In Vitro Treatment of Melanoma Brain Metastasis by Simultaneously Targeting the MAPK and PI3K Signaling Pathways

Abstract
:1. Introduction
2. Results and Discussion
2.1. BRAF and PTEN Status of the H1_DL2 Melanoma Brain Metastasis Cell Line
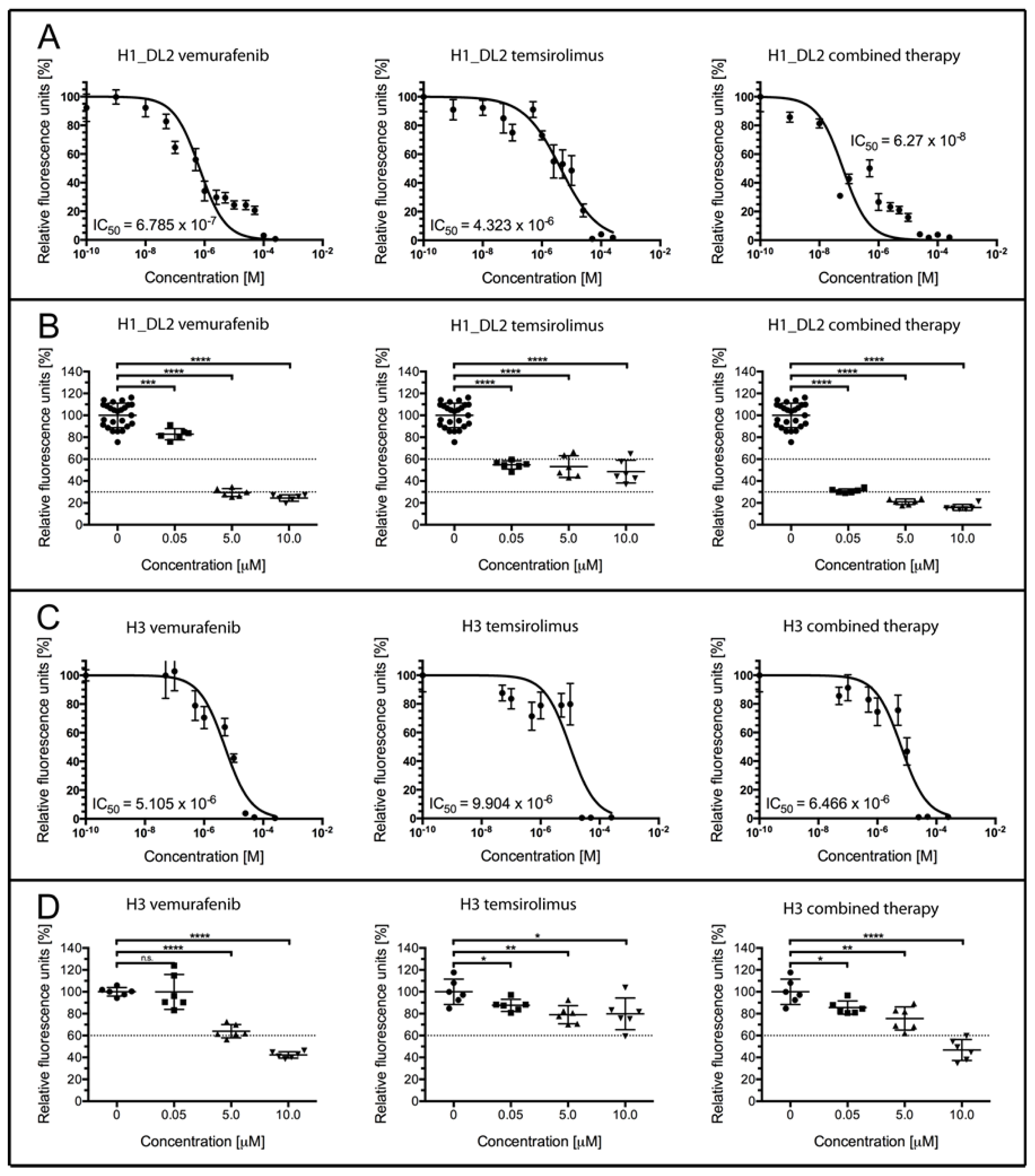
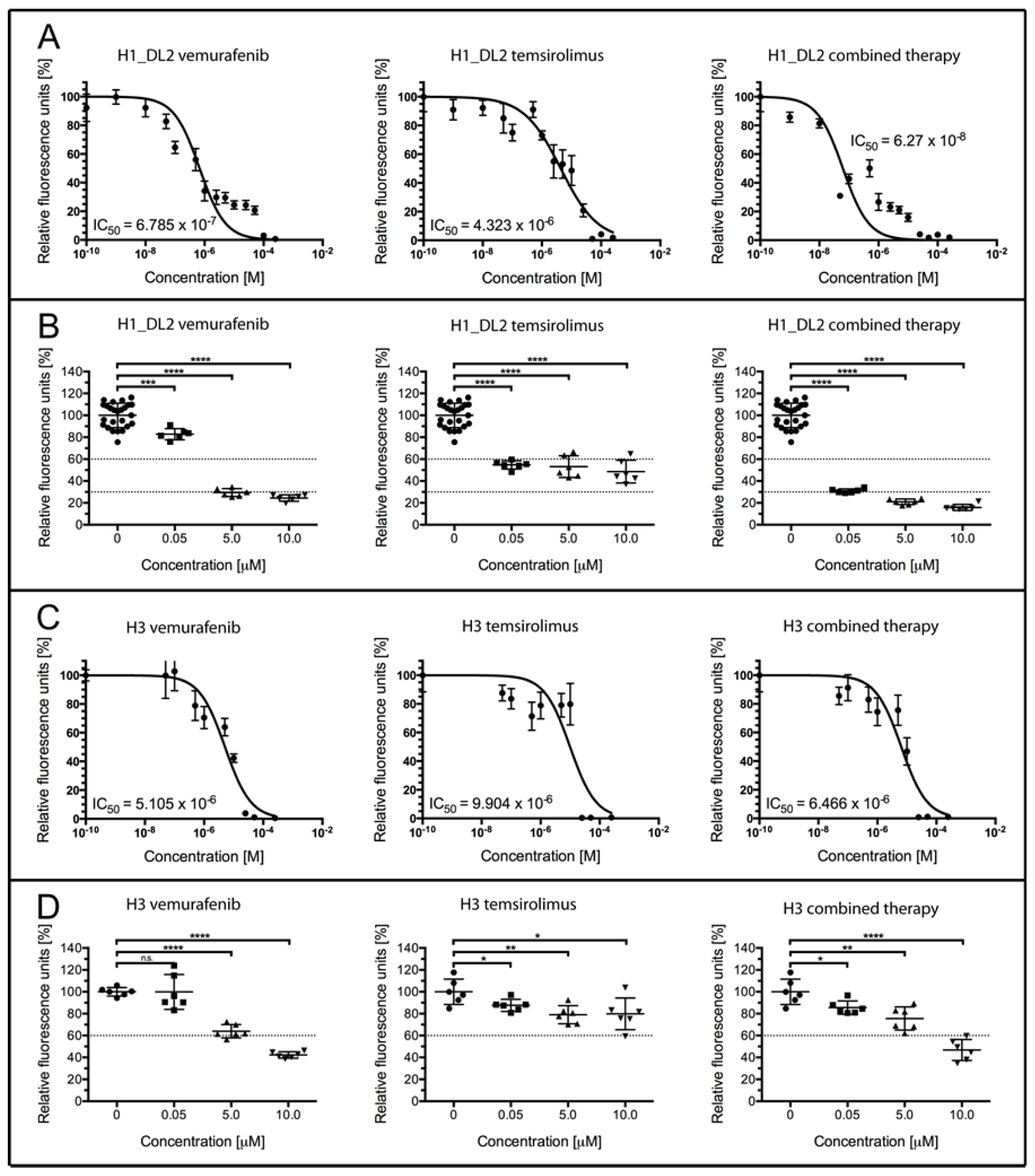
2.2. Treatment with Vemurafenib and Temsirolimus Induces Anti-Proliferative Effects in H1_DL2 and H3 Cell Lines Grown as Monolayers
2.3. Treatment with Vemurafenib Induces Anti-Migratory Effects in the H1_DL2 Cell Line Grown as Monolayers
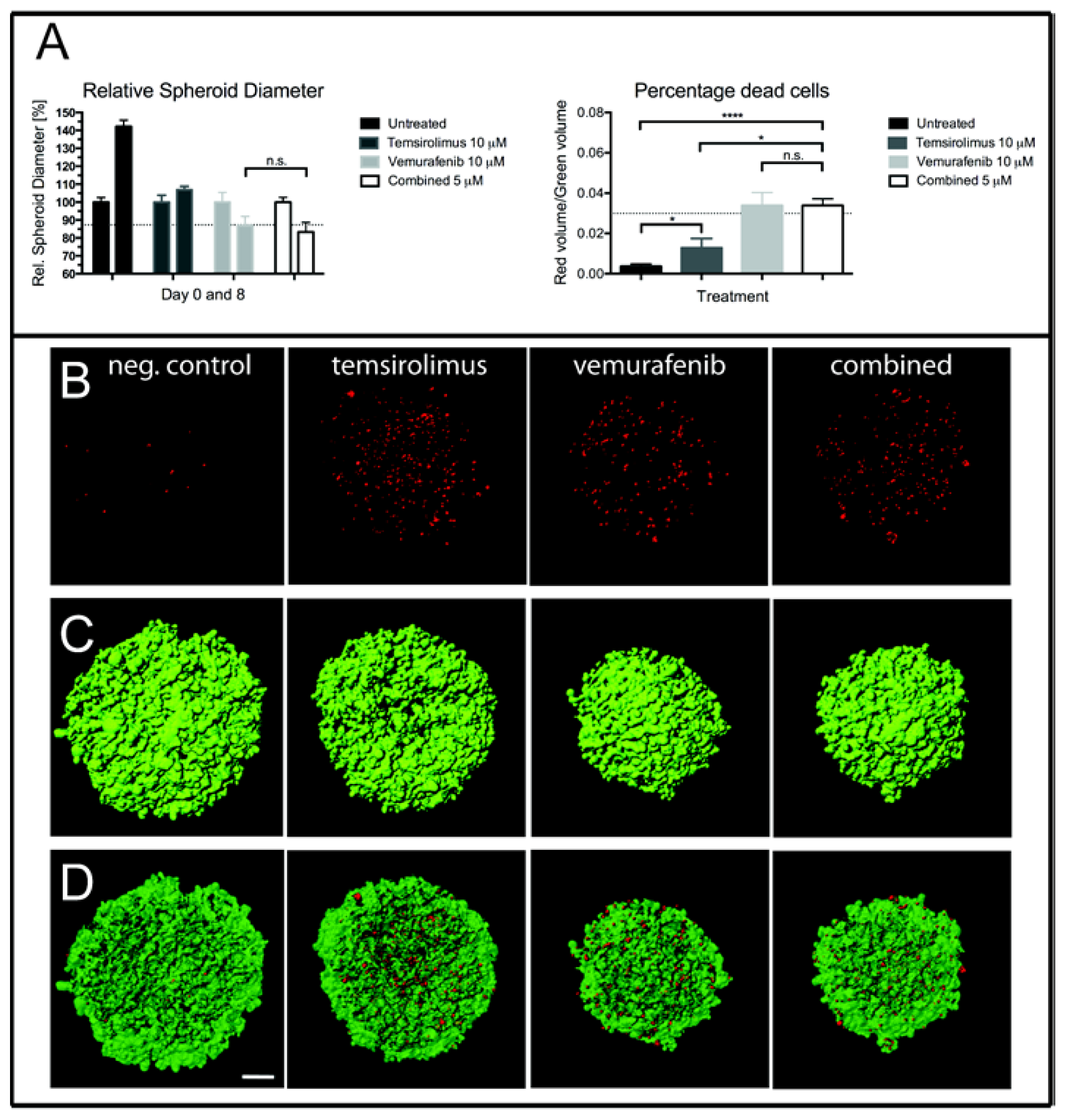
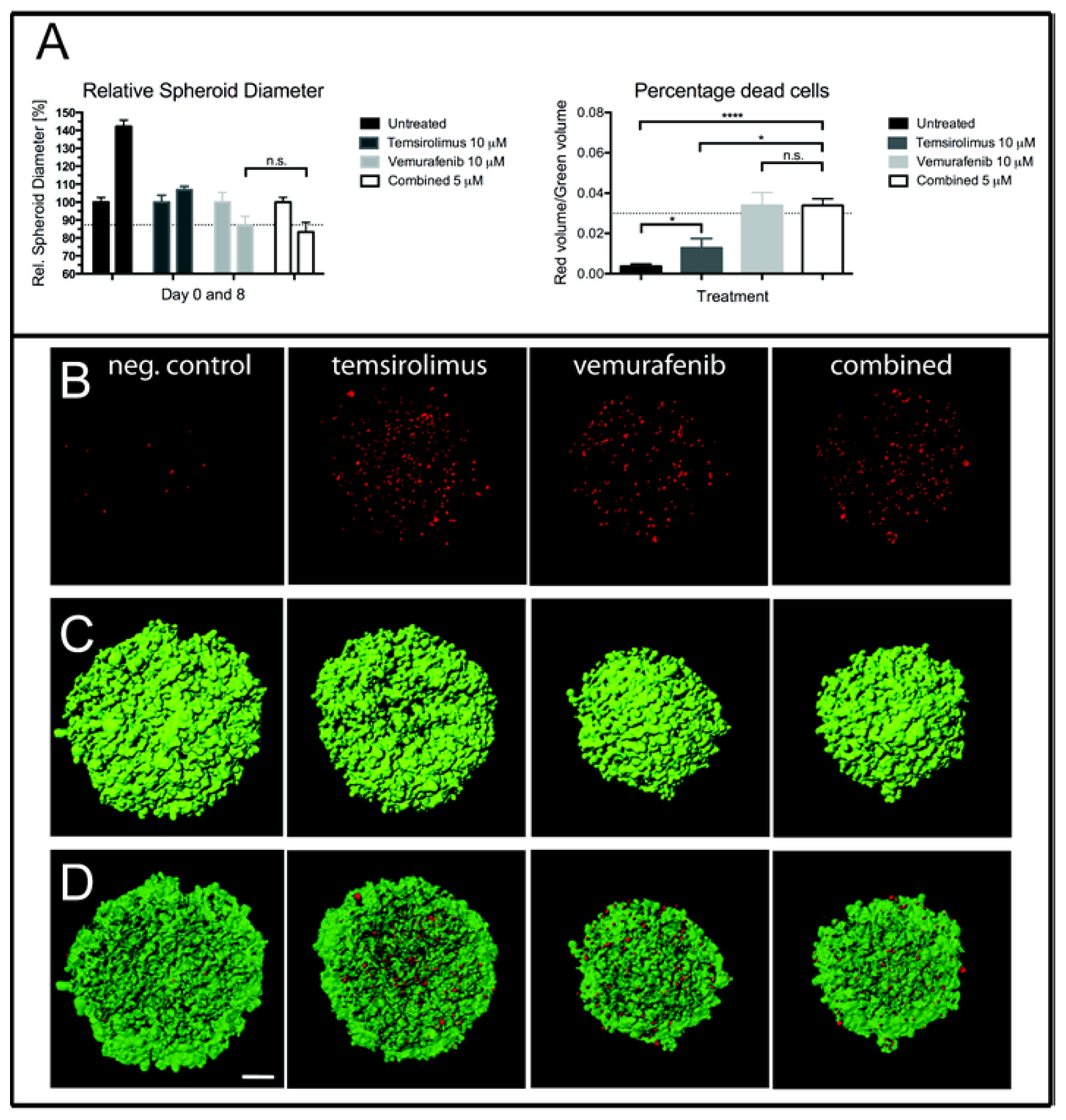
2.4. Treatment of H1_DL2 Spheroids with Vemurafenib and Temsirolimus Inhibits Spheroid Growth and Increases Cell Death
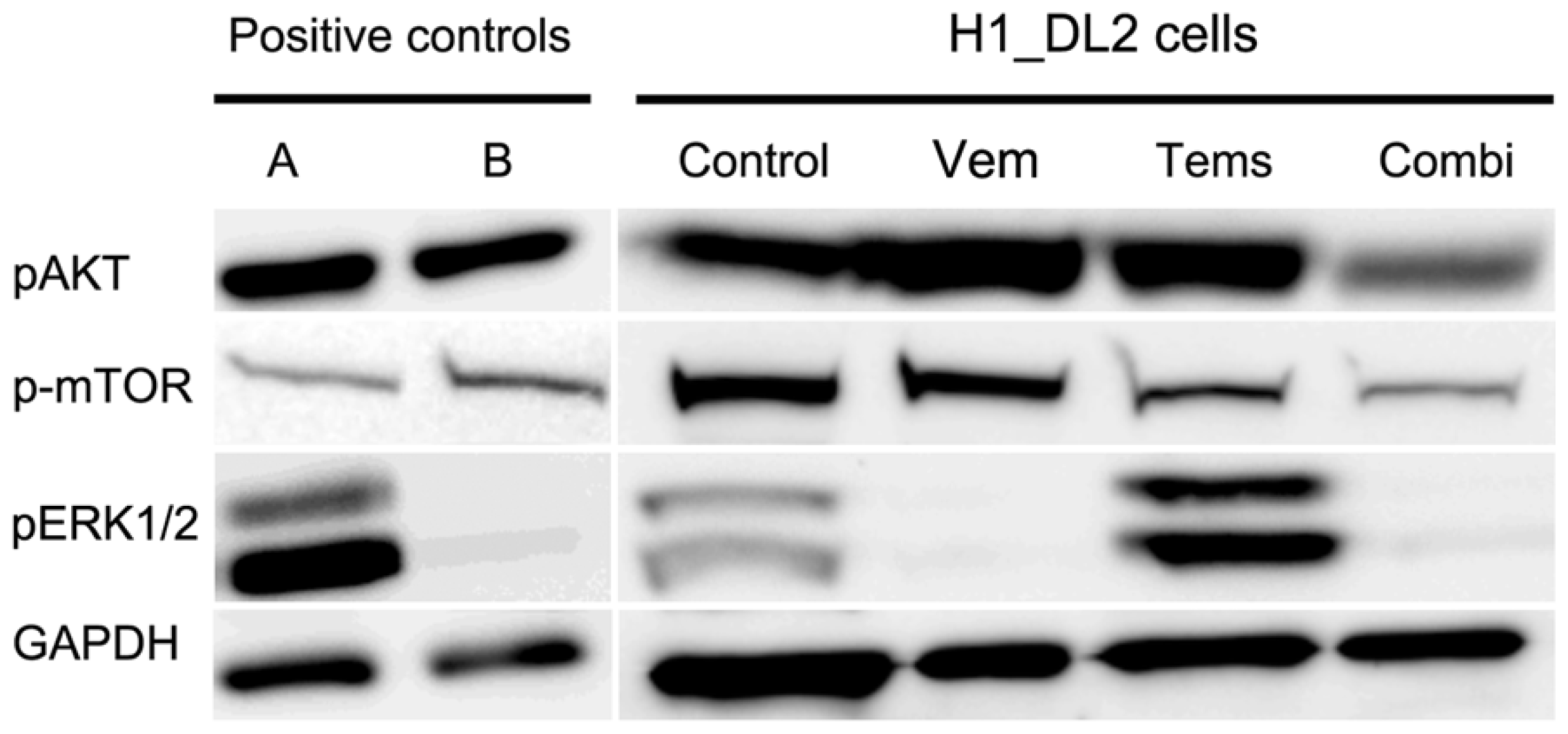
2.5. Western Blots Suggest Drug-Related Inhibition of the MAPK (Mitogen-Activated Protein Kinase) and PI3K (Phosphoinositide 3-Kinase) Pathways
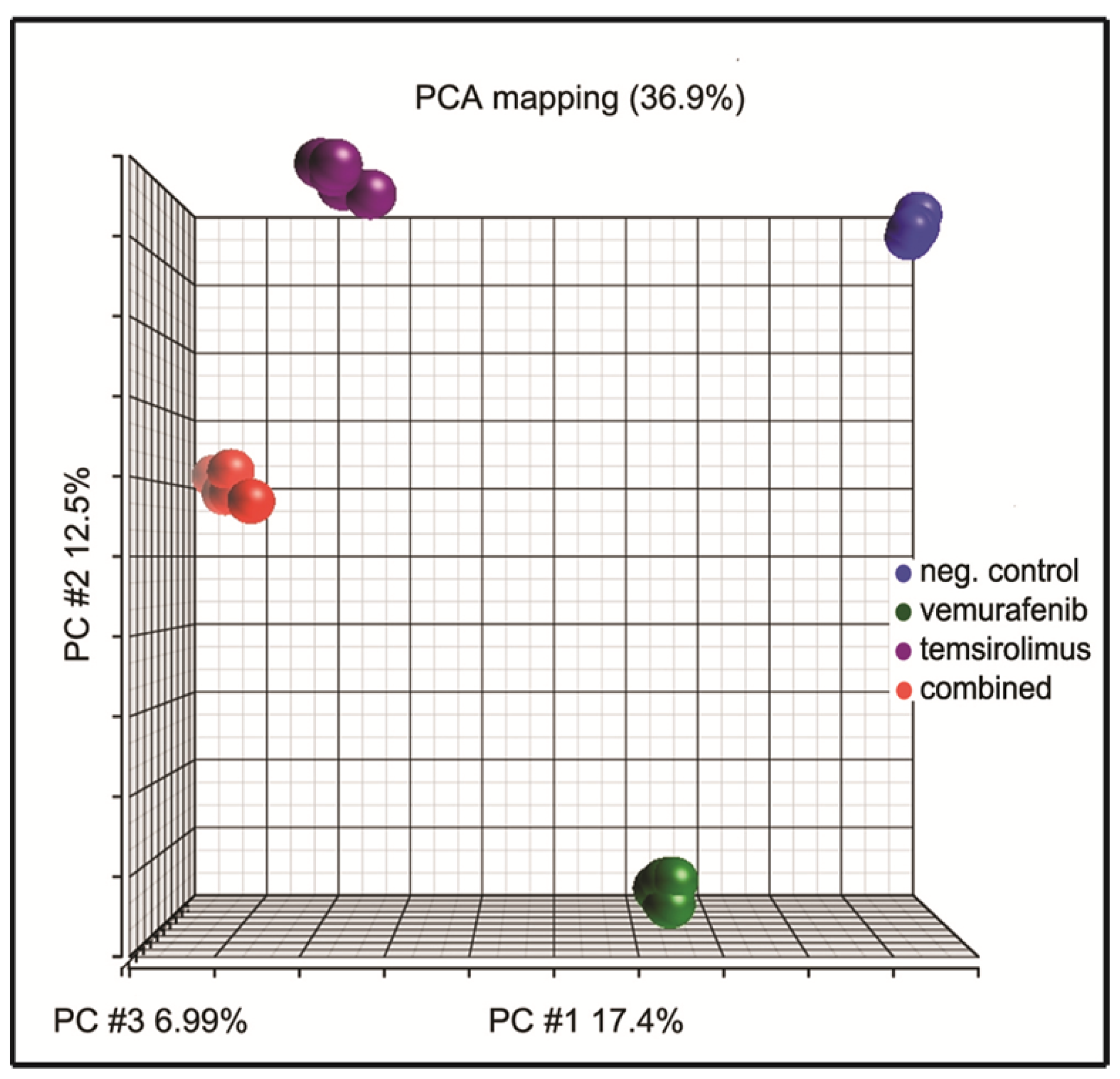
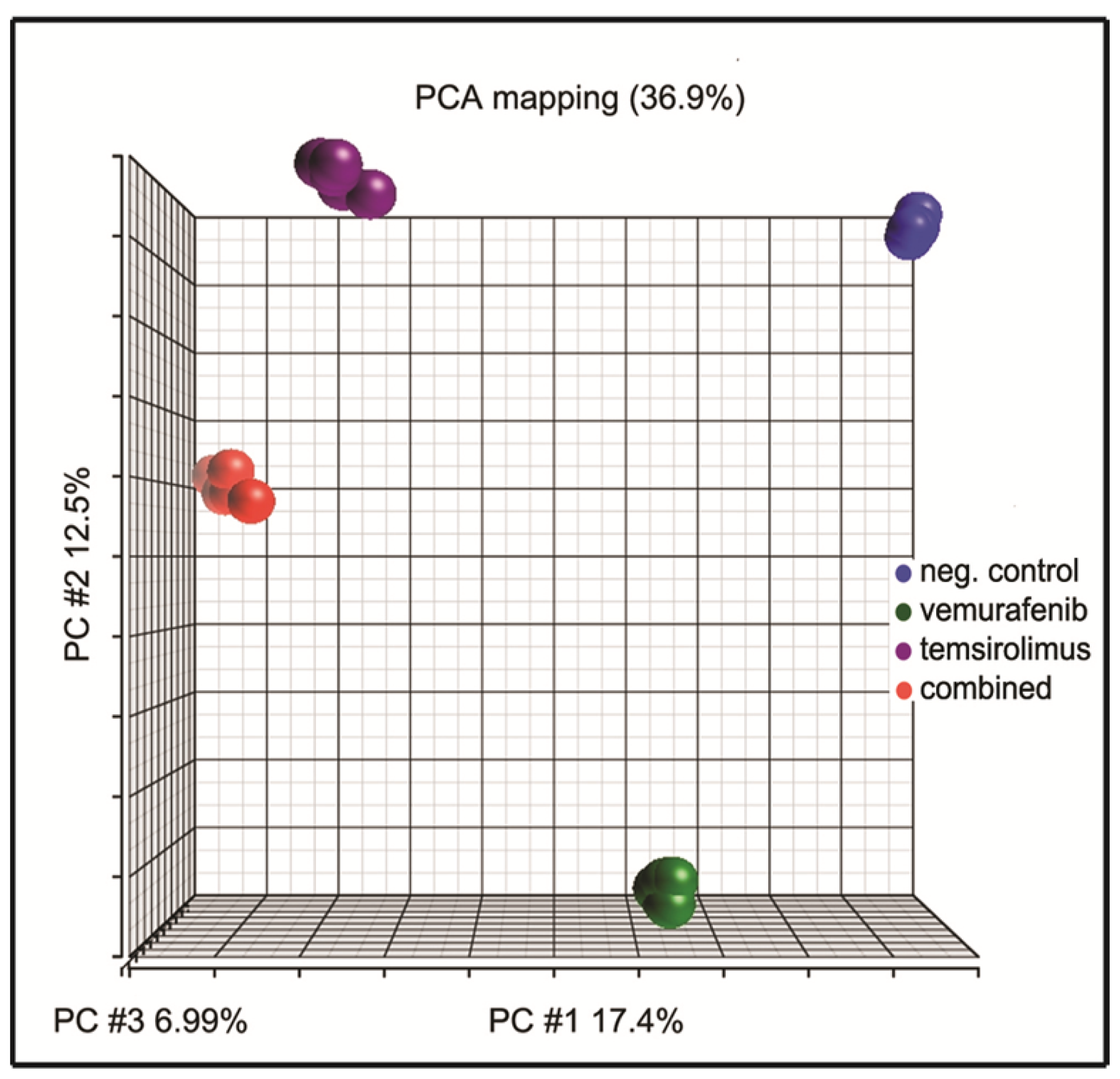
2.6. Principal Component Analysis of Gene Expression Arrays Shows Alterations in Gene Clustering after Treatment
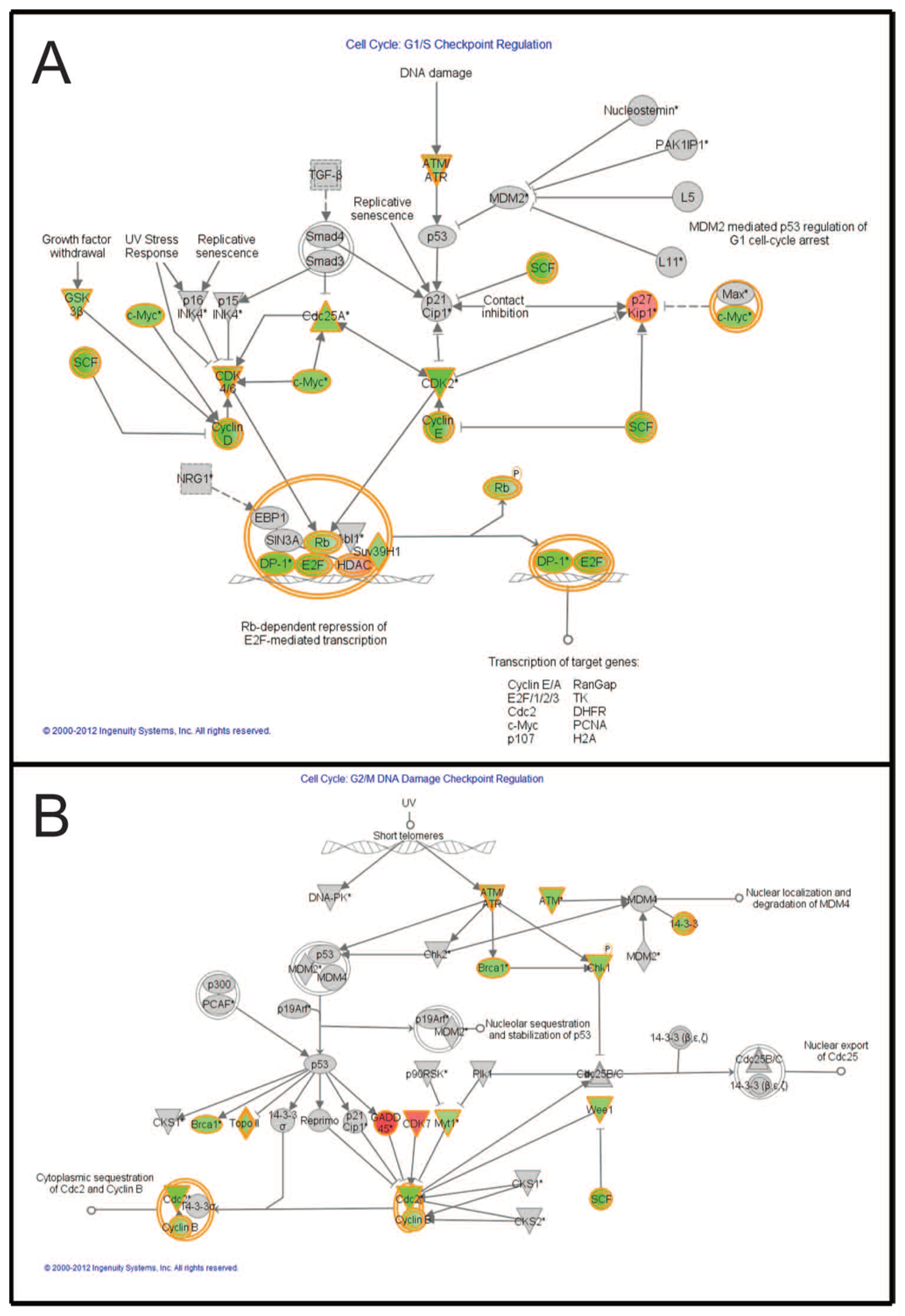
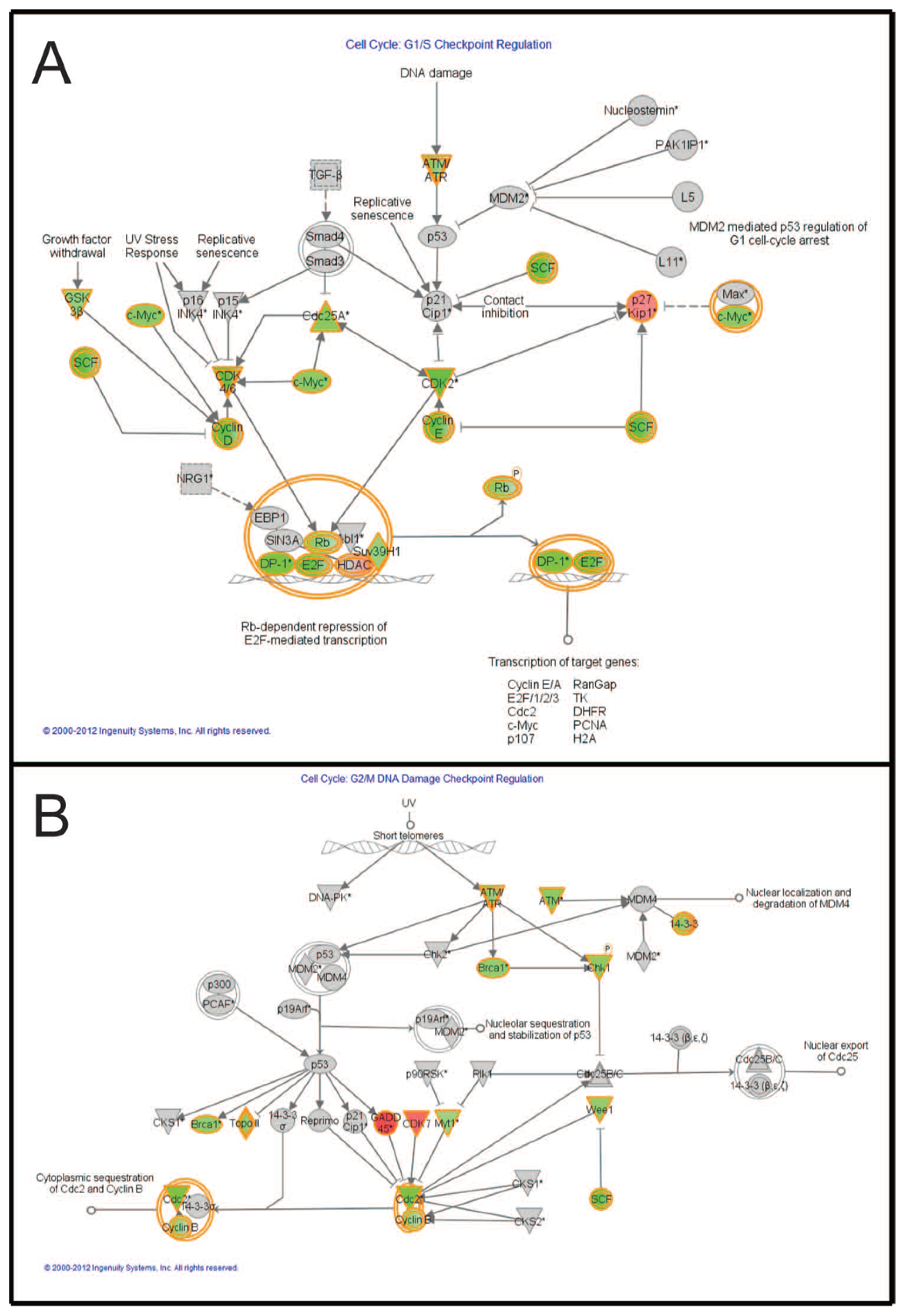
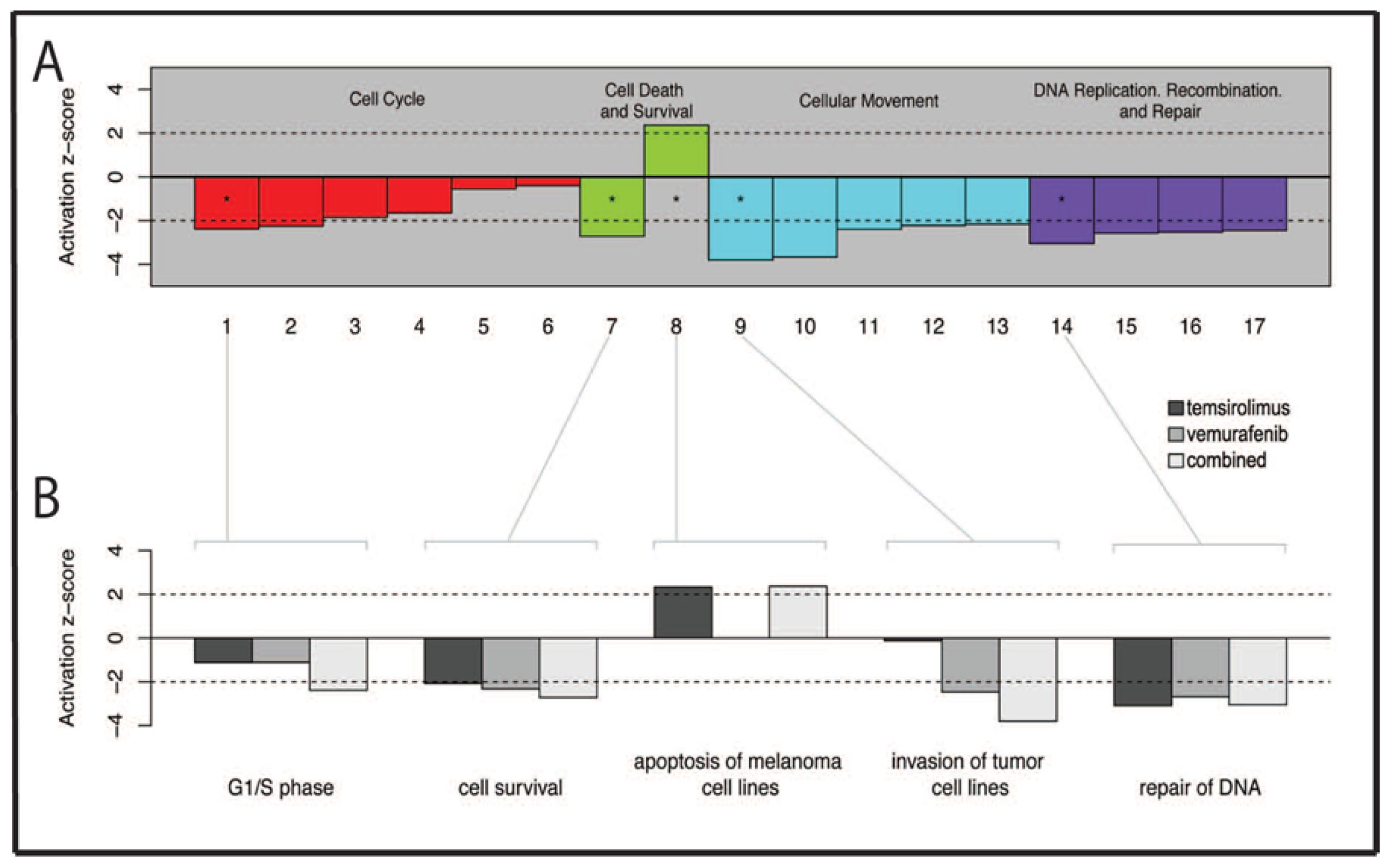
2.7. Pathways Affecting Cell Cycle Checkpoint Regulation Are Affected by Combined Treatment
2.8. Several Genes Related to Cell Survival and Invasion Are Affected by Treatment
2.9. Discussion
3. Materials and Methods
3.1. Cell Line and Cell Culture
3.2. BRAF Gene Mutation Status
3.3. PTEN Deletion Status
