A Comprehensive Insight into Tetracycline Resistant Bacteria and Antibiotic Resistance Genes in Activated Sludge Using Next-Generation Sequencing


Abstract
:1. Introduction
2. Results and Discussion
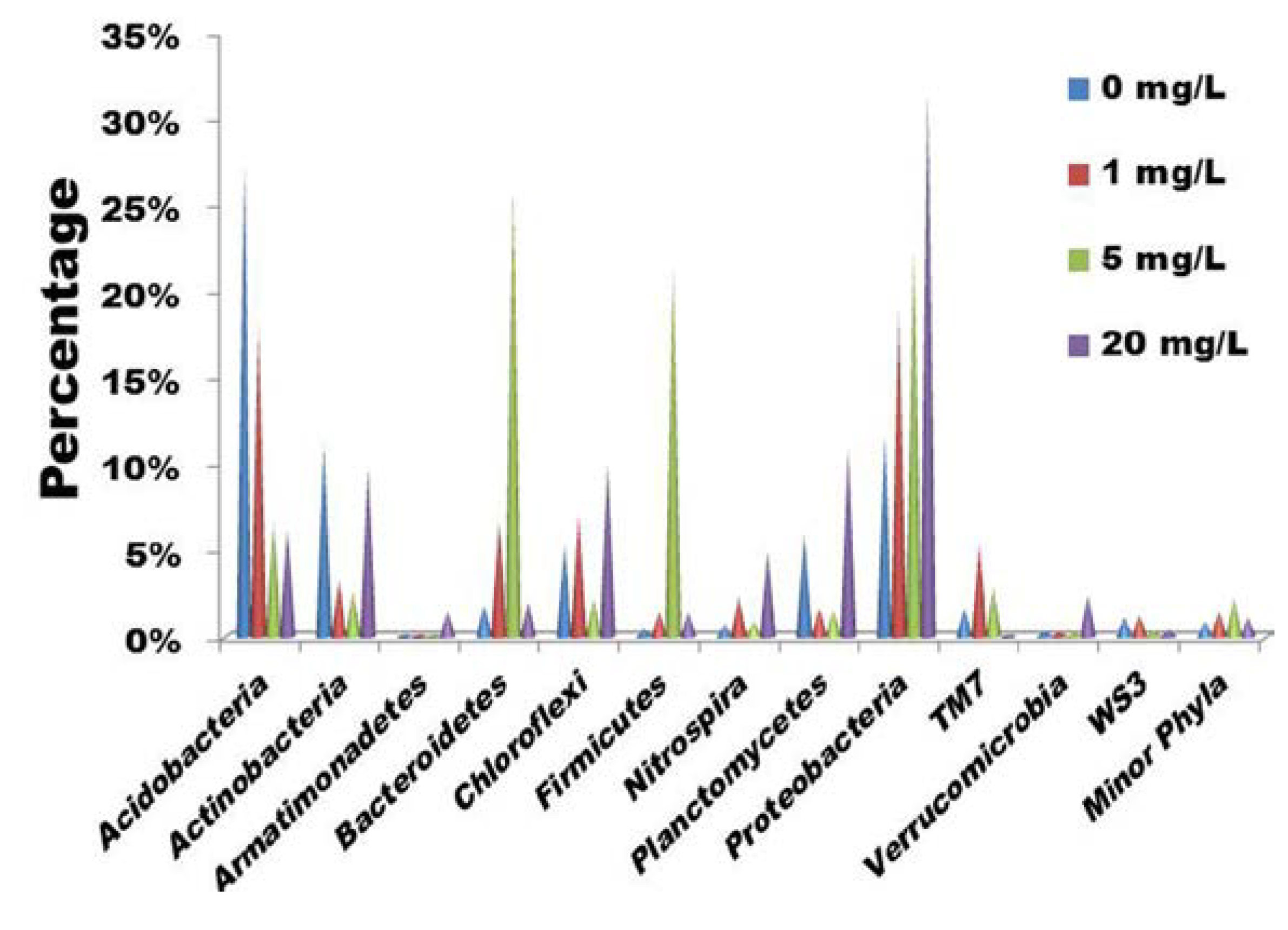
2.1. Bacterial Community Shift under Tetracycline Stress

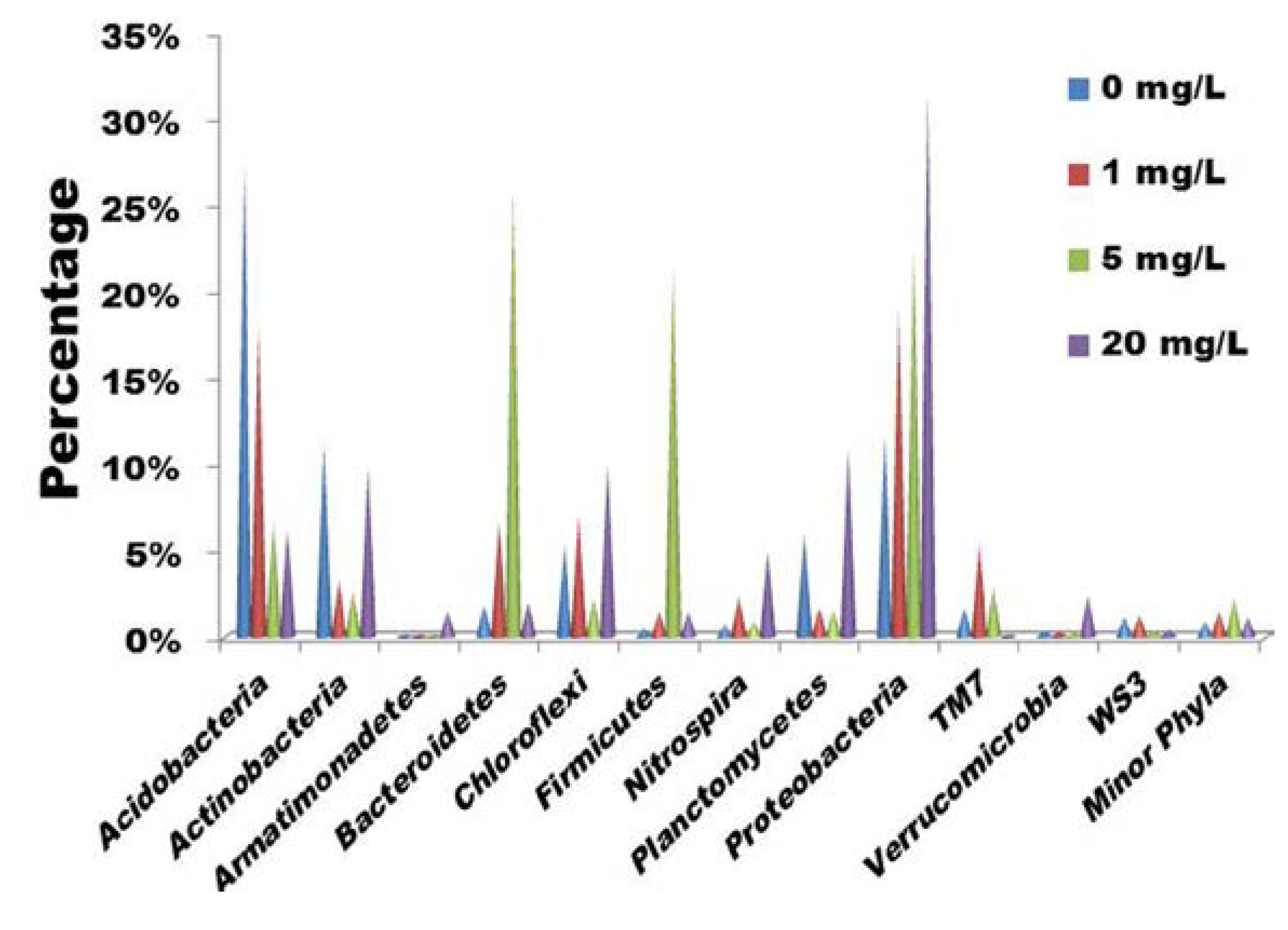{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Tetracycline Concentrations | No. of Raw Sequences | Observed OTUs | Chao 1 | Shannon Index |
|---|---|---|---|---|
| 0 mg/L | 7097 | 1112 | 1562 | 5.94 |
| 1 mg/L | 13,351 | 1306 | 1988 | 6.19 |
| 5 mg/L | 9306 | 1692 | 2899 | 6.60 |
| 20 mg/L | 12,802 | 1347 | 1975 | 6.35 |
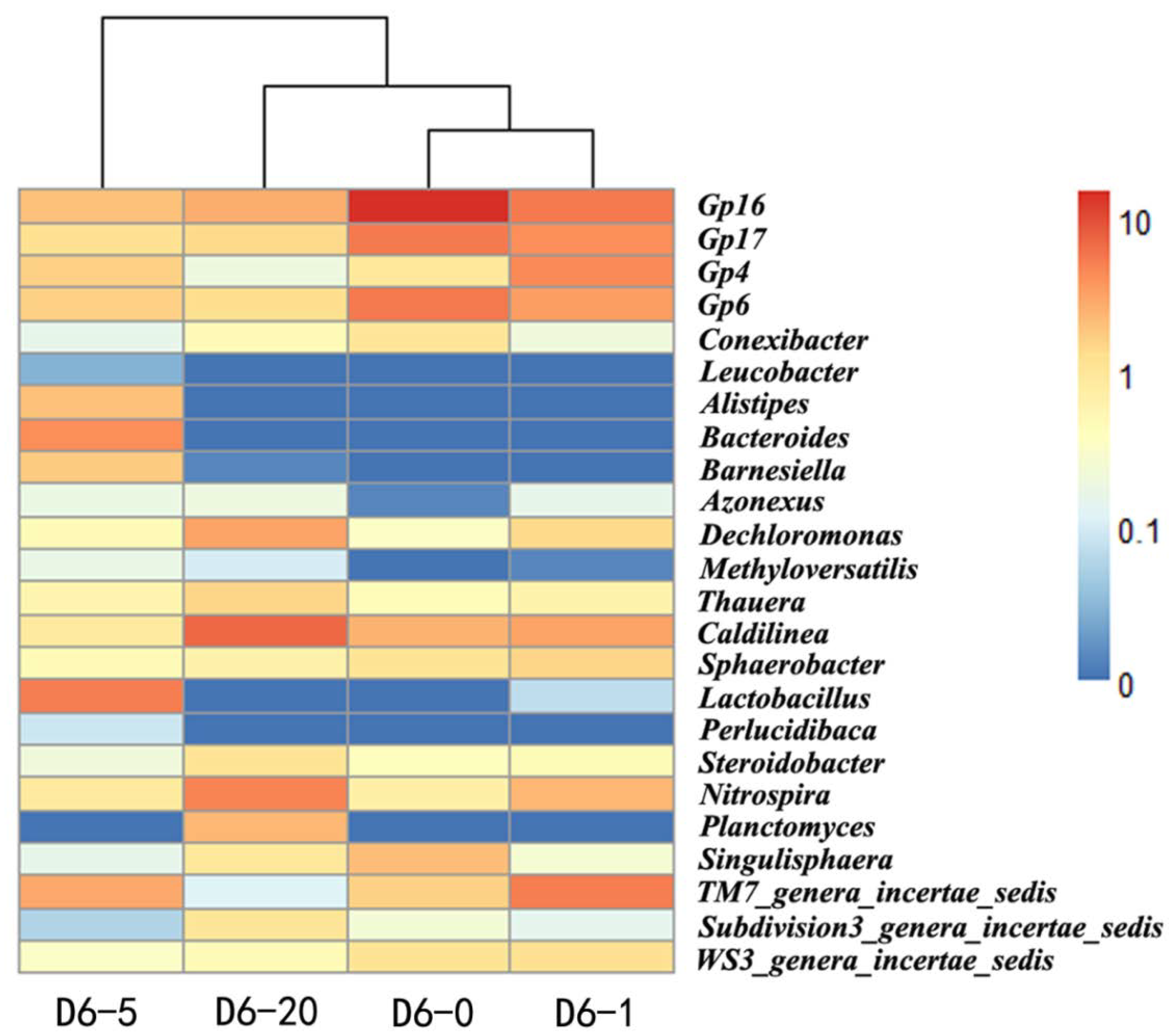
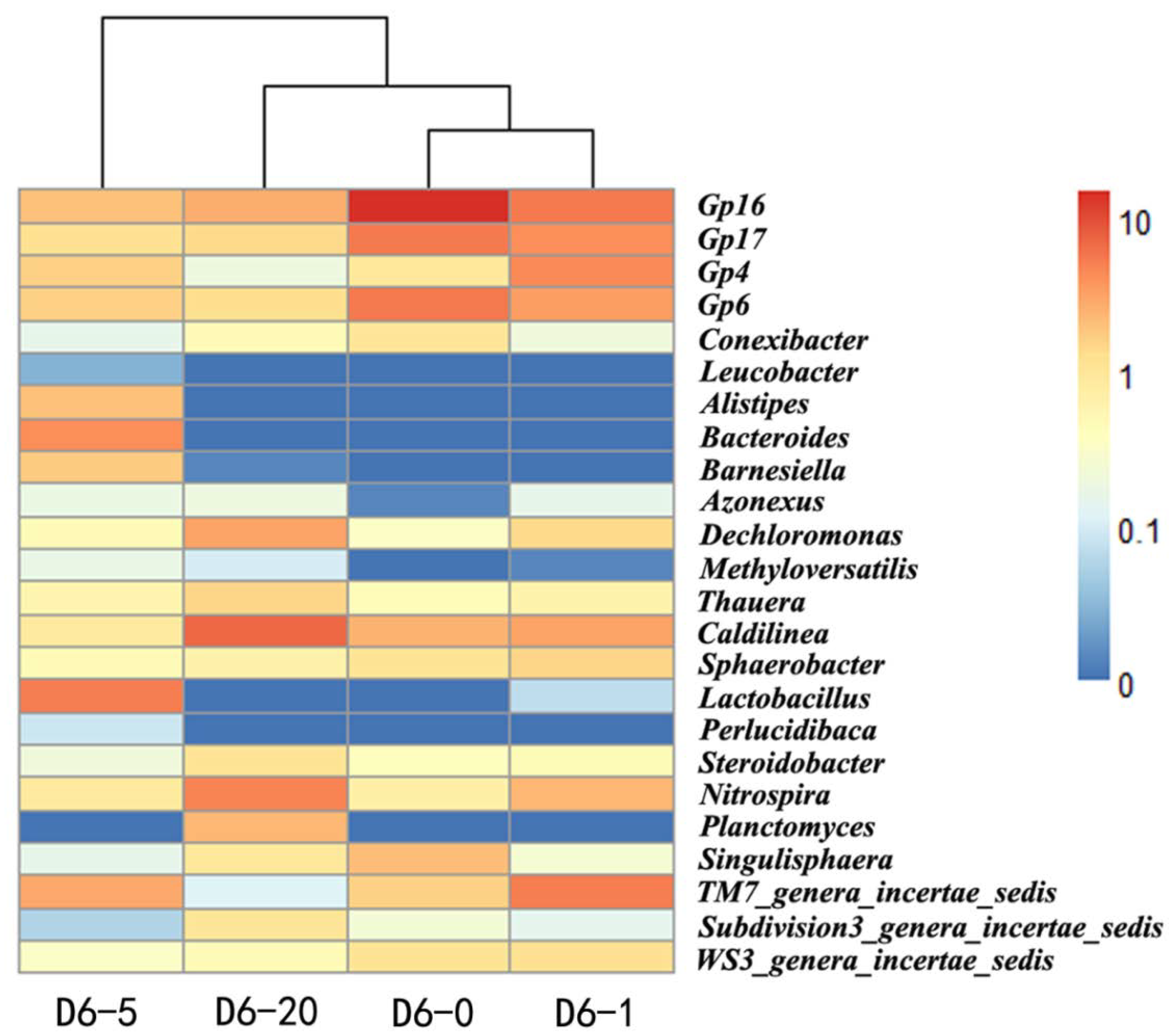
2.2. Identification of TRB in the Sludge
| Phylum | Class | Genus | D6-0 | D6-1 | D6-5 | D6-20 |
|---|---|---|---|---|---|---|
| Proteobacteria | Betaproteobacteria | Sulfuritalea | 0.01% | 0.10% | 0.10% | 0.54% |
| Armatimonadetes | Armatimonadia | Armatimonas | ND | ND | 0.01% | 0.39% |
| Verrucomicrobia | Verrucomicrobiae | Prosthecobacter | ND | ND | 0.01% | 0.37% |
| Proteobacteria | Alphaproteobacteria | Hyphomicrobium | ND | 0.11% | 0.13% | 0.34% |
| Proteobacteria | Betaproteobacteria | Azonexus | 0.01% | 0.15% | 0.18% | 0.20% |
| Chloroflexi | Anaerolineae | Longilinea | ND | 0.01% | 0.01% | 0.15% |
| Proteobacteria | Alphaproteobacteria | Novosphingobium | 0.04% | 0.04% | 0.07% | 0.13% |
| Proteobacteria | Alphaproteobacteria | Paracoccus | ND | 0.01% | 0.01% | 0.11% |
| Proteobacteria | Alphaproteobacteria | Rhodobacter | ND | 0.01% | 0.03% | 0.10% |
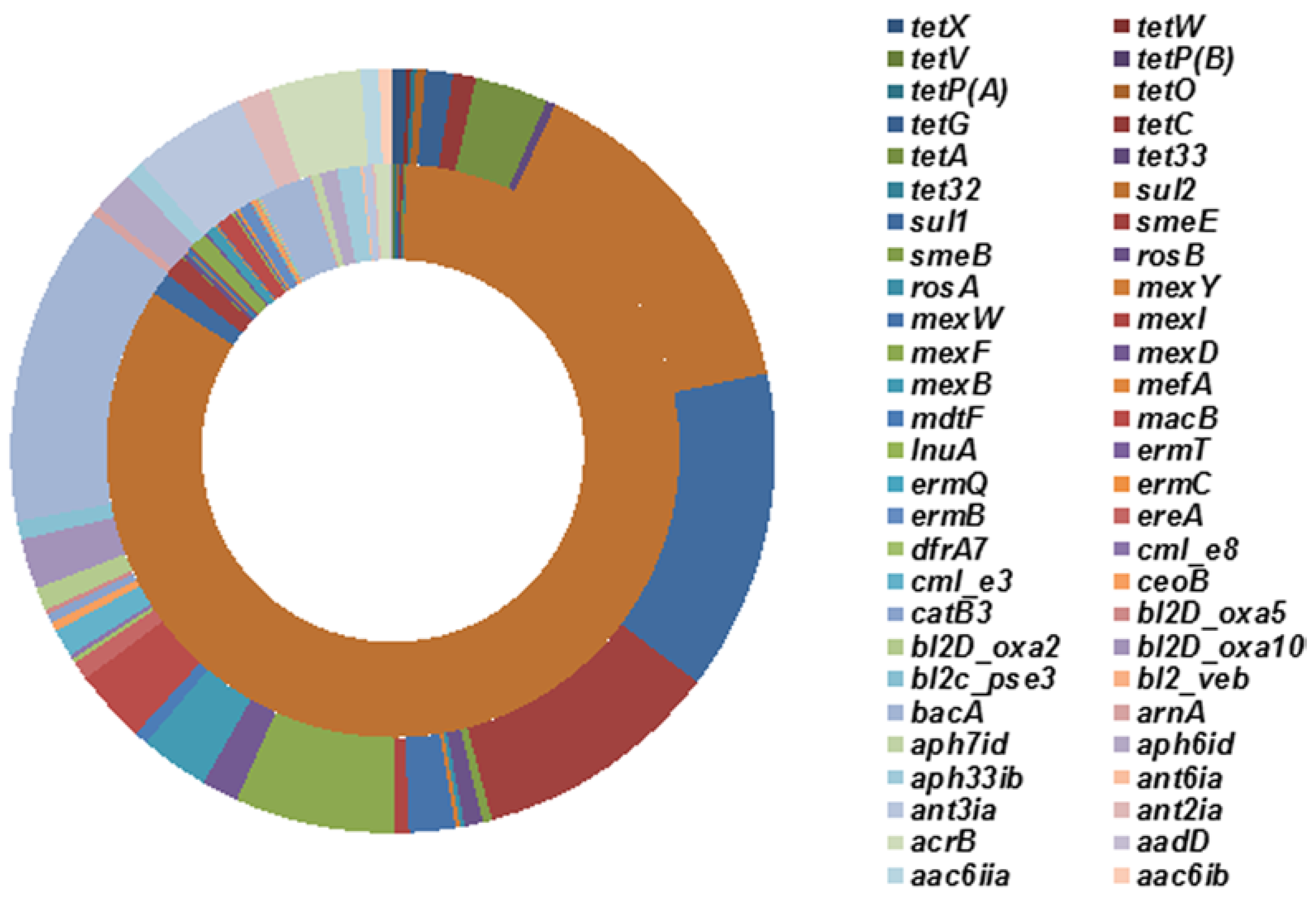
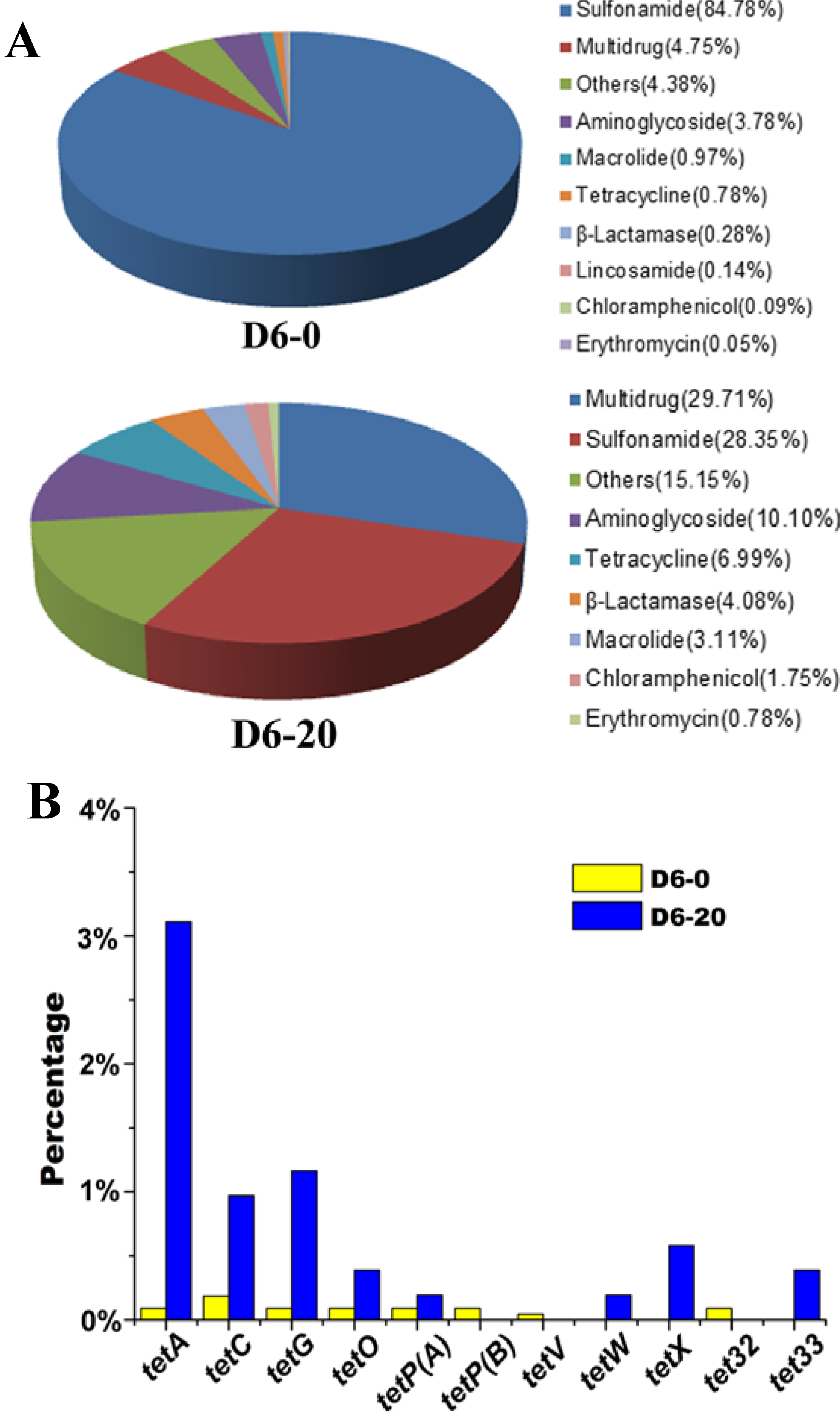
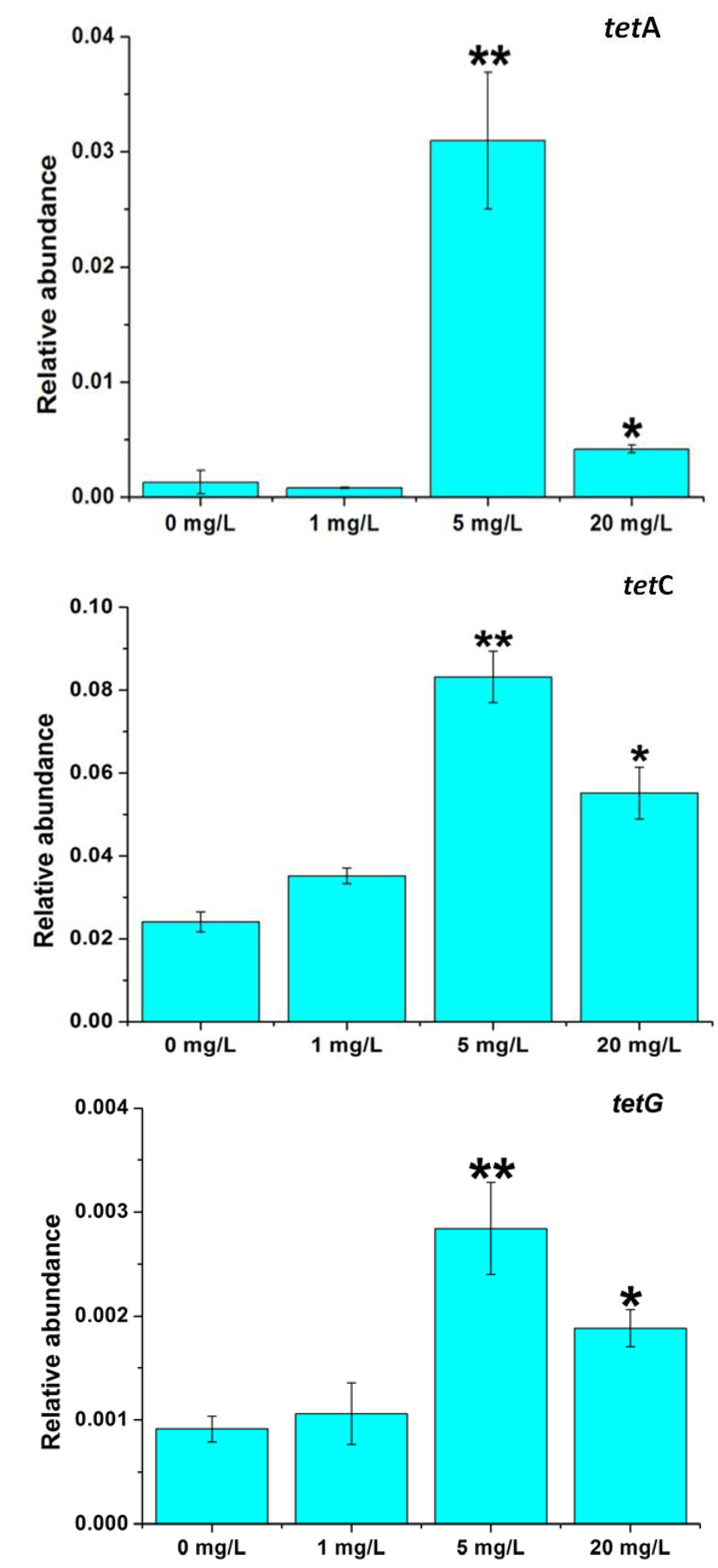
2.3. Effects of Tetracycline Stress on the Abundance and Diversity of ARGs
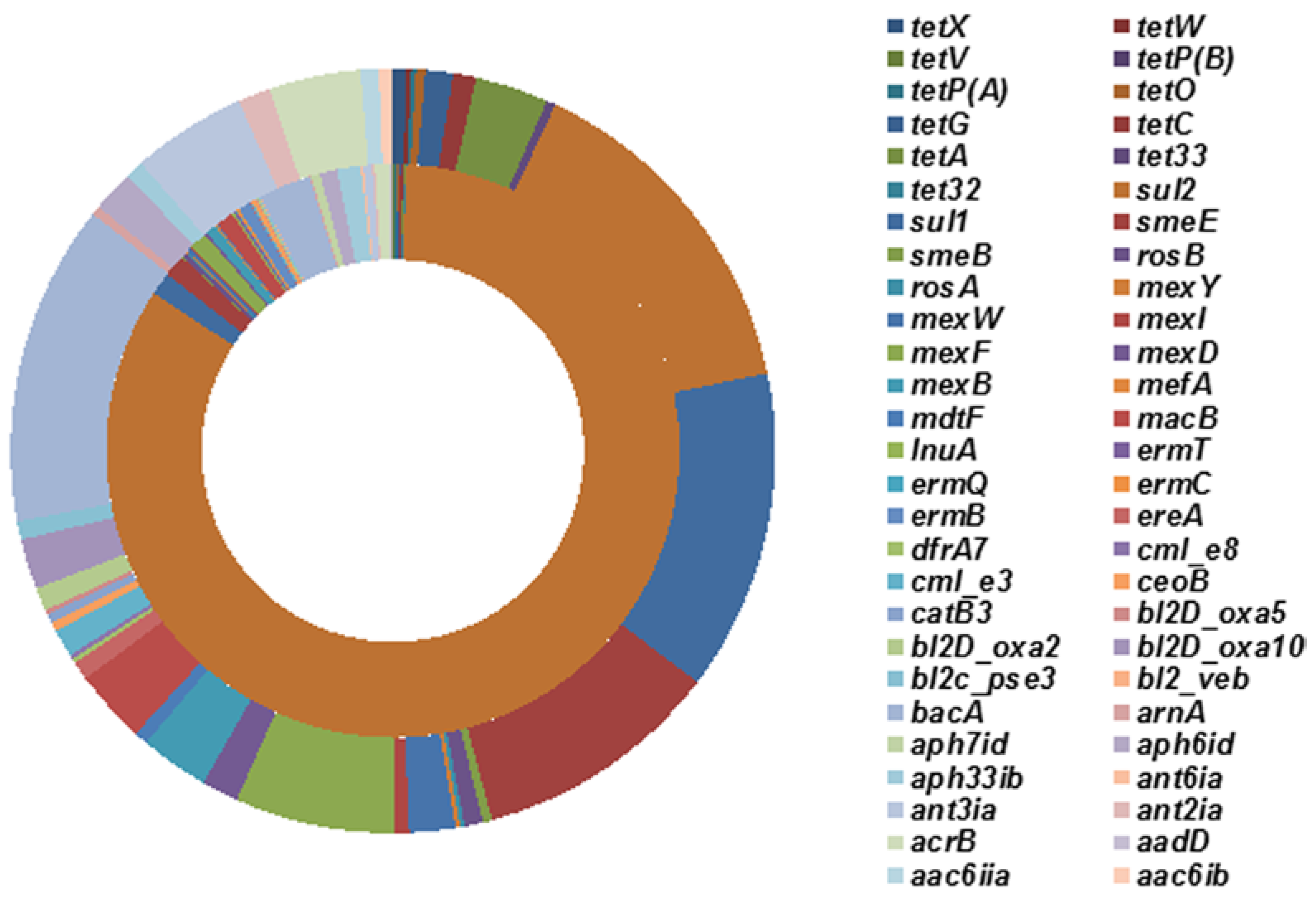
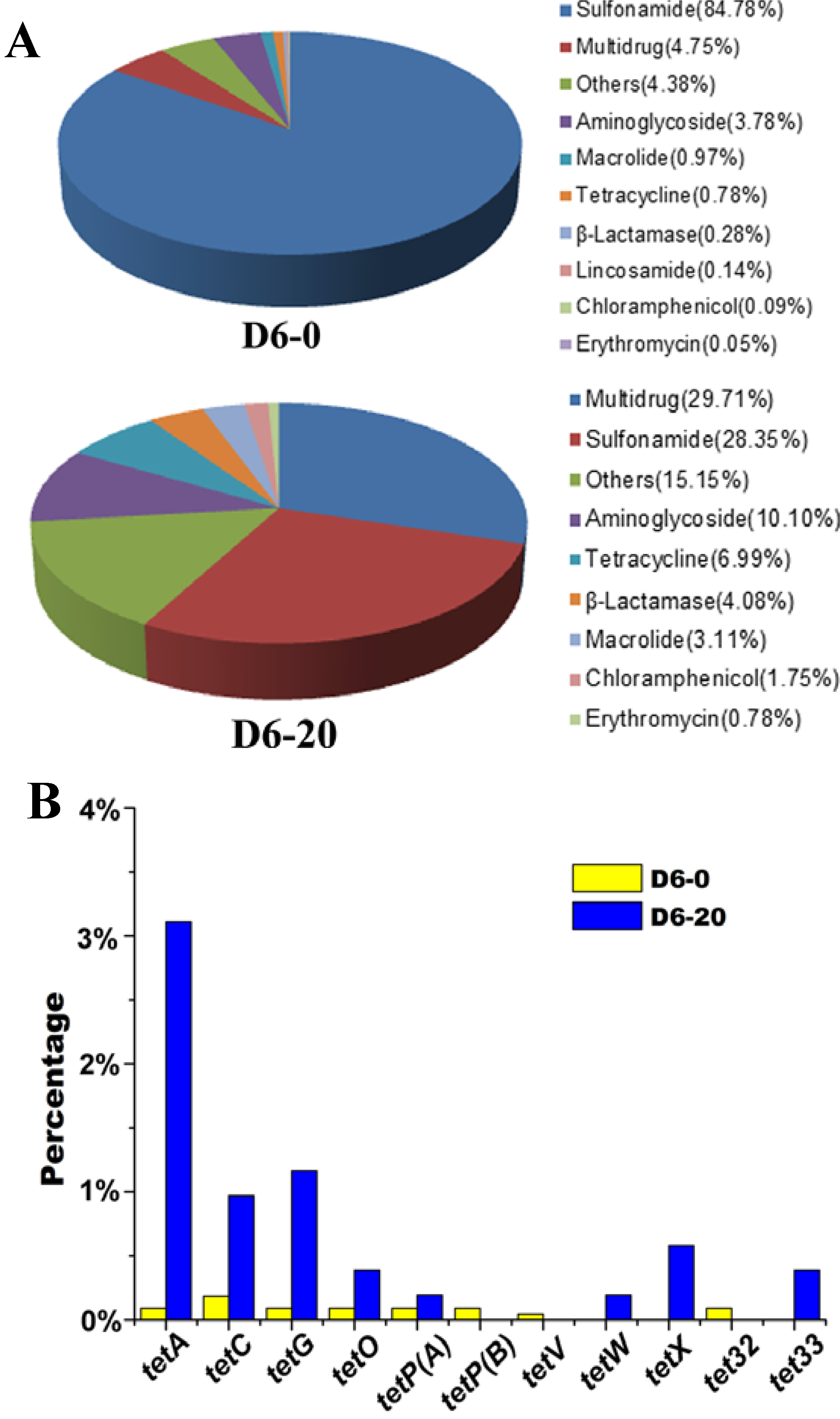

