The Structure and Dynamics of BmR1 Protein from Brugia malayi: In Silico Approaches


Abstract
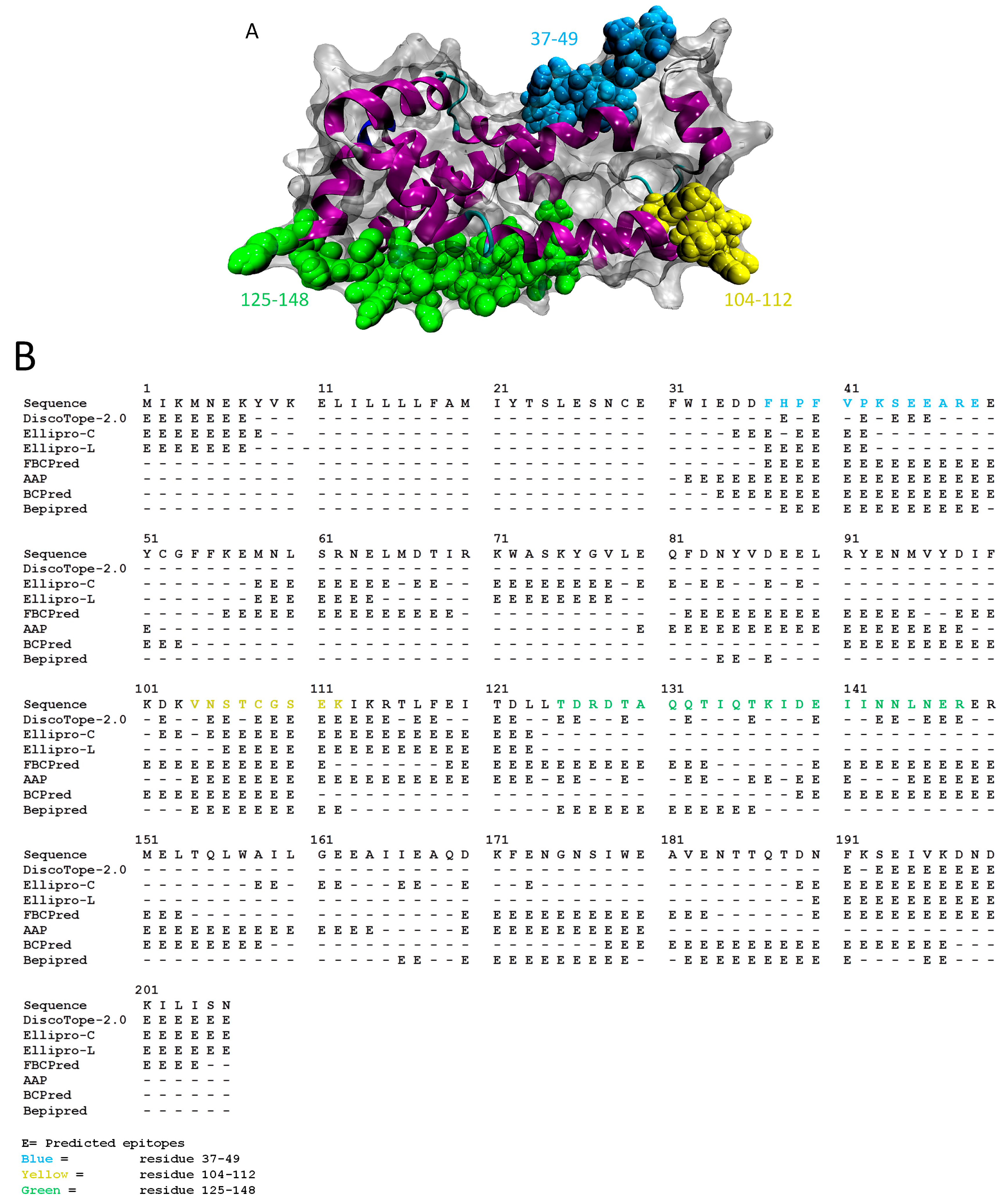
:1. Introduction
2. Results and Discussion
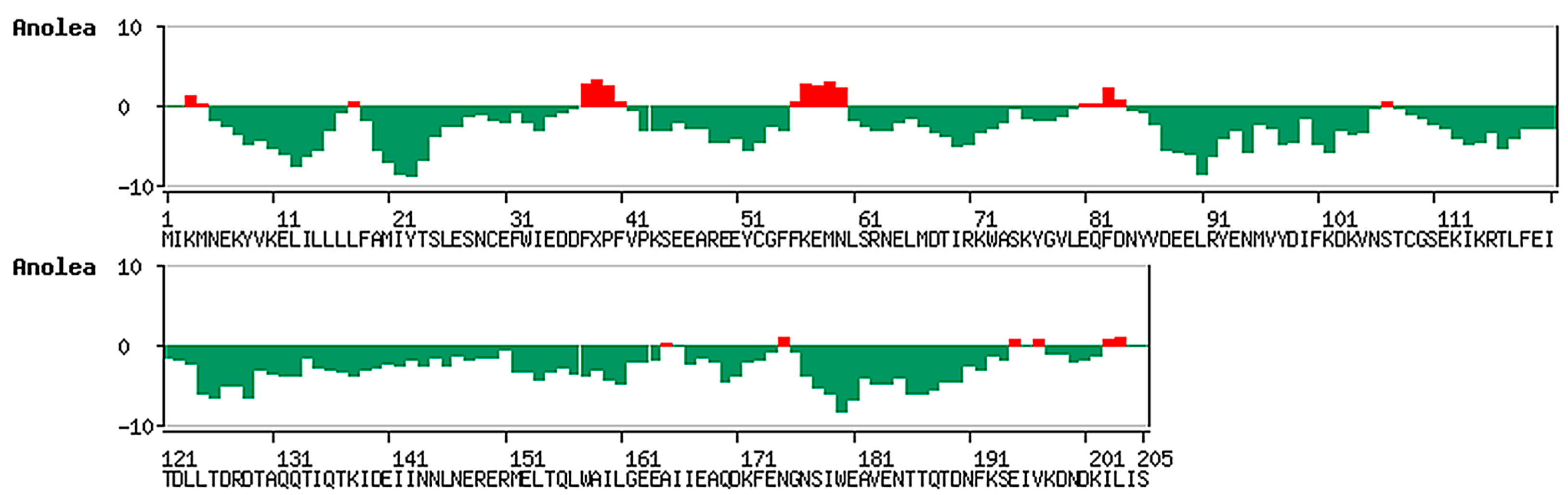
2.1. Results

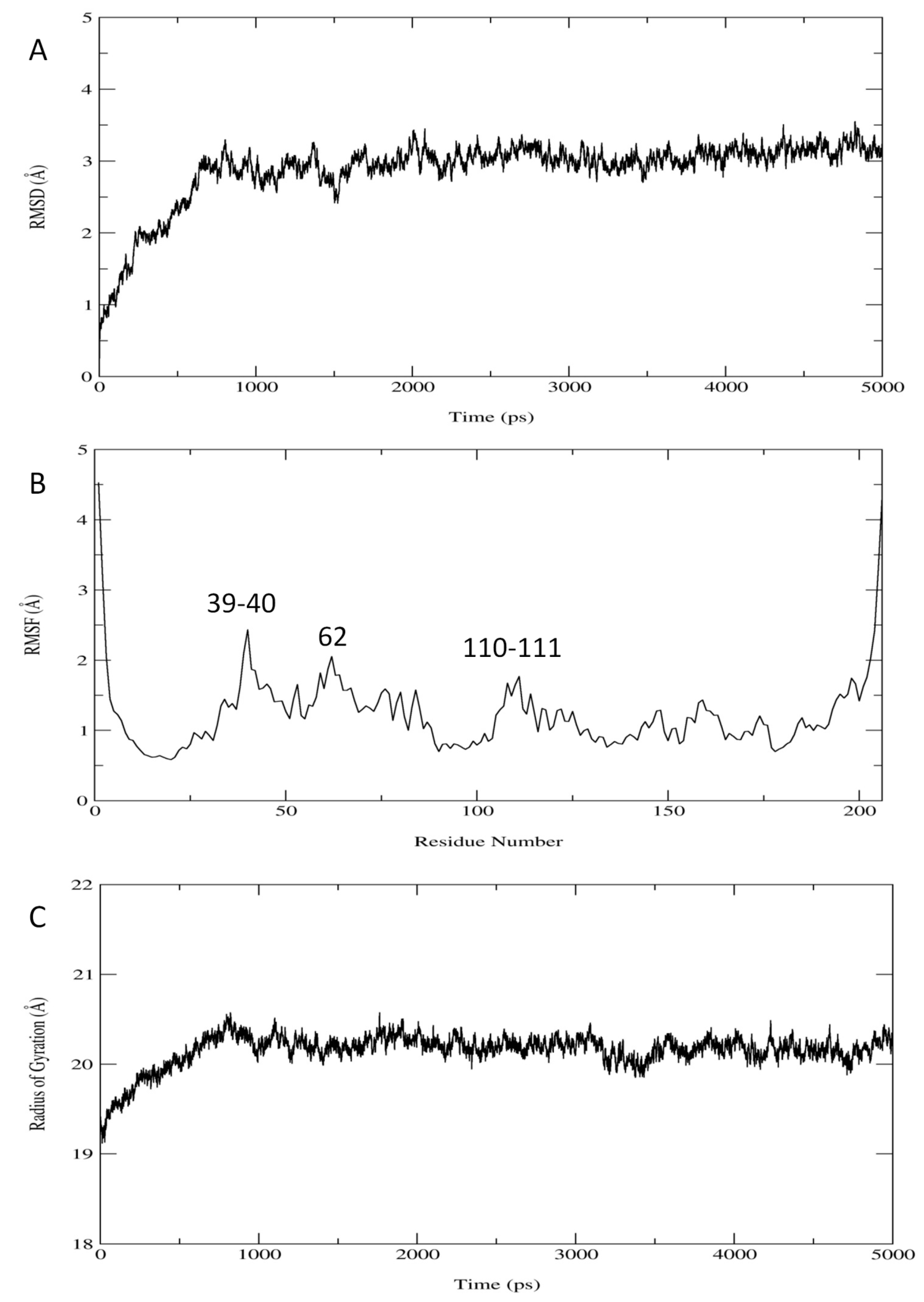{kind=link}
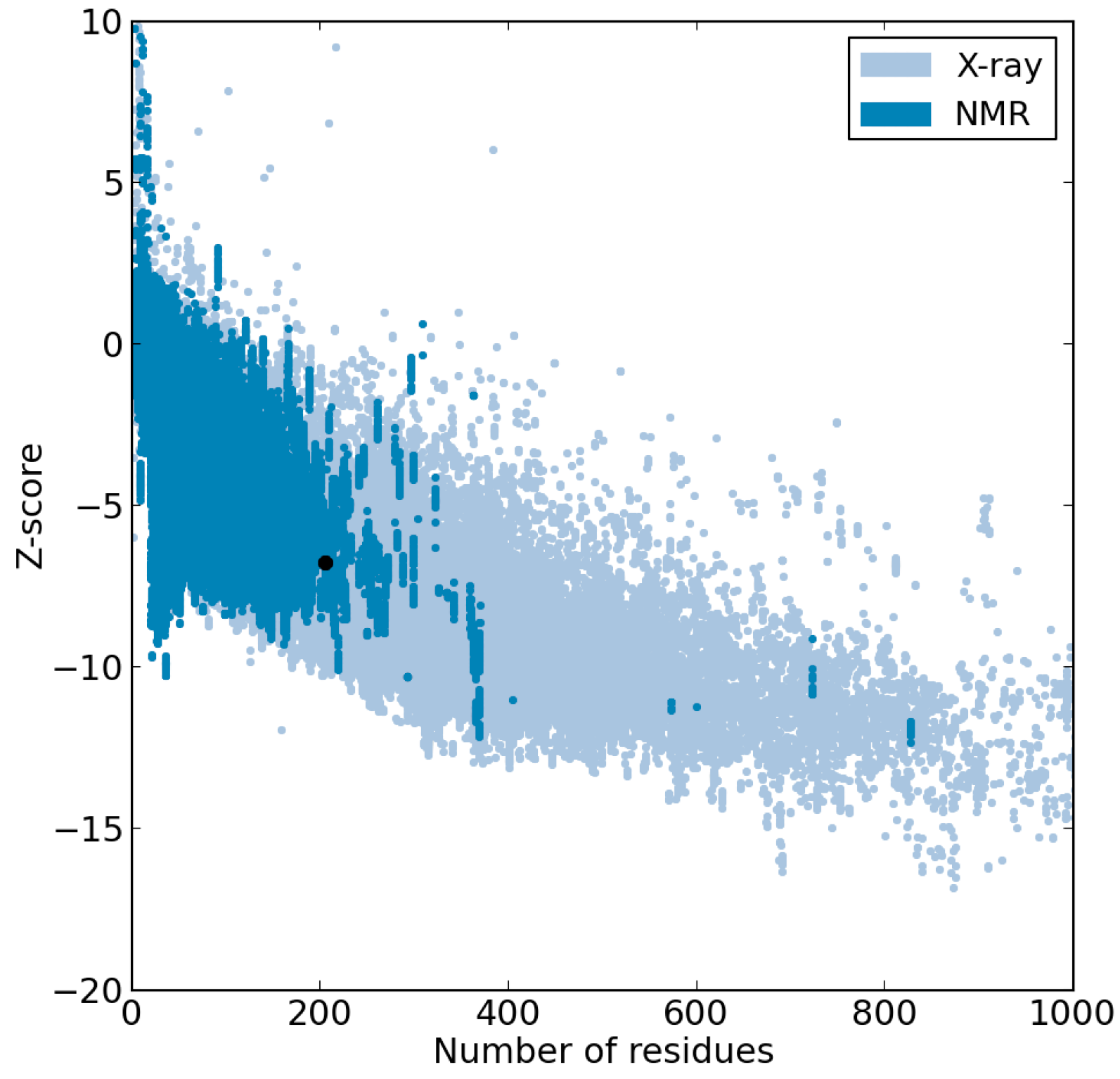{kind=link}
{kind=link}
{kind=link}
| 1 | 11 | 21 | 31 | |
|---|---|---|---|---|
| Sequence | M I K M N E K Y V K | E L I L L L L F A M | I Y T S L E S N C E | F W I E D D F H P F |
| PSIPRED | - - - H H H H H H H | H H H H H H H H H H | H H H H H H H - - B | B B B - - - - - - - |
| Jpred3 | - - - - - H H H H H | H H H H H H H H H H | H H H - - - - - - B | B B B - - - - - - - |
| SSpro | - - - - - H H H H H | H H H H H H H H H H | H H H H H H H - - - | B B B B - - - - - - |
| PORTER | - - - - - H H H H H | H H H H H H H H H H | H H H H H H H - - - | H H H - - - - - - - |
| STRIDE | - - - - H H H H H H | H H H H H H H H H H | H H H H H H H H H H | H H H H H - - - - - |
| 41 | 51 | 61 | 71 | |
| Sequence | V P K S E E A R E E | Y C G F F K E M N L | S R N E L M D T I R | K W A S K Y G V L E |
| PSIPRED | - - - - H H H H H H | H H H H H H H - - - | - H H H H H H H H H | H H H H H H H H H H |
| Jpred3 | - - - - - - H H H H | H H H H H H H - - - | - H H H H H H H H H | H H H - - - - H H H |
| SSpro | - - - - H H H H H H | H H H H H H H H H - | - H H H H H H H H H | H H H H H - - H H H |
| PORTER | - - - - H H H H H H | H H H H H H H H - - | - H H H H H H H H H | H H H H H H H H H H |
| STRIDE | - - H H H H H H H H | H H H H H H H H - - | - - - - H H H H H H | H H H H H H - H H H |
| 81 | 91 | 101 | 111 | |
| Sequence | Q F D N Y V D E E L | R Y E N M V Y D I F | K D K V N S T C G S | E K I K R T L F E I |
| PSIPRED | H H H H H H H H H H | H H H H H H H H H H | H H H H - - - - - H | H H H H H H H H H H |
| Jpred3 | H H H H H H - - - - | - - - - H H H H H H | H H - - - - - - - - | H H H H H H H H H H |
| SSpro | H H - - - - H H H H | H H H H H H H H H H | H - - - - - - - - H | H H H H H H H H H H |
| PORTER | H H H H H H H H H H | H H H H H H H H H H | H H H H - - - - - H | H H H H H H H H H H |
| STRIDE | H H H H H H - - - H | H H H H H H H H H H | H H H H H H H - - - | - H H H H H H H H H |
| 121 | 131 | 141 | 151 | |
| Sequence | T D L L T D R D T A | Q Q T I Q T K I D E | I I N N L N E R E R | M E L T Q L W A I L |
| PSIPRED | H H H H - - H H H H | H H H H H H H H H H | H H H - - - H H H H | H H H H H H H H H H |
| Jpred3 | H H H H - - - H H H | H H H H H H H H H H | H H H H - - - - - H | H H H H H H H H H H |
| SSpro | H H H H - - H H H H | H H H H H H H H H H | H H H H H H H H H H | H H H H H H H H H H |
| PORTER | H H H H - - H H H H | H H H H H H H H H H | H H H H H H H H H H | H H H H H H H H H H |
| STRIDE | H H H - H H H H H H | H H H H H H H H H H | H H H H - - H H H H | H H H H H H H H H H |
| 161 | 171 | 181 | 191 | |
| Sequence | G E E A I I E A Q D | K F E N G N S I W E | A V E N T T Q T D N | F K S E I V K D N D |
| PSIPRED | H H H H H H H H H H | H H H - - - H H H H | H H H - - - - - - - | H H H H H H - - - - |
| Jpred3 | - - H H H H H H H H | H H - - - - - H H H | H H H - - - - - - - | - H H H B - - - - - |
| SSpro | H H H H H H H H H H | H - - - - - - B B B | B B B - - - - - - - | - - H H H - - - - - |
| PORTER | H H H H H H H H H H | H H H - - - H H H H | H H H H - - - - - - | - H H H H H - - - - |