3.4. In Vitro Drug Treatment
3.5. Resazurin Cell Proliferation Assay
3.6. Cell Migration Assay
3.7. Spheroid Growth and Cytotoxicity
3.8. Western Blot Analysis
3.9. RNA Extraction after Drug Treatment of Monolayer Cultures
3.10. Illumina HT-12 Array; RNA Preparation, Labeling and Microarray Hybridization
3.11. Microarray Analysis
3.12. Statistics
4. Conclusions
Acknowledgments
Conflicts of Interest
- Author ContributionsI.D. performed monolayer cell growth experiments, in vitro drug treatments, western blots, spheroid growth assays, RNA extraction, data analysis, made figures and prepared the manuscript. S.H. did monolayer cell growth experiments, western blots, data analysis, made figures and contributed to the manuscript. D.S. performed DNA copy number analysis, made figures and contributed to the manuscript. J.K.V. performed microarray experiments, principal component analysis, functional analysis, made figures and contributed to the manuscript. E.S. did the wound healing assays, data analysis, contributed to making figures and movies and contributed to the manuscript. H.A.D. performed spheroid growth assays, including confocal imaging, data analysis, preparation of figures and contributed to the manuscript. K.O.S. transfected the cell lines with reporter genes, contributed to the resazurin assay and making figures and contributed to the manuscript. R.B. contributed to the design of this study, the data analysis and the writing of the manuscript. F.T. was responsible for the design of this study, contributed to all the experiments, including data analysis, made figures and finalized the manuscript.