3. Materials and Methods
3.1. Batch Experiments
3.2. DNA Extraction and PCR
3.3. Quantitative Real-Time PCR
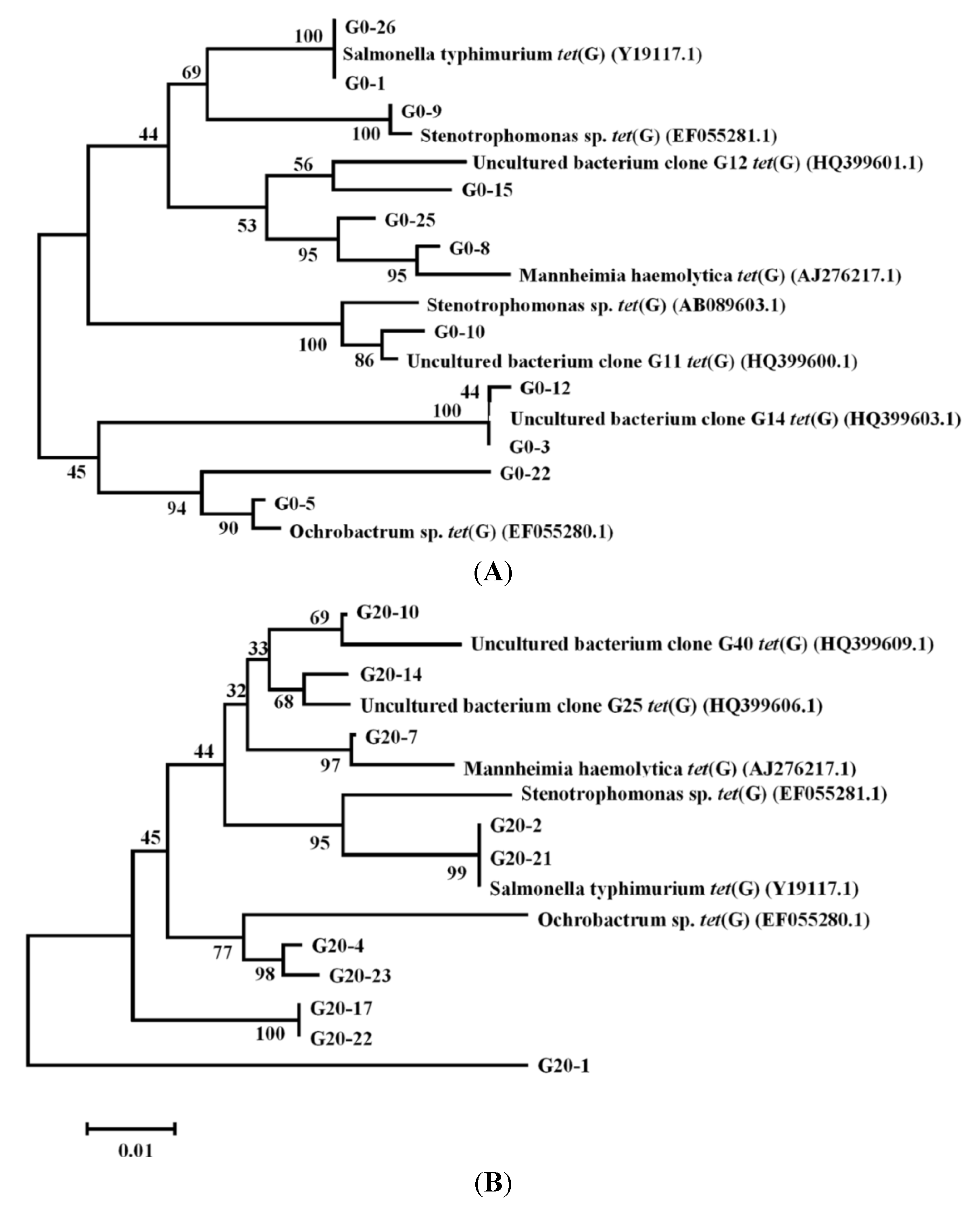
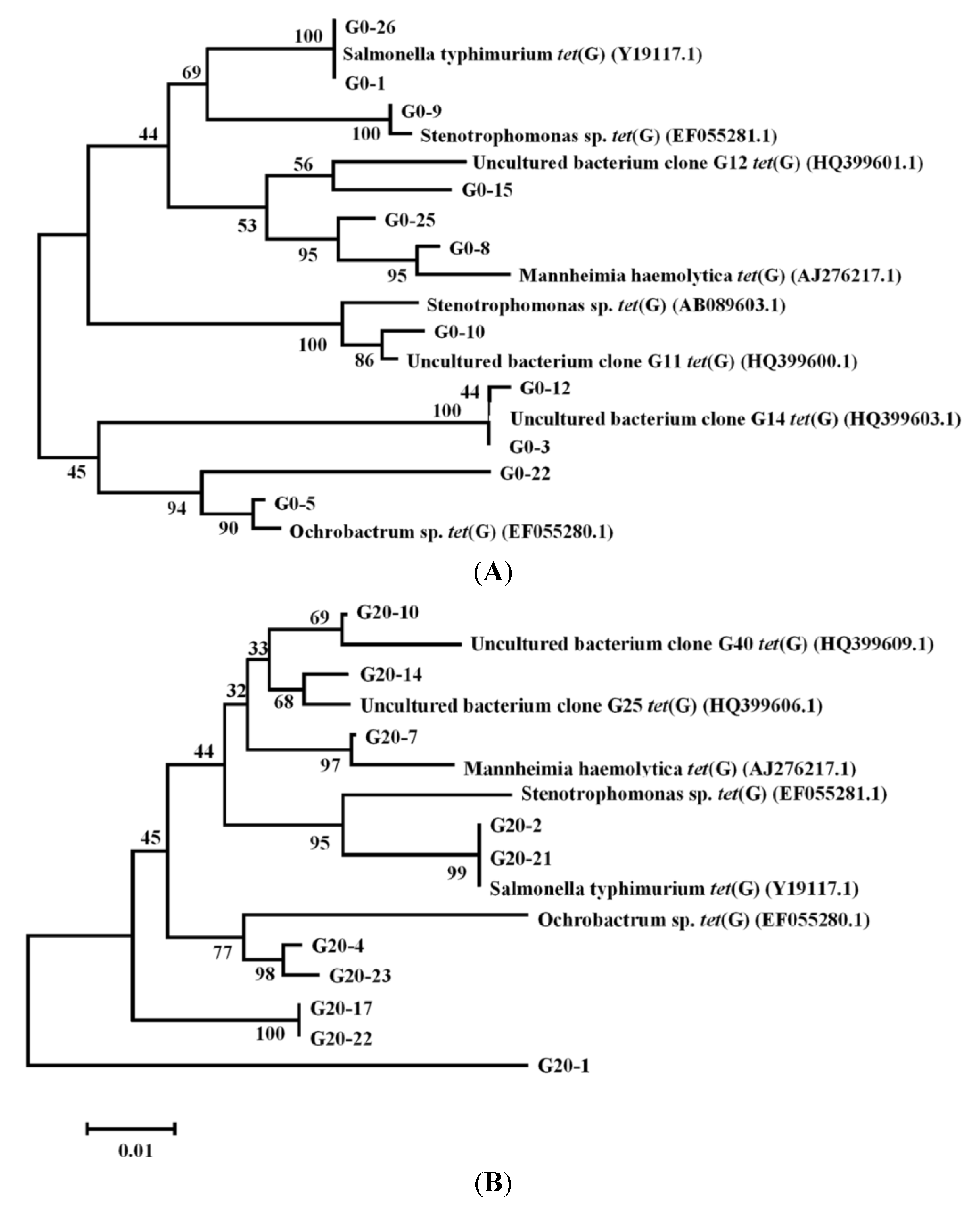
3.4. Cloning and Phylogenetic Analysis of tetG
3.5. 454 Pyrosequencing
3.6. Illumina High-Throughput Sequencing
3.7. Bioinformatics Analysis
4. Conclusions
Supplementary Files
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Kümmerer, K. Resistance in the environment. J. Antimicrob. Chemoth. 2004, 54, 311–320. [Google Scholar]
- Levy, S.B.; Marshall, B. Antibacterial resistance worldwide: Causes, challenges and responses. Nat. Med. 2004, 10, 122–129. [Google Scholar]
- Walsh, T.R.; Weeks, J.; Livermore, D.M.; Toleman, M.A. Dissemination of NDM-1 positive bacteria in the New Delhi environment and its implications for human health: An environmental point prevalence study. Lancet Infect. Dis. 2011, 11, 355–362. [Google Scholar]
- Sarmah, A.K.; Meyer, M.T.; Boxall, A. A global perspective on the use, sales, exposure pathways, occurrence, fate and effects of veterinary antibiotics (VAs) in the environmt. Chemosphere 2006, 65, 725–759. [Google Scholar] [CrossRef]
- Kim, H.; Hong, Y.; Park, J.-E.; Sharma, V.K.; Cho, S.-I. Sulfonamides and tetracyclines in livestock wastewater. Chemosphere 2013, 91, 888–894. [Google Scholar] [CrossRef]
- Ben, W.; Qiang, Z.; Adams, C.; Zhang, H.; Chen, L. Simultaneous determination of sulfonamides, tetracyclines and tiamulin in swine wastewater by solid-phase extraction and liquid chromatography-mass spectrometry. J. Chromatogr. A 2008, 1202, 173–180. [Google Scholar]
- Li, B.; Zhang, T.; Xu, Z.; Fang, H.H.P. Rapid analysis of 21 antibiotics of multiple classes in municipal wastewater using ultra performance liquid chromatography-tandem mass spectrometry. Anal. Chim. Acta 2009, 645, 64–72. [Google Scholar] [CrossRef]