| STRIDE | - H H H H H H H H H | H H H H - - - H H H | H H H H - - - - - - | - - - H H H H H H H |
| 201 | ||||
| Sequence | K I L I S N | |||
| PSIPRED | B B B - - - | |||
| Jpred3 | B B B B - - | |||
| SSpro | B B B B - - | |||
| PORTER | - B B - - - | |||
| STRIDE | - - - - - - |
| 1 | 11 | 21 | 31 | |
|---|---|---|---|---|
| Sequence | M I K M N E K Y V K | E L I L L L L F A M | I Y T S L E S N C E | F W I E D D F H P F |
| MODELLER9v9 | - - - - - - H H H - | H H H H H H H H H H | H H H H H H - - - - | - - - - - - - - - - |
| QUARK | - - - - - H H H H H | H H H H H H H H H H | H H H H H H H - - - | - - - - - - - - - - |
| Robetta | - - H H H H H H H H | H H H H H H H H H H | H H H H H H H - - - | - - - - - - - - - - |
| Rosetta | - - - - H H H H H H | H H H H H H H H H H | H H H H H H H H H H | H H H H H - - - - - |
| I-TASSER | - - - - - - H H H H | H H H H H H H H H - | - - H H H H H H H H | H H - - - - - - - - |
| Bhageerath | - - - - - H H H H H | H H H H H H H - - - | - - - - - - - - - - | - - - - - - - - - - |
| 41 | 51 | 61 | 71 | |
| Sequence | V P K S E E A R E E | Y C G F F K E M N L | S R N E L M D T I R | K W A S K Y G V L E |
| MODELLER9v9 | - - - - H H H H H H | H H H H H H H H H H | H H H H H H H H H H | H H H H H H - H H H |
| QUARK | - - - - H H H H H H | H H H H H H H H - - | - H H H H H H H H H | H H H H H H - H H H |
| Robetta | - - - H H H H H H H | H H H H H H H H - - | - H H H H H H H H H | H H H H H H - H H H |
| Rosetta | - H H H H H H H H H | H H H H H H H H - - | - - - - H H H H H H | H H H H H - - H H H |
| I-TASSER | H H H H H H H H H H | H H H H H H H H - - | - H H H H H H H H H | H - - - H H H H H H |
| Bhageerath | - H H H H H H H H H | H H H H H H H H H H | H H H H - - - - - - | - - H H H H H H - H |
| 81 | 91 | 101 | 111 | |
| Sequence | Q F D N Y V D E E L | R Y E N M V Y D I F | K D K V N S T C G S | E K I K R T L F E I |
| MODELLER9v9 | H H H H H H H H H H | H H H – H H H H H H | H H H H H H - - - H | H H - - H H H H H H |
| QUARK | H H H H H H H H H H | H H H H H H H H H H | H H H H H - - - - H | H H H H H H H H H H |
| Robetta | H H H H H H H H H H | H H H H H H H H H H | H H H H H H - - - H | H H H H H H H H H H |
| Rosetta | H H H H H H H H H H | H H H H H H H H H H | H H H H H H - - - H | H H H H H H H H H H |
| I-TASSER | H H H H H H H H - - | - H H H H H H H H H | H H H H H - - - - - | H H H H H H H H H H |
| Bhageerath | H H H H H H H H H H | H H H H H H H H H - | - - H H H H H H H H | H H H H H H H - - - |
| 121 | 131 | 141 | 151 | |
| Sequence | T D L L T D R D T A | Q Q T I Q T K I D E | I I N N L N E R E R | M E L T Q L W A I L |
| MODELLER9v9 | H H H H H - H H H H | H H H H H H H H - H | H H H H H H H - H H | H H H H H H H H H H |
| QUARK | H H H H - - H H H H | H H H H H H H H H H | H H H H - - H H H H | H H H H H H H H H H |
| Robetta | H H H H H – H H H H | H H H H H H H H H H | H H H H - - H H H H | H H H H H H H H H H |
| Rosetta | H H H H H H H H H H | H H H H H H H H H H | H H H H - - H H H H | H H H H H H H H H H |
| I-TASSER | H H H H - - H H H H | H H H H H H H H H H | H H H - - - H H H H | H H H H H H H H H H |
| Bhageerath | - - - - - H H H H H | H H H H H H H H H H | - - - - - - - - - - | - - - - H H H H H H |
| 161 | 171 | 181 | 191 | |
| Sequence | G E E A I I E A Q D | K F E N G N S I W E | A V E N T T Q T D N | F K S E I V K D N D |