References
- Brose, M.S.; Volpe, P.; Feldman, M.; Kumar, M.; Rishi, I.; Gerrero, R.; Einhorn, E.; Herlyn, M.; Minna, J.; Nicholson, A.; et al. BRAF and RAS mutations in human lung cancer and melanoma. Cancer Res 2002, 62, 6997–7000. [Google Scholar]
- Margolin, K.; Longmate, J.; Baratta, T.; Synold, T.; Christensen, S.; Weber, J.; Gajewski, T.; Quirt, I.; Doroshow, J.H. CCI-779 in metastatic melanoma: A phase II trial of the California Cancer Consortium. Cancer 2005, 104, 1045–1048. [Google Scholar]
- Fonkem, E.; Uhlmann, E.J.; Floyd, S.R.; Mahadevan, A.; Kasper, E.; Eton, O.; Wong, E.T. Melanoma brain metastasis: Overview of current management and emerging targeted therapies. Expert Rev. Neurother 2012, 12, 1207–1215. [Google Scholar]
- Thorsen, F.; Fite, B.; Mahakian, L.M.; Seo, J.W.; Qin, S.; Harrison, V.; Johnson, S.; Ingham, E.; Caskey, C.; Sundstrøm, T.; et al. Multimodal imaging enables early detection and characterization of changes in tumor permeability of brain metastases. J. Control. Release 2013, 172, 812–822. [Google Scholar]
- Tosoni, A.; Ermani, M.; Brandes, A.A. The pathogenesis and treatment of brain metastases: A comprehensive review. Critical Rev. Oncol. Hematol 2004, 52, 199–215. [Google Scholar]
- Carbonell, W.S.; Ansorge, O.; Sibson, N.; Muschel, R. The vascular basement membrane as “soil” in brain metastasis. PLoS One 2009, 4, e5857. [Google Scholar]
