity in Hepatocellular Carcinoma" International Journal of Molecular Sciences 15, no. 6: 11142-11160. https://doi.org/10.3390/ijms150611142