- Gao, P.; Munir, M.; Xagoraraki, I. Correlation of tetracycline and sulfonamide antibiotics with corresponding resistance genes and resistant bacteria in a conventional municipal wastewater treatment plant. Sci. Total Environ. 2012, 421, 173–183. [Google Scholar]
- Taviani, E.; Ceccarelli, D.; Lazaro, N.; Bani, S.; Cappuccinelli, P.; Colwell, R.R.; Colombo, M.M. Environmental Vibrio spp., isolated in Mozambique, contain a polymorphic group of integrative conjugative elements and class 1 integrons. FEMS Microbiol. Ecol. 2008, 64, 45–54. [Google Scholar] [CrossRef]
- Zhang, T.; Zhang, M.; Zhang, X.-X.; Fang, H.H. Tetracycline resistance genes and tetracycline resistant lactose-fermenting Enterobacteriaceae in activated sludge of sewage treatment plants. Environ. Sci. Technol. 2009, 43, 3455–3460. [Google Scholar]
- Zhang, X.-X.; Zhang, T. Occurrence, abundance, and diversity of tetracycline resistance genes in 15 sewage treatment plants across China and other global locations. Environ. Sci. Technol. 2011, 45, 2598–2604. [Google Scholar] [CrossRef]
- Kim, S.; Jensen, J.N.; Aga, D.S.; Weber, A.S. Tetracycline as a selector for resistant bacteria in activated sludge. Chemosphere 2007, 66, 1643–1651. [Google Scholar] [CrossRef]
- Li, B.; Zhang, X.-X.; Guo, F.; Wu, W.; Zhang, T. Characterization of tetracycline resistant bacterial community in saline activated sludge using batch stress incubation with high-throughput sequencing analysis. Water Res. 2013, 47, 4207–4216. [Google Scholar]
- Zhang, W.; Huang, M.-H.; Qi, F.-F.; Sun, P.-Z.; van Ginkel, S.W. Effect of trace tetracycline concentrations on the structure of a microbial community and the development of tetracycline resistance genes in sequencing batch reactors. Bioresour. Technol. 2013, 150, 9–14. [Google Scholar] [CrossRef]