| MODELLER9v9 | H H H H H H H H H H | H H H H H H H H - H | H H - - - - - - - - | - - H H H - - - - - |
| QUARK | H H H H H H H H H H | H H H H - - - H H H | H H H - - - - H H H | H H H H H H H - - - |
| Robetta | H H H H H H H H H H | H H H H - - - H H H | H H H - H H H H H H | - - - - H H H H - - |
| Rosetta | - H H H H H H H H H | H H H H - - - H H H | H H H H - - - - - - | - - - H H H H H H - |
| I-TASSER | - - - - - H H H H H | H H H - - - - - H H | H H H H H H H H - - | H H H H H H - - - - |
| Bhageerath | H H H H H H H H H H | H H H H - - - - - - | - - - - - - - - - - | - - - - - - - - - - |
| 201 | ||||
| Sequence | K I L I S N | |||
| MODELLER9v9 | - - - - - - | |||
| QUARK | - - - - - - | |||
| Robetta | - H H H - - | |||
| Rosetta | - - - - - - | |||
| I-TASSER | - - - - - - | |||
| Bhageerath | - - - - - - |
| Name | Approach | Ramachandran Plot [17] | VERIFY3D [18] (%) | ERRAT [19] (%) | |
|---|---|---|---|---|---|
| Residues in Most Favored Region (%) | Residues in Disallow Region | ||||
| MODELLER 9v9 [10] | Comparative modelling | 87.3 | - | 16.4 | 58.6 |
| CPHmodels 3.0 [20] | Comparative modelling | 79.6 | - | 48.3 | 93.3 |
| QUARK [11] | Ab initio | 92.9 | - | 83.6 | 95.5 |
| Robetta [12,13] | Ab initio | 97.5 | - | 73.9 | 91.4 |
| Rosetta [14] | Ab initio | 98.5 | - | 71.0 | 97.0 |
| I-TASSER [15] | Threading & ab initio | 80.7 | Ser 61 | 55.6 | 96.0 |
| Bhageerath [16] | Ab initio | 84.3 | Met 20 Ser 24 Ile 142 Asp 102 Asn 176 | 33.3 | 61.4 |


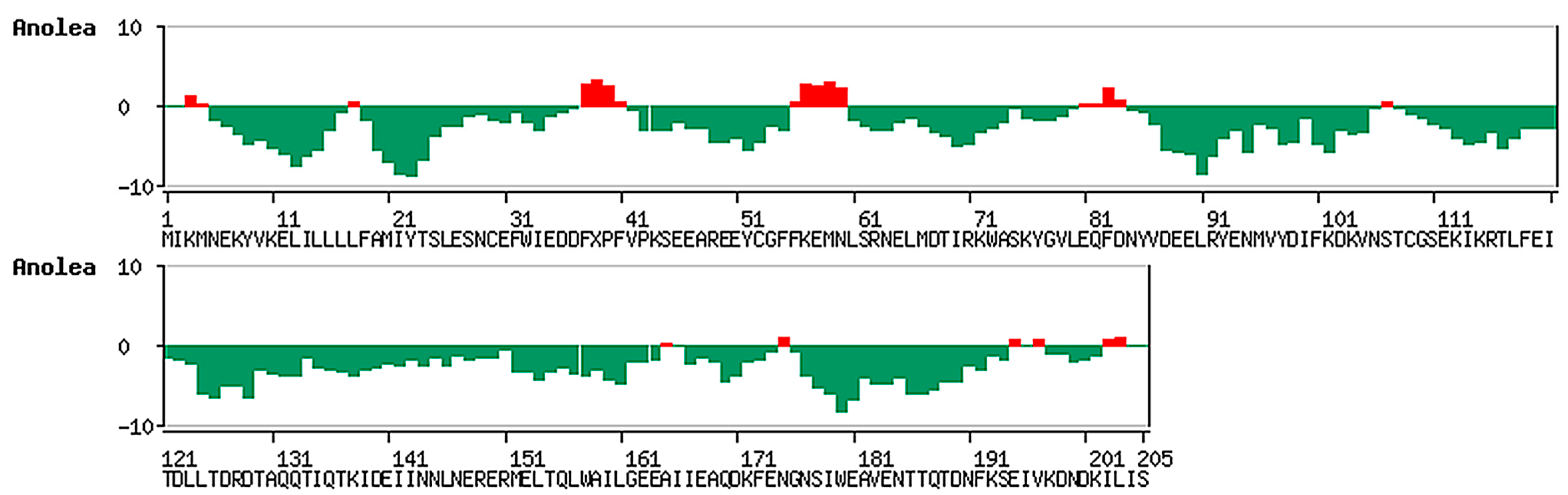

2.2. Discussion
3. Methods
3.1. Sequence Analysis
3.2. Structural Prediction and Evaluation
3.3. Minimization and Molecular Dynamics Simulation
4. Conclusions
Supplementary Files
Acknowledgments
Author Contributions
Conflicts of Interest
References
- World Health Organization. Global programme to eliminate lymphatic filariasis. Wkly. Epidemiol. Rec. 2011, 86, 518–588. [Google Scholar]
- Noordin, R.; Aziz, R.; Ravindran, B. Homologs of the Brugia malayi diagnostic antigen BmR1 are present in other filarial parasites but induce different humoral immune responses. Filaria J. 2004, 3, 10. [Google Scholar] [CrossRef]
- Noordin, R.; Itoh, M.; Kimura, E.; Abdul Rahman, R.; Ravindran, B.; Mahmud, R.; Supali, T.; Weerasooriya, M. Multicentre evaluations of two new rapid IgG4 tests (WB rapid and panLF rapid) for detection of lymphatic filariasis. Filaria J. 2007, 6, 9. [Google Scholar] [CrossRef]
- Abdul Rahman, R.; Hwen-Yee, C.; Noordin, R. Pan LF-ELISA using BmR1 and BmSXP recombinant antigens for detection of lymphatic filariasis. Filaria J. 2007, 6, 10. [Google Scholar] [CrossRef]
- Buchan, D.W.A.; Ward, S.M.; Lobley, A.E.; Nugent, T.C.O.; Bryson, K.; Jones, D.T. Protein annotation and modelling servers at University College London. Nucleic Acids Res. 2010, 38, W563–W568. [Google Scholar] [CrossRef]
- Cole, C.; Barber, J.D.; Barton, G.J. The Jpred 3 secondary structure prediction server. Nucleic Acids Res. 2008, 36, W197–W201. [Google Scholar] [CrossRef]
- Cheng, J.; Randall, A.Z.; Sweredoski, M.J.; Baldi, P. SCRATCH: A protein structure and structural feature prediction server. Nucleic Acids Res. 2005, 33, W72–W76. [Google Scholar] [CrossRef]
- Pollastri, G.; McLysaght, A. Porter: A new, accurate server for protein secondary structure prediction. Bioinformatics 2005, 21, 1719–1720. [Google Scholar] [CrossRef]