- Yajima, I.; Kumasaka, M.Y.; Thang, N.D.; Goto, Y.; Takeda, K.; Yamanoshita, O.; Iida, M.; Ohgami, N.; Tamura, H.; Kawamoto, Y.; et al. RAS/RAF/MEK/ERK and PI3K/PTEN/AKT signaling in malignant melanoma progression and therapy. Dermatol. Res. Pract 2012. [Google Scholar] [CrossRef]
- Nathanson, K.L. Using genetics and genomics strategies to personalize therapy for cancer: Focus on melanoma. Biochem. Pharmacol 2010, 80, 755–761. [Google Scholar]
- Pacheco, I.; Buzea, C.; Tron, V. Towards new therapeutic approaches for malignant melanoma. Expert Rev. Mol. Med 2011, 13. [Google Scholar] [CrossRef]
- Davies, H.; Bignell, G.R.; Cox, C.; Stephens, P.; Edkins, S.; Clegg, S.; Teague, J.; Woffendin, H.; Garnett, M.J.; Bottomley, W.; et al. Mutations of the BRAF gene in human cancer. Nature 2002, 417, 949–954. [Google Scholar]
- Inamdar, G.S.; Madhunapantula, SV.; Robertson, G.P. Targeting the MAPK pathway in melanoma: Why some approaches succeed and other fail. Biochem. Pharmacol 2010, 80, 624–637. [Google Scholar]
- Chapman, P.B.; Hauschild, A.; Robert, C.; Haanen, J.B.; Ascierto, P.; Larkin, J.; Dummer, R.; Garbe, C.; Testori, A.; Maio, M.; et al. Improved survival with Vemurafenib in melanoma with BRAF V600E mutation. N. Engl. J. Med 2011, 364, 2507–2516. [Google Scholar]
- Davies, M.A.; Stemke-Hale, K.; Lin, E.; Tellez, C.; Deng, W.; Gopal, Y.N.; Woodman, S.E.; Calderone, T.C.; Ju, Z.; Lazar, A.J.; et al. Integrated molecular and clinical analysis of AKT activation in metastatic melanoma. Clin. Cancer Res 2009, 15, 7538–7546. [Google Scholar]