- Walsh, F.; Cooke, N.M.; Smith, S.G.; Moran, G.P.; Cooke, F.J.; Ivens, A.; Wain, J.; Rogers, T.R. Comparison of two DNA microarrays for detection of plasmid-mediated antimicrobial resistance and virulence factor genes in clinical isolates of Enterobacteriaceae and non-Enterobacteriaceae. Int. J. Antimicrob. Agent 2010, 35, 593–598. [Google Scholar] [CrossRef]
- Roesch, L.F.; Fulthorpe, R.R.; Riva, A.; Casella, G.; Hadwin, A.K.; Kent, A.D.; Daroub, S.H.; Camargo, F.A.; Farmerie, W.G.; Triplett, E.W. Pyrosequencing enumerates and contrasts soil microbial diversity. ISME J. 2007, 1, 283–290. [Google Scholar]
- Qin, J.; Li, R.; Raes, J.; Arumugam, M.; Burgdorf, K.S.; Manichanh, C.; Nielsen, T.; Pons, N.; Levenez, F.; Yamada, T. A human gut microbial gene catalogue established by metagenomic sequencing. Nature 2010, 464, 59–65. [Google Scholar] [Green Version]
- Håvelsrud, O.; Haverkamp, T.; Kristensen, T.; Jakobsen, K.; Rike, A. Metagenomic and geochemical characterization of pockmarked sediments overlaying the Troll petroleum reservoir in the North Sea. BMC Microbiol. 2012, 12, 203. [Google Scholar] [Green Version]
- Ye, L.; Zhang, T.; Wang, T.; Fang, Z. Microbial structures, functions, and metabolic pathways in wastewater treatment bioreactors revealed using high-throughput sequencing. Environ. Sci. Technol. 2012, 46, 13244–13252. [Google Scholar]
- Li, D.; Qi, R.; Yang, M.; Zhang, Y.; Yu, T. Bacterial community characteristics under long-term antibiotic selection pressures. Water Res. 2011, 45, 6063–6073. [Google Scholar] [CrossRef]
- Czárán, T.L.; Hoekstra, R.F.; Pagie, L. Chemical warfare between microbes promotes biodiversity. Proc. Natl. Acad. Sci. USA 2002, 99, 786–790. [Google Scholar]
- Lozada, M.; Figuerola, E.L.M.; Itria, R.F.; Erijman, L. Replicability of dominant bacterial populations after long-term surfactant-enrichment in lab-scale activated sludge. Environ. Microbiol. 2006, 8, 625–638. [Google Scholar] [CrossRef]
- Kobashi, Y.; Hasebe, A.; Nishio, M.; Uchiyama, H. Diversity of tetracycline resistance genes in bacteria isolated from various agricultural environments. Microbes Environ. 2007, 22, 44–51. [Google Scholar] [CrossRef]