- Heinig, M.; Frishman, D. STRIDE: A web server for secondary structure assignment from known atomic coordinates of proteins. Nucleic Acids Res. 2004, 32, W500–W502. [Google Scholar] [CrossRef]
- Martí-Renom, M.A.; Stuart, A.C.; Fiser, A.; Sánchez, R.; Melo, F.; Šali, A. Comparative protein structure modeling of genes and genomes. Annu. Rev. Biophys. Biomol. Struct. 2000, 29, 291–325. [Google Scholar] [CrossRef]
- Xu, D.; Zhang, J.; Roy, A.; Zhang, Y. Automated protein structure modeling in CASP9 by I-TASSER pipeline combined with QUARK-based ab initio folding and FG-MD-based structure refinement. Proteins Struct. Funct. Bioinform. 2011, 79 (Suppl. S10), 147–160. [Google Scholar] [CrossRef] [Green Version]
- Chivian, D.; Kim, D.E.; Malmström, L.; Bradley, P.; Robertson, T.; Murphy, P.; Strauss, C.E.M.; Bonneau, R.; Rohl, C.A.; Baker, D. Automated prediction of CASP-5 structures using the Robetta server. Proteins Struct. Funct. Bioinform. 2003, 53, 524–533. [Google Scholar] [CrossRef]
- Chivian, D.; Kim, D.E.; Malmström, L.; Schonbrun, J.; Rohl, C.A.; Baker, D. Prediction of CASP6 structures using automated Robetta protocols. Proteins Struct. Funct. Bioinform. 2005, 61, 157–166. [Google Scholar] [CrossRef]
- Bonneau, R.; Tsai, J.; Ruczinski, I.; Chivian, D.; Rohl, C.; Strauss, C.E.M.; Baker, D. Rosetta in CASP4: Progress in ab initio protein structure prediction. Proteins Struct. Funct. Bioinform. 2001, 45, 119–126. [Google Scholar] [CrossRef]
- Zhang, Y. I-TASSER server for protein 3D structure prediction. BMC Bioinform. 2008, 9, 40. [Google Scholar] [CrossRef]
- Jayaram, B.; Bhushan, K.; Shenoy, S.R.; Narang, P.; Bose, S.; Agrawal, P.; Sahu, D.; Pandey, V. Bhageerath: An energy based web enabled computer software suite for limiting the search space of tertiary structures of small globular proteins. Nucleic Acids Res. 2006, 34, 6195–6204. [Google Scholar]
- Laskowski, R.A.; MacArthur, M.W.; Moss, D.S.; Thornton, J.M. PROCHECK: A program to check the stereochemical quality of protein structures. J. Appl. Crystallogr. 1993, 26, 283–291. [Google Scholar] [CrossRef]
- Eisenberg, D.; Lüthy, R.; Bowie, J.U. VERIFY3D: Assessment of protein models with three-dimensional profiles. Methods Enzymol. 1997, 277, 396–404. [Google Scholar] [CrossRef]
- Colovos, C.; Yeates, T.O. Verification of protein structures: Patterns of nonbonded atomic interactions. Protein Sci. 1993, 2, 1511–1519. [Google Scholar] [CrossRef]
- Nielsen, M.; Lundegaard, C.; Lund, O.; Petersen, T.N. CPHmodels-3.0—Remote homology modeling using structure-guided sequence profiles. Nucleic Acids Res. 2010, 38, W576–W581. [Google Scholar] [CrossRef]
- Wiederstein, M.; Sippl, M.J. ProSA-web: Interactive web service for the recognition of errors in three-dimensional structures of proteins. Nucleic Acids Res. 2007, 35, W407–W410. [Google Scholar] [CrossRef]
- Melo, F.; Feytmans, E. Assessing protein structures with a non-local atomic interaction energy. J. Mol. Biol. 1998, 277, 1141–1152. [Google Scholar] [CrossRef]
- Bordoli, L.; Kiefer, F.; Arnold, K.; Benkert, P.; Battey, J.; Schwede, T. Protein structure homology modeling using SWISS-MODEL workspace. Nat. Protoc. 2008, 4, 1–13. [Google Scholar] [CrossRef]
- Offredi, F.; Dubail, F.; Kischel, P.; Sarinski, K.; Stern, A.S.; van de Weerdt, C.; Hoch, J.C.; Prosperi, C.; François, J.M.; Mayo, S.L.; et al. De novo backbone and sequence design of an idealized α/β-barrel protein: Evidence of stable tertiary structure. J. Mol. Biol. 2003, 325, 163–174. [Google Scholar] [CrossRef]