- Rubinstein, J.C.; Sznol, M.; Pavlick, A.C.; Ariyan, S.; Cheng, E.; Bacchiocchi, A.; Kluger, H.M.; Narayan, D.; Halaban, R. Incidence of the V600K mutation among melanoma patients with BRAF mutations, and potential therapeutic response to the specific BRAF inhibitor PLX4032. J. Transl. Med 2010, 8, 67:1–67:3. [Google Scholar]
- Dhomen, N.; Marais, R. BRAF signaling and targeted therapies in melanoma. Hematol. Oncol. Clin. N. Am 2009, 23, 529–545. [Google Scholar]
- Vultur, A.; Villanueva, J.; Herlyn, M. Targeting BRAF in Advanced Melanoma: A First Step toward Manageable Disease. Clin. Cancer Res 2011, 17, 1658–1663. [Google Scholar]
- Paraiso, K.H.T.; Xiang, Y.; Rebecca, V.W.; Abel, E.V.; Chen, Y.A.; Munko, A.C.; Wood, E.; Fedorenko, I.V.; Sondak, V.K.; Anderson, A.R.A.; et al. PTEN loss confers BRAF inhibitor resistance to melanoma cells through the suppression of BIM expression. Cancer Res 2011, 71, 2750–2760. [Google Scholar]
- Tsao, H.; Goel, V.; Wu, H.; Yang, G.; Haluska, F.G. Genetic interaction between NRAS and BRAF mutations and PTEN/MMAC1 inactivation in melanoma. J. Invest. Dermatol 2004, 122, 337–341. [Google Scholar]
- Tsao, H.; Zhang, X.; Fowlkes, K.; Haluska, F.G. Relative reciprocity of NRAS and PTEN/MMAC1 alterations in cutaneous melanoma cell lines. Cancer Res 2000, 60, 1800–1804. [Google Scholar]
- Villanueva, J.; Vultur, A.; Lee, J.T.; Somasundaram, R.; Fukunaga-Kalabis, M.; Cipolla, A.K.; Wubbenhorst, B.; Xu, X.; Gimotty, P.A.; Kee, D.; et al. Acquired resistance to BRAF inhibitors mediated by a RAF kinase switch in melanoma can be overcome by cotargeting MEK and IGF-1R/PI3K. Cancer Cell 2010, 18, 683–695. [Google Scholar]