- Li, D.; Yu, T.; Zhang, Y.; Yang, M.; Li, Z.; Liu, M.; Qi, R. Antibiotic resistance characteristics of environmental bacteria from an oxytetracycline production wastewater treatment plant and the receiving river. Appl. Environ. Microb. 2010, 76, 3444–3451. [Google Scholar]
- Deng, Y.; Zhang, Y.; Gao, Y.; Li, D.; Liu, R.; Liu, M.; Zhang, H.; Hu, B.; Yu, T.; Yang, M. Microbial community compositional analysis for series reactors treating high level antibiotic wastewater. Environ. Sci. Technol. 2011, 46, 795–801. [Google Scholar]
- Vacková, L.; Srb, M.; Stloukal, R.; Wanner, J. Comparison of denitrification at low temperature using encapsulated Paracoccus denitrificans, Pseudomonas fluorescens and mixed culture. Bioresour. Technol. 2011, 102, 4661–4666. [Google Scholar]
- Kojima, H.; Fukui, M. Sulfuritalea hydrogenivorans gen. nov., sp. nov., a facultative autotroph isolated from a freshwater lake. Int. J. Syst. Evol. Microbiol. 2011, 61, 1651–1655. [Google Scholar] [CrossRef]
- Quan, Z.-X.; Im, W.-T.; Lee, S.-T. Azonexus caeni sp. nov., a denitrifying bacterium isolated from sludge of a wastewater treatment plant. Int. J. Syst. Evol. Microbiol. 2006, 56, 1043–1046. [Google Scholar]
- Kim, J.K.; Lee, B.-K.; Kim, S.-H.; Moon, J.-H. Characterization of denitrifying photosynthetic bacteria isolated from photosynthetic sludge. Aquacult. Eng. 1999, 19, 179–193. [Google Scholar]
- McDonald, I.R.; Doronina, N.V.; Trotsenko, Y.A.; McAnulla, C.; Murrell, J.C. Hyphomicrobium chloromethanicum sp. nov. and Methylobacterium chloromethanicum sp. nov., chloromethane-utilizing bacteria isolated from a polluted environment. Int. J. Syst. Evol. Microbiol. 2001, 51, 119–122. [Google Scholar]