- Von Mering, C.; Jensen, L.J.; Snel, B.; Hooper, S.D.; Krupp, M.; Foglierini, M.; Jouffre, N.; Huynen, M.A.; Bork, P. STRING: Known and predicted protein–protein associations, integrated and transferred across organisms. Nucleic Acids Res. 2005, 33, D433–D437. [Google Scholar]
- Sigrist, C.J.A.; de Castro, E.; Cerutti, L.; Cuche, B.A.; Hulo, N.; Bridge, A.; Bougueleret, L.; Xenarios, I. New and continuing developments at PROSITE. Nucleic Acids Res. 2013, 41, D344–D347. [Google Scholar] [CrossRef]
- Laskowski, R.A.; Watson, J.D.; Thornton, J.M. ProFunc: A server for predicting protein function from 3D structure. Nucleic Acids Res. 2005, 33, W89–W93. [Google Scholar] [CrossRef]
- Faucher, F.; Wallace, S.S.; Doublié, S. The C-terminal lysine of Ogg2 DNA glycosylases is a major molecular determinant for guanine/8-oxoguanine distinction. J. Mol. Biol. 2010, 397, 46–56. [Google Scholar] [CrossRef]
- Faucher, F.; Duclos, S.; Bandaru, V.; Wallace, S.S.; Doublié, S. Crystal structures of two archaeal 8-oxoguanine DNA glycosylases provide structural insight into guanine/8-oxoguanine distinction. Structure 2009, 17, 703–712. [Google Scholar] [CrossRef]
- Golovin, A.; Henrick, K. MSDmotif: Exploring protein sites and motifs. BMC Bioinform. 2008, 9, 312. [Google Scholar] [CrossRef]
- Gasteiger, E.; Gattiker, A.; Hoogland, C.; Ivanyi, I.; Appel, R.D.; Bairoch, A. ExPASy: The proteomics server for in-depth protein knowledge and analysis. Nucleic Acids Res. 2003, 31, 3784–3788. [Google Scholar] [CrossRef]
- Ponomarenko, J.; Bui, H.-H.; Li, W.; Fusseder, N.; Bourne, P.; Sette, A.; Peters, B. ElliPro: A new structure-based tool for the prediction of antibody epitopes. BMC Bioinform. 2008, 9, 514. [Google Scholar] [CrossRef]
- Yasser, E.-M.; Dobbs, D.; Honavar, V. Predicting flexible length linear B-cell epitopes. Comput. Syst. Bioinform. Conf. 2008, 7, 121–132. [Google Scholar]
- Chen, J.; Liu, H.; Yang, J.; Chou, K.C. Prediction of linear B-cell epitopes using amino acid pair antigenicity scale. Amino Acids 2007, 33, 423–428. [Google Scholar] [CrossRef]
- Larsen, J.; Lund, O.; Nielsen, M. Improved method for predicting linear B-cell epitopes. Immunome Res. 2006, 2, 2. [Google Scholar] [CrossRef]
- Kringelum, J.V.; Lundegaard, C.; Lund, O.; Nielsen, M. Reliable B cell epitope predictions: Impacts of method development and improved benchmarking. PLoS Comput. Biol. 2012, 8, e1002829. [Google Scholar] [CrossRef]
- Haste Andersen, P.; Nielsen, M.; Lund, O. Prediction of residues in discontinuous B-cell epitopes using protein 3D structures. Protein Sci. 2006, 15, 2558–2567. [Google Scholar] [CrossRef]
- Konc, J.; Janežič, D. ProBiS-2012: Web server and web services for detection of structurally similar binding sites in proteins. Nucleic Acids Res. 2012, 40, W214–W221. [Google Scholar] [CrossRef]
- Eswar, N.; Webb, B.; Marti-Renom, M.A.; Madhusudhan, M.S.; Eramian, D.; Shen, M.; Pieper, U.; Sali, A. Comparative protein structure modeling using Modeller. In Current Protocols in Bioinformatics; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2002. [Google Scholar]
- Choong, Y.S.; Lim, T.S.; Chew, A.L.; Aziah, I.; Ismail, A. Structural and functional studies of a 50 kda antigenic protein from Salmonella enterica serovar Typhi. J. Mol. Graph. Model. 2011, 29, 834–842. [Google Scholar] [CrossRef]