- Mittapalli, R.K.; Vaidhyanathan, S.; Dudek, A.Z.; Elmquist, W.F. Mechanisms limiting distribution of the threonine-protein kinase B-RaF(V600E) inhibitor dabrafenib to the brain: Implications for the treatment of melanoma brain metastases. J. Pharmacol. Exp. Ther 2013, 344, 655–664. [Google Scholar]
- Livingstone, E.; Zimmer, L.; Piel, S.; Schadendorf, D. PLX4032: Does it keep its promise for metastatic melanoma treatment? Expert Opin. Investig. Drugs 2010, 19, 1439–1449. [Google Scholar]
- Shao, Y.; Aplin, A.E. BH3-only protein silencing contributes to acquired resistance to PLX4720 in human melanoma. Cell Death Differ 2012, 19, 2029–2039. [Google Scholar]
- Balakan, O.; Süner, A.; Yiğiter, R.; Balakan, T.; Sirikçi, A.; Sevinç, A. Long-term survival in metastatic malignant melanoma: Ipilimumab followed by vemurafenib in a patient with brain metastasis. Internal Med 2012, 51, 2819–2823. [Google Scholar]
- Dummer, R.; Rinderknecht, J.; Goldinger, S.M.; Wagner, I.; Mitchell, L.; Veronese, M.L.; Nick, S.; Hilfiker, P.; Gobbi, S. An open-label pilot study of vemurafenib in previously treated metastatic melanoma patients with brain metastases. Proceedings of 2011 ASCO Annual Meeting, Chicago, IL, USA, 3–7 June 2011.
- Forschner, A.; Niessner, H.; Bauer, J.; Bender, B.; Garbe, C.; Meier, F. Successful treatment with vemurafenib in BRAF V600K-positive cerebral melanoma metastasis. JAMA Dermatol 2013, 149, 642–644. [Google Scholar]
- Kolar, G.R.; Miller-Thomas, M.M.; Schmidt, R.E.; Simpson, J.R.; Rich, K.M.; Linette, G.P. Neoadjuvant treatment of a solitary melanoma brain metastasis with vemurafenib. J. Clin. Oncol 2013, 31. [Google Scholar] [CrossRef]