- Tiirola, M.A.; Männistö, M.K.; Puhakka, J.A.; Kulomaa, M.S. Isolation and characterization of Novosphingobium sp. strain MT1, a dominant polychlorophenol-degrading strain in a groundwater bioremediation system. Appl. Environ. Microbiol. 2002, 68, 173–180. [Google Scholar]
- Yuan, J.; Lai, Q.; Zheng, T.; Shao, Z. Novosphingobium indicum sp. nov., a polycyclic aromatic hydrocarbon-degrading bacterium isolated from a deep-sea environment. Int. J. Syst. Evol. Microbiol. 2009, 59, 2084–2088. [Google Scholar] [CrossRef]
- Liu, Z.-P.; Wang, B.-J.; Liu, Y.-H.; Liu, S.-J. Novosphingobium taihuense sp. nov., a novel aromatic-compound-degrading bacterium isolated from Taihu Lake, China. Int. J. Syst. Evol. Microbiol. 2005, 55, 1229–1232. [Google Scholar] [CrossRef]
- Fujii, K.; Satomi, M.; Morita, N.; Motomura, T.; Tanaka, T.; Kikuchi, S. Novosphingobium tardaugens sp. nov., an oestradiol-degrading bacterium isolated from activated sludge of a sewage treatment plant in Tokyo. Int. J. Syst. Evol. Microbiol. 2003, 53, 47–52. [Google Scholar] [CrossRef]
- Kristiansson, E.; Fick, J.; Janzon, A.; Grabic, R.; Rutgersson, C.; Weijdegård, B.; Söderström, H.; Larsson, D.J. Pyrosequencing of antibiotic-contaminated river sediments reveals high levels of resistance and gene transfer elements. PLoS One 2011, 6, 1–7. [Google Scholar]
- Stevens, D.L.; Higbee, J.W.; Oberhofer, T.R.; Everett, E.D. Antibiotic susceptibilities of human isolates of Pasteurella. multocida. Antimicrob. Agents Chemother. 1979, 16, 322–324. [Google Scholar] [CrossRef]
- Alonso, A.; Campanario, E.; Martinez, J.L. Emergence of multidrug-resistant mutants is increased under antibiotic selective pressure in Pseudomonas aeruginosa. Microbiol-Sgm 1999, 145, 2857–2862. [Google Scholar]
- Auerbach, E.A.; Seyfried, E.E.; McMahon, K.D. Tetracycline resistance genes in activated sludge wastewater treatment plants. Water Res. 2007, 41, 1143–1151. [Google Scholar] [CrossRef]