- Errami, M.; Geourjon, C.; Deléage, G. Detection of unrelated proteins in sequences multiple alignments by using predicted secondary structures. Bioinformatics 2003, 19, 506–512. [Google Scholar] [CrossRef]
- Hansen, S.F.; Bettler, E.; Wimmerová, M.; Imberty, A.; Lerouxel, O.; Breton, C. Combination of several bioinformatics approaches for the identification of new putative glycosyltransferases in arabidopsis. J. Proteome Res. 2008, 8, 743–753. [Google Scholar]
- Moult, J.; Fidelis, K.; Kryshtafovych, A.; Tramontano, A. Critical assessment of methods of protein structure prediction (CASP)—Round ix. Proteins Struct. Funct. Bioinform. 2011, 79, 1–5. [Google Scholar]
- Eswar, N.; Eramian, D.; Webb, B.; Shen, M.-Y.; Sali, A. Protein structure modeling with MODELLER. In Structural Proteomics; Kobe, B., Guss, M., Huber, T., Eds.; Humana Press: New York City, NY, USA, 2008; Volume 426, pp. 145–159. [Google Scholar]
- Barh, D.; Barve, N.; Gupta, K.; Chandra, S.; Jain, N.; Tiwari, S.; Leon-Sicairos, N.; Canizalez-Roman, A.; Rodrigues dos Santos, A.; Hassan, S.S.; et al. Exoproteome and secretome derived broad spectrum novel drug and vaccine candidates in Vibrio cholerae targeted by piper betel/derived compounds. PLoS One 2013, 8, e52773. [Google Scholar] [CrossRef]
- Arnold, K.; Bordoli, L.; Kopp, J.; Schwede, T. The SWISS-MODEL workspace: A web-based environment for protein structure homology modelling. Bioinformatics 2006, 22, 195–201. [Google Scholar]
- Bates, P.A.; Kelley, L.A.; MacCallum, R.M.; Sternberg, M.J.E. Enhancement of protein modeling by human intervention in applying the automatic programs 3D-JIGSAW and 3D-PSSM. Proteins Struct. Funct. Bioinform. 2001, 45, 39–46. [Google Scholar] [CrossRef]
- Lambert, C.; Léonard, N.; de Bolle, X.; Depiereux, E. ESyPred3D: Prediction of proteins 3D structures. Bioinformatics 2002, 18, 1250–1256. [Google Scholar] [CrossRef]
- Combet, C.; Jambon, M.; Deléage, G.; Geourjon, C. Geno3D: Automatic comparative molecular modelling of protein. Bioinformatics 2002, 18, 213–214. [Google Scholar] [CrossRef]
- Ahmadi Adl, A.; Nowzari-Dalini, A.; Xue, B.; Uversky, V.N.; Qian, X. Accurate prediction of protein structural classes using functional domains and predicted secondary structure sequences. J. Biomol. Struct. Dyn. 2012, 29, 1127–1137. [Google Scholar] [CrossRef]
- Lin, H.-N.; Sung, T.-Y.; Ho, S.-Y.; Hsu, W.-L. Improving protein secondary structure prediction based on short subsequences with local structure similarity. BMC Genomics 2010, 11, S4. [Google Scholar]
- Lee, L.; Leopold, J.L.; Frank, R.L. In Protein secondary structure prediction using blast and exhaustive rt-rico, the search for optimal segment length and threshold. In Proceedings of the 2012 IEEE Symposium on Computational Intelligence in Bioinformatics and Computational Biology (CIBCB), San Diego, CA, USA, 9–12 May 2012; pp. 35–42.
- Garofalo, A.; Kläger, S.L.; Rowlinson, M.-C.; Nirmalan, N.; Klion, A.; Allen, J.E.; Kennedy, M.W.; Bradley, J.E. The FAR proteins of filarial nematodes: Secretion, glycosylation and lipid binding characteristics. Mol. Biochem. Parasitol. 2002, 122, 161–170. [Google Scholar] [CrossRef]
- Notredame, C.; Higgins, D.G.; Heringa, J. T-Coffee: A novel method for fast and accurate multiple sequence alignment. J. Mol. Biol. 2000, 302, 205–217. [Google Scholar] [CrossRef]