- Dummer, R.; Goldinger, S.M.; Turtschi, C.P.; Eggmann, N.B.; Michielin, O.; Mitchell, L.; Veronese, L.; Hilfiker, P.R.; Felderer, L.; Rinderknecht, J.D. Vemurafenib in patients with BRAF (V600) mutation-positive melanoma with symptomatic brain metastases: Final results of an open-label pilot study. Eur. J. Cancer 2014, 50, 611–621. [Google Scholar]
- Kuhn, J.G.; Chang, S.M.; Wen, P.Y.; Cloughesy, T.F.; Greenberg, H.; Schiff, D.; Conrad, C.; Fink, K.L.; Robins, H.I.; MEHTA, M.; et al. Pharmacokinetic and Tumor Distribution Characteristics of Temsirolimus in Patients with Recurrent Malignant Glioma. Clin. Cancer Res 2007, 13, 7401–7406. [Google Scholar]
- Davies, M.A.; Fox, P.S.; Papadopoulos, N.E.; Bedikian, A.Y.; Hwu, W.J.; Lazar, A.J.; Prieto, V.G.; Culotta, K.S.; Madden, T.L.; Xu, Q.; et al. Phase I Study of the Combination of Sorafenib and Temsirolimus in Patients with Metastatic Melanoma. Clin. Cancer Res 2012, 18, 1120–1128. [Google Scholar]
- Sundstrom, T.; Daphu, I.; Wendelbo, I.; Hodneland, E.; Lundervold, A.; Immervoll, H.; Skaftnesmo, K.O.; Babic, M.; Jendelova, P.; Syková, E.; et al. Automated tracking of nanoparticle-labeled melanoma cells improves the predictive power of a brain metastasis model. Cancer Res 2013, 73, 2445–2456. [Google Scholar]
- Hwang, S. Comparison and evaluation of pathway-level aggregation methods of gene expression data. BMC Genomics 2012, 13. [Google Scholar] [CrossRef]
- Ingenuity. Available online: http://www.ingenuity.com accessed on 1 March 2014.