- Laureti, L.; Matic, I.; Gutierrez, A. Bacterial responses and genome instability induced by subinhibitory concentrations of antibiotics. Antibiotics 2013, 2, 100–114. [Google Scholar] [CrossRef]
- Li, B.; Zhang, T. Biodegradation and adsorption of antibiotics in the activated sludge process. Environ. Sci. Technol. 2010, 44, 3468–3473. [Google Scholar] [CrossRef]
- López-Gutiérrez, J.C.; Henry, S.; Hallet, S.; Martin-Laurent, F.; Catroux, G.; Philippot, L. Quantification of a novel group of nitrate-reducing bacteria in the environment by real-time PCR. J. Microbiol. Meth. 2004, 57, 399–407. [Google Scholar]
- Tamura, K.; Peterson, D.; Peterson, N.; Stecher, G.; Nei, M.; Kumar, S. MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol. Biol. Evol. 2011, 28, 2731–2739. [Google Scholar] [CrossRef]
- Meyer, F.; Paarmann, D.; D’Souza, M.; Olson, R.; Glass, E.M.; Kubal, M.; Paczian, T.; Rodriguez, A.; Stevens, R.; Wilke, A.; et al. The metagenomics RAST server–a public resource for the automatic phylogenetic and functional analysis of metagenomes. BMC Bioinform. 2008, 9, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Cole, J.R.; Wang, Q.; Cardenas, E.; Fish, J.; Chai, B.; Farris, R.J.; Kulam-Syed-Mohideen, A.S.; McGarrell, D.M.; Marsh, T.; Garrity, G.M. The ribosomal database project: Improved alignments and new tools for rRNA analysis. Nucleic Acids Res. 2009, 37, 141–145. [Google Scholar]
- Claesson, M.J.; O’Sullivan, O.; Wang, Q.; Nikkila, J.; Marchesi, J.R.; Smidt, H.; de Vos, W.M.; Ross, R.P.; O’Toole, P.W. Comparative analysis of pyrosequencing and a phylogenetic microarray for exploring microbial community structures in the human distal intestine. PLoS One 2009, 4, 1–15. [Google Scholar]
- Huse, S.M.; Welch, D.M.; Morrison, H.G.; Sogin, M.L. Ironing out the wrinkles in the rare biosphere through improved OTU clustering. Environ. Microbiol. 2010, 12, 1889–1898. [Google Scholar] [CrossRef]