- Cheng, X.; Xiang, Y.; Xie, H.; Xu, C.-L.; Xie, T.-F.; Zhang, C.; Li, Y. Molecular characterization and functions of fatty acid and retinoid binding protein gene (Ab-far-1) in Aphelenchoides besseyi. PLoS One 2013, 8, e66011. [Google Scholar]
- Jennifer, L.B.; Amber, E.F. The potential role of binding proteins in human parasitic infections: An in-depth look at the novel family of nematode-specific fatty acid and retinol binding proteins. In Binding Protein; Abdelmohsen, K., Ed.; InTech: Rijeka, Croatia, 2012. [Google Scholar]
- Fairfax, K.C.; Vermeire, J.J.; Harrison, L.M.; Bungiro, R.D.; Grant, W.; Husain, S.Z.; Cappello, M. Characterisation of a fatty acid and retinol binding protein orthologue from the hookworm Ancylostoma ceylanicum. Int. J. Parasitol. 2009, 39, 1561–1571. [Google Scholar]
- Moreno, Y.; Geary, T.G. Stage- and gender-specific proteomic analysis of Brugia malayi excretory-secretory products. PLoS Negl. Trop. Dis. 2008, 2, e326. [Google Scholar] [CrossRef]
- Conesa, A.; Götz, S.; García-Gómez, J.M.; Terol, J.; Talón, M.; Robles, M. Blast2GO: A universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 2005, 21, 3674–3676. [Google Scholar] [CrossRef]
- Bennuru, S.; Semnani, R.; Meng, Z.; Ribeiro, J.M.C.; Veenstra, T.D.; Nutman, T.B. Brugia malayi excreted/secreted proteins at the host/parasite interface: Stage- and gender-specific proteomic profiling. PLoS Negl. Trop. Dis. 2009, 3, e410. [Google Scholar] [CrossRef]
- Jordanova, R.; Groves, M.R.; Kostova, E.; Woltersdorf, C.; Liebau, E.; Tucker, P.A. Fatty acid- and retinoid-binding proteins have distinct binding pockets for the two types of cargo. J. Biol. Chem. 2009, 284, 35818–35826. [Google Scholar]
- Kennedy, M.W.; Garside, L.H.; Goodrick, L.E.; McDermott, L.; Brass, A.; Price, N.C.; Kelly, S.M.; Cooper, A.; Bradley, J.E. The Ov20 protein of the parasitic nematode Onchocerca volvulus: A structurally novel class of small helix-rich retinol-binding proteins. J. Biol. Chem. 1997, 272, 29442–29448. [Google Scholar] [CrossRef]
- Novotný, J.; Handschumacher, M.; Haber, E.; Bruccoleri, R.E.; Carlson, W.B.; Fanning, D.W.; Smith, J.A.; Rose, G.D. Antigenic determinants in proteins coincide with surface regions accessible to large probes (antibody domains). Proc. Natl. Acad. Sci. USA 1986, 83, 226–230. [Google Scholar] [CrossRef]
- Barlow, D.J.; Edwards, M.S.; Thornton, J.M. Continuous and discontinuous protein antigenic determinants. Nature 1986, 322, 747–748. [Google Scholar] [CrossRef]
- Kulkarni-Kale, U.; Bhosle, S.; Kolaskar, A.S. CEP: A conformational epitope prediction server. Nucleic Acids Res. 2005, 33, W168–W171. [Google Scholar] [CrossRef]
- Wang, H.-W.; Lin, Y.-C.; Pai, T.-W.; Chang, H.-T. Prediction of B-cell linear epitopes with a combination of support vector machine classification and amino acid propensity identification. J. Biomed. Biotechnol. 2011, 2011, 12. [Google Scholar]
- Marchler-Bauer, A.; Bryant, S.H. CD-search: Protein domain annotations on the fly. Nucleic Acids Res. 2004, 32, W327–W331. [Google Scholar] [CrossRef]
- Marchler-Bauer, A.; Lu, S.; Anderson, J.B.; Chitsaz, F.; Derbyshire, M.K.; DeWeese-Scott, C.; Fong, J.H.; Geer, L.Y.; Geer, R.C.; Gonzales, N.R.; et al. CDD: A conserved domain database for the functional annotation of proteins. Nucleic Acids Res. 2011, 39, D225–D229. [Google Scholar] [CrossRef]
- Quevillon, E.; Silventoinen, V.; Pillai, S.; Harte, N.; Mulder, N.; Apweiler, R.; Lopez, R. InterProScan: Protein domains identifier. Nucleic Acids Res. 2005, 33, W116–W120. [Google Scholar] [CrossRef]