- Bollag, G.; Hirth, P.; Tsai, J.; Zhang, J.; Ibrahim, P.N.; Cho, H.; Spevak, W.; Zhang, C.; Zhang, Y.; Habets, G.; et al. Clinical efficacy of a RAF inhibitor needs broad target blockade in BRAF-mutant melanoma. Nature 2010, 467, 596–599. [Google Scholar]
- Flaherty, K.T.; Puzanov, I.; Kim, K.B.; Ribas, A.; McArthur, G.A.; Sosman, J.A.; O’Dwyer, P.J.; Lee, R.J.; Grippo, J.F.; Nolop, K.; et al. Inhibition of mutated, activated BRAF in metastatic melanoma. N. Engl. J. Med 2010, 363, 809–819. [Google Scholar]
- Tsai, J.; Lee, J.T.; Wang, W.; Zhang, J.; Cho, H.; Mamo, S.; Bremer, R.; Gillette, S.; Kong, J.; Haass, N.K.; et al. Discovery of a selective inhibitor of oncogenic B-Raf kinase with potent antimelanoma activity. Proc. Natl. Acad. Sci. USA 2008, 105, 3041–3046. [Google Scholar]
- Abraham, R.T.; Eng, C.H. Mammalian target of rapamycin as a therapeutic target in oncology. Expert Opin. Ther. Targets 2008, 12, 209–222. [Google Scholar]
- LoPiccolo, J.; Blumenthal, G.M.; Bernstein, W.B.; Dennis, P.A. Targeting the PI3K/Akt/mTOR pathway: Effective combinations and clinical considerations. Drug Resist. Updat 2008, 11, 32–50. [Google Scholar]
- Lázár, V.; Ecsedi, S.; Szöllosi, A.G.; Tóth, R.; Vízkeleti, L.; Rákosy, Z.; Bégány, A.; Adány, R.; Balázs, M. Characterization of candidate gene copy number alterations in the 11q13 region along with BRAF and NRAS mutations in human melanoma. Mod. Pathol 2009, 22, 1367–1378. [Google Scholar]
- Yuan, T.L.; Cantley, L.C. PI3K pathway alterations in cancer: Variations on a theme. Oncogene 2008, 27, 5497–5510. [Google Scholar]
- Li, L.; Price, J.E.; Fan, D.; Zhang, R.D.; Bucana, C.D.; Fidler, I.J. Correlation of growth capacity of human tumor cells in hard agarose with their in vivo proliferative capacity at specific metastatic sites. J. Natl. Cancer Inst 1989, 81, 1406–1412. [Google Scholar]
- Fecher, L.A.; Cummings, S.D.; Keefe, M.J.; Alani, R.M. Toward a molecular classification of melanoma. J. Clin. Oncol 2007, 25, 1606–1620. [Google Scholar]
- Nambiar, S.; Mirmohammadsadegh, A.; Doroudi, R.; Gustrau, A.; Marini, A.; Roeder, G.; Ruzicka, T.; Hengge, U.R. Signaling networks in cutaneous melanoma metastasis identified by complementary DNA microarrays. Arch. Dermatol 2005, 141, 165–173. [Google Scholar]
- Wang, J.; Daphu, I.; Pedersen, P.H.; Miletic, H.; Hovland, R.; Mørk, S.; Bjerkvig, R.; Tiron, C.; McCormack, E.; Micklem, D.; et al. A novel brain metastases model developed in immunodeficient rats closely mimics the growth of metastatic brain tumours in patients. Neuropathol. Appl. Neurobiol 2011, 37, 189–205. [Google Scholar]
- Leibniz-Institut DSMZ-DEUTSCHE Sammlung von Mikroorganisemen und Zellkulturen GmbH. Available online: http://www.dsmz.de accessed on 13 May 2014.
- O’Brien, J.; Wilson, I.; Orton, T.; Pognan, F. Investigation of the Alamar Blue (resazurin) fluorescent dye for the assessment of mammalian cell cytotoxicity. Eur. J. Biochem 2000, 267, 5421–5426. [Google Scholar]
- Jin, J.-L.; Gong, J.; Yin, T.-J.; Lu, Y.-J.; Xia, J.-J.; Xie, Y.-Y.; Di, Y.; He, L.; Guo, J.-L.; Sun, J.; et al. PTD4-apoptin protein and dacarbazine show a synergistic antitumor effect on B16-F1 melanoma in vitro and in vivo. Eur. J. Pharmacol 2011, 654, 17–25. [Google Scholar]
- Ivascu, A.; Kubbies, M. Rapid generation of single-tumor spheroids for high-throughput cell function and toxicity analysis. J. Biomol. Screen 2006, 11, 922–932. [Google Scholar]
- Talasila, K.M.; Soentgerath, A.; Euskirchen, P.; Rosland, G.V.; Wang, J.; Huszthy, P.C.; Prestegarden, L.; Skaftnesmo, K.O.; Sakariassen, P.O.; Eskilsson, E.; et al. EGFR wild-type amplification and activation promote invasion and development of glioblastoma independent of angiogenesis. Acta Neuropathol 2013, 125, 683–698. [Google Scholar]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Concentration (M) | Temsirolimus (% Survival) | Vemurafenib (% Survival) | Combined (% Survival) | CDI |
|---|---|---|---|---|
| 0 | 100 ± 2.2 | 100 ± 2.2 | 100 ± 2.2 | - |
| 0.05 | 54.7 ± 1.5 | 82.8 ± 2.1 | 31.0 ± 0.8 | 0.68 |
| 5 | 53.1 ± 4.0 | 29.5 ± 1.5 | 21.0 ± 1.1 | 0.66 |
| 10 | 48.6 ± 4.2 | 24.4 ± 1.2 | 15.9 ± 1.1 | 1.34 |
| Concentration (M) | Temsirolimus (% Survival) | Vemurafenib (% Survival) | Combined (% Survival) | CDI |
|---|---|---|---|---|
| 0 | 100 ± 4.7 | 100 ± 4.7 | 100 ± 4.7 | - |
| 0.05 | 87.6 ± 2.3 | 99.9 ± 6.5 | 85.6 ± 2.5 | 0.98 |
| 5 | 79.0 ± 3.4 | 64.0 ± 2.5 | 75.6 ± 4.3 | 1.50 |
| 10 | 79.8 ± 5.9 | 42.3 ± 1.2 | 46.8 ± 3.9 | 1.39 |
| Bar Number | Downstream Functions |
|---|---|
| 1 | G1/S phase |
| 2 | S phase |
| 3 | M phase |
| 4 | G1 phase |
| 5 | G2/M phase |
| 6 | G2 phase |
| 7 | Cell survival |
| 8 | Apoptosis of melanoma cell lines |
| 9 | Invasion of tumor cell lines |
| 10 | Cell movement of tumor cell lines |
| 11 | Chemotaxis of PBMCs |
| 12 | Cell movement of carcinoma cell lines |
| 13 | Cytokinesis of tumor cell lines |
| 14 | Repair of DNA |
| 15 | Checkpoint control |
| 16 | Alignment of chromosomes |
| 17 | Chromosomal congression of chromosomes |
© 2014 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Daphu, I.; Horn, S.; Stieber, D.; Varughese, J.K.; Spriet, E.; Dale, H.A.; Skaftnesmo, K.O.; Bjerkvig, R.; Thorsen, F. In Vitro Treatment of Melanoma Brain Metastasis by Simultaneously Targeting the MAPK and PI3K Signaling Pathways. Int. J. Mol. Sci. 2014, 15, 8773-8794. https://doi.org/10.3390/ijms15058773
Daphu I, Horn S, Stieber D, Varughese JK, Spriet E, Dale HA, Skaftnesmo KO, Bjerkvig R, Thorsen F. In Vitro Treatment of Melanoma Brain Metastasis by Simultaneously Targeting the MAPK and PI3K Signaling Pathways. International Journal of Molecular Sciences. 2014; 15(5):8773-8794. https://doi.org/10.3390/ijms15058773
Chicago/Turabian StyleDaphu, Inderjit, Sindre Horn, Daniel Stieber, Jobin K. Varughese, Endy Spriet, Hege Avsnes Dale, Kai Ove Skaftnesmo, Rolf Bjerkvig, and Frits Thorsen. 2014. "In Vitro Treatment of Melanoma Brain Metastasis by Simultaneously Targeting the MAPK and PI3K Signaling Pathways" International Journal of Molecular Sciences 15, no. 5: 8773-8794. https://doi.org/10.3390/ijms15058773