- Roeselers, G.; Mittge, E.K.; Stephens, W.Z.; Parichy, D.M.; Cavanaugh, C.M.; Guillemin, K.; Rawls, J.F. Evidence for a core gut microbiota in the zebrafish. ISME J. 2011, 5, 1595–1608. [Google Scholar] [CrossRef]
- Haas, B.J.; Gevers, D.; Earl, A.M.; Feldgarden, M.; Ward, D.V.; Giannoukos, G.; Ciulla, D.; Tabbaa, D.; Highlander, S.K.; Sodergren, E. Chimeric 16S rRNA sequence formation and detection in Sanger and 454-pyrosequenced PCR amplicons. Genome Res. 2011, 21, 494–504. [Google Scholar] [CrossRef]
- Zhang, T.; Shao, M-F.; Ye, L. 454 Pyrosequencing reveals bacterial diversity of activated sludge from 14 sewage treatment plants. ISME J. 2012, 6, 1137–1147. [Google Scholar] [CrossRef]
- Wang, Q.; Garrity, G.M.; Tiedje, J.M.; Cole, J.R. Naive bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl. Environ. Microbiol. 2007, 73, 5261–5267. [Google Scholar] [CrossRef]
- Liu, B.; Pop, M. ARDB-Antibiotic Resistance Genes Database. Nucleic Acids Res. 2009, 37, 443–447. [Google Scholar] [CrossRef]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Huang, K.; Tang, J.; Zhang, X.-X.; Xu, K.; Ren, H. A Comprehensive Insight into Tetracycline Resistant Bacteria and Antibiotic Resistance Genes in Activated Sludge Using Next-Generation Sequencing. Int. J. Mol. Sci. 2014, 15, 10083-10100. https://doi.org/10.3390/ijms150610083
Huang K, Tang J, Zhang X-X, Xu K, Ren H. A Comprehensive Insight into Tetracycline Resistant Bacteria and Antibiotic Resistance Genes in Activated Sludge Using Next-Generation Sequencing. International Journal of Molecular Sciences. 2014; 15(6):10083-10100. https://doi.org/10.3390/ijms150610083
Chicago/Turabian StyleHuang, Kailong, Junying Tang, Xu-Xiang Zhang, Ke Xu, and Hongqiang Ren. 2014. "A Comprehensive Insight into Tetracycline Resistant Bacteria and Antibiotic Resistance Genes in Activated Sludge Using Next-Generation Sequencing" International Journal of Molecular Sciences 15, no. 6: 10083-10100. https://doi.org/10.3390/ijms150610083