- Letunic, I.; Doerks, T.; Bork, P. Smart 6: Recent updates and new developments. Nucleic Acids Res. 2009, 37, D229–D232. [Google Scholar] [CrossRef]
- Finn, R.D.; Mistry, J.; Tate, J.; Coggill, P.; Heger, A.; Pollington, J.E.; Gavin, O.L.; Gunasekaran, P.; Ceric, G.; Forslund, K.; et al. The Pfam protein families database. Nucleic Acids Res. 2010, 38, D211–D222. [Google Scholar] [CrossRef]
- Lee, J.H.; Im, Y.J.; Bae, J.; Kim, D.; Kim, M.-K.; Kang, G.B.; Lee, D.-S.; Eom, S.H. Crystal structure of Thermus caldophilus phosphoglycerate kinase in the open conformation. Biochem. Biophys. Res. Commun. 2006, 350, 1044–1049. [Google Scholar] [CrossRef]
- Kim, Y.; Joachimiak, G.; Ye, Z.; Binkowski, T.A.; Zhang, R.; Gornicki, P.; Callahan, S.M.; Hess, W.R.; Haselkorn, R.; Joachimiak, A. Structure of transcription factor HetR required for heterocyst differentiation in cyanobacteria. Proc. Natl. Acad. Sci. USA 2011, 108, 10109–10114. [Google Scholar] [CrossRef]
- Fu, Z.; Voynova, N.E.; Herdendorf, T.J.; Miziorko, H.M.; Kim, J.-J.P. Biochemical and structural basis for feedback inhibition of mevalonate kinase and isoprenoid metabolism. Biochemistry 2008, 47, 3715–3724. [Google Scholar] [CrossRef]
- Kim, D.E.; Chivian, D.; Baker, D. Protein structure prediction and analysis using the Robetta server. Nucleic Acids Res. 2004, 32, W526–W531. [Google Scholar]
- Salomon-Ferrer, R.; Case, D.A.; Walker, R.C. An overview of the Amber biomolecular simulation package. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2013, 3, 198–210. [Google Scholar] [CrossRef]
- Berendsen, H.J.C.; Postma, J.P.M.; van Gunsteren, W.F.; DiNola, A.; Haak, J.R. Molecular dynamics with coupling to an external bath. J. Chem. Phys. 1984, 81, 3684–3690. [Google Scholar] [CrossRef]
- Ryckaert, J.-P.; Ciccotti, G.; Berendsen, H. Numerical integration of the cartesian equations of motion of a system with constraints: Molecular dynamics of n-alkanes. J. Comput. Phys. 1977, 23, 327–341. [Google Scholar] [CrossRef]
- Yang, J.; Roy, A.; Zhang, Y. Protein-ligand binding site recognition using complementary binding-specific substructure comparison and sequence profile alignment. Bioinformatics 2013, 29, 2588–2595. [Google Scholar] [CrossRef]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Khor, B.Y.; Tye, G.J.; Lim, T.S.; Noordin, R.; Choong, Y.S. The Structure and Dynamics of BmR1 Protein from Brugia malayi: In Silico Approaches. Int. J. Mol. Sci. 2014, 15, 11082-11099. https://doi.org/10.3390/ijms150611082
Khor BY, Tye GJ, Lim TS, Noordin R, Choong YS. The Structure and Dynamics of BmR1 Protein from Brugia malayi: In Silico Approaches. International Journal of Molecular Sciences. 2014; 15(6):11082-11099. https://doi.org/10.3390/ijms150611082
Chicago/Turabian StyleKhor, Bee Yin, Gee Jun Tye, Theam Soon Lim, Rahmah Noordin, and Yee Siew Choong. 2014. "The Structure and Dynamics of BmR1 Protein from Brugia malayi: In Silico Approaches" International Journal of Molecular Sciences 15, no. 6: 11082-11099. https://doi.org/10.3390/ijms150611082
APA StyleKhor, B. Y., Tye, G. J., Lim, T. S., Noordin, R., & Choong, Y. S. (2014). The Structure and Dynamics of BmR1 Protein from Brugia malayi: In Silico Approaches. International Journal of Molecular Sciences, 15(6), 11082-11099. https://doi.org/10.3390/ijms150611082