Role of Mitochondria in Nonalcoholic Fatty Liver Disease



Abstract
:1. Introduction
2. Mechanisms Leading to NAFLD
2.1. Source of Fatty Acids (FAs) in the Liver
2.1.1. Diet
2.1.2. Adipose Tissue Lipolysis
2.1.3. De Novo Lipogenesis
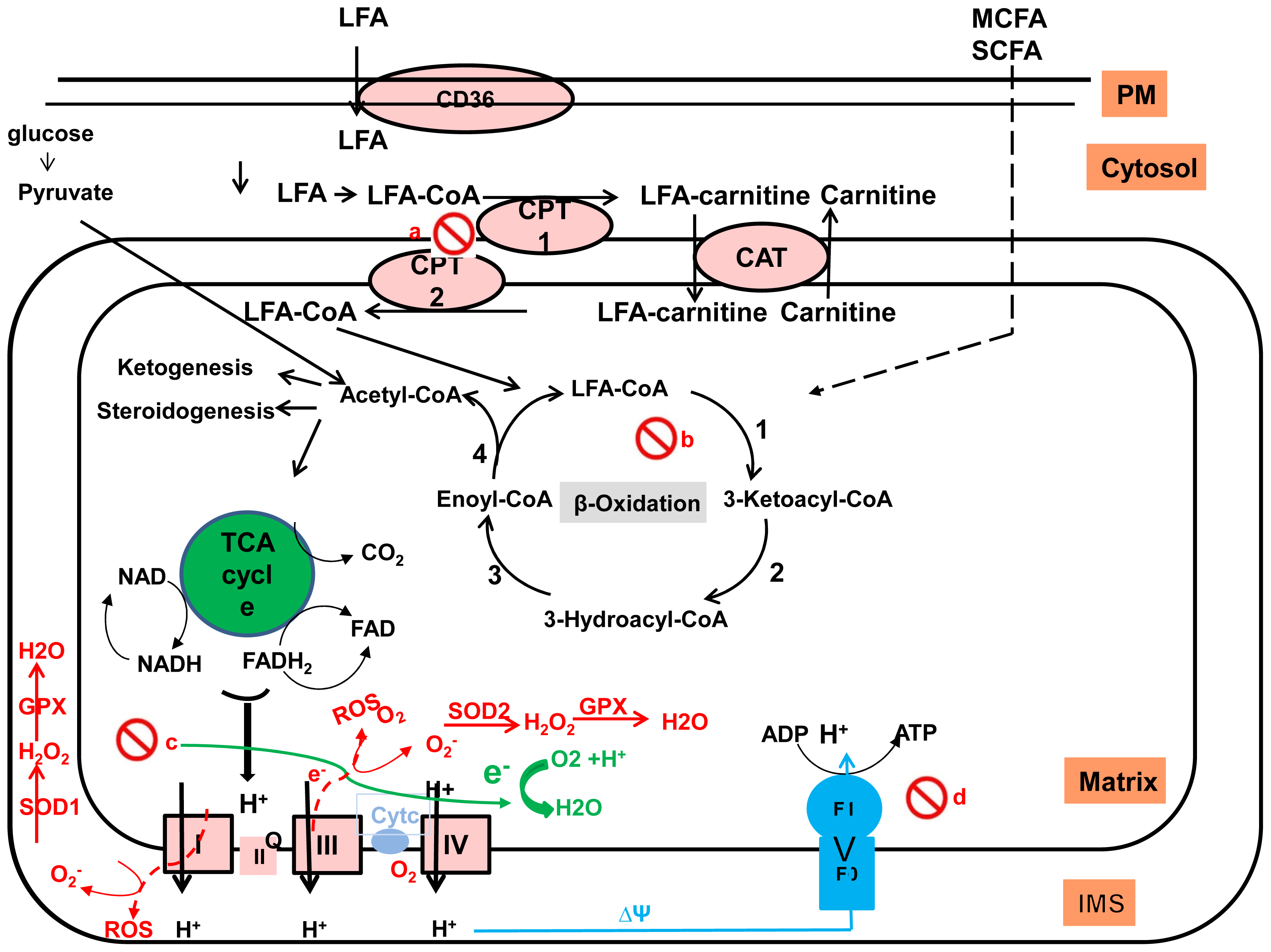
2.2. Fatty Acid Uptake by the Liver
2.3. Disposal of Hepatic FFAs
2.3.1. Secretion of Hepatic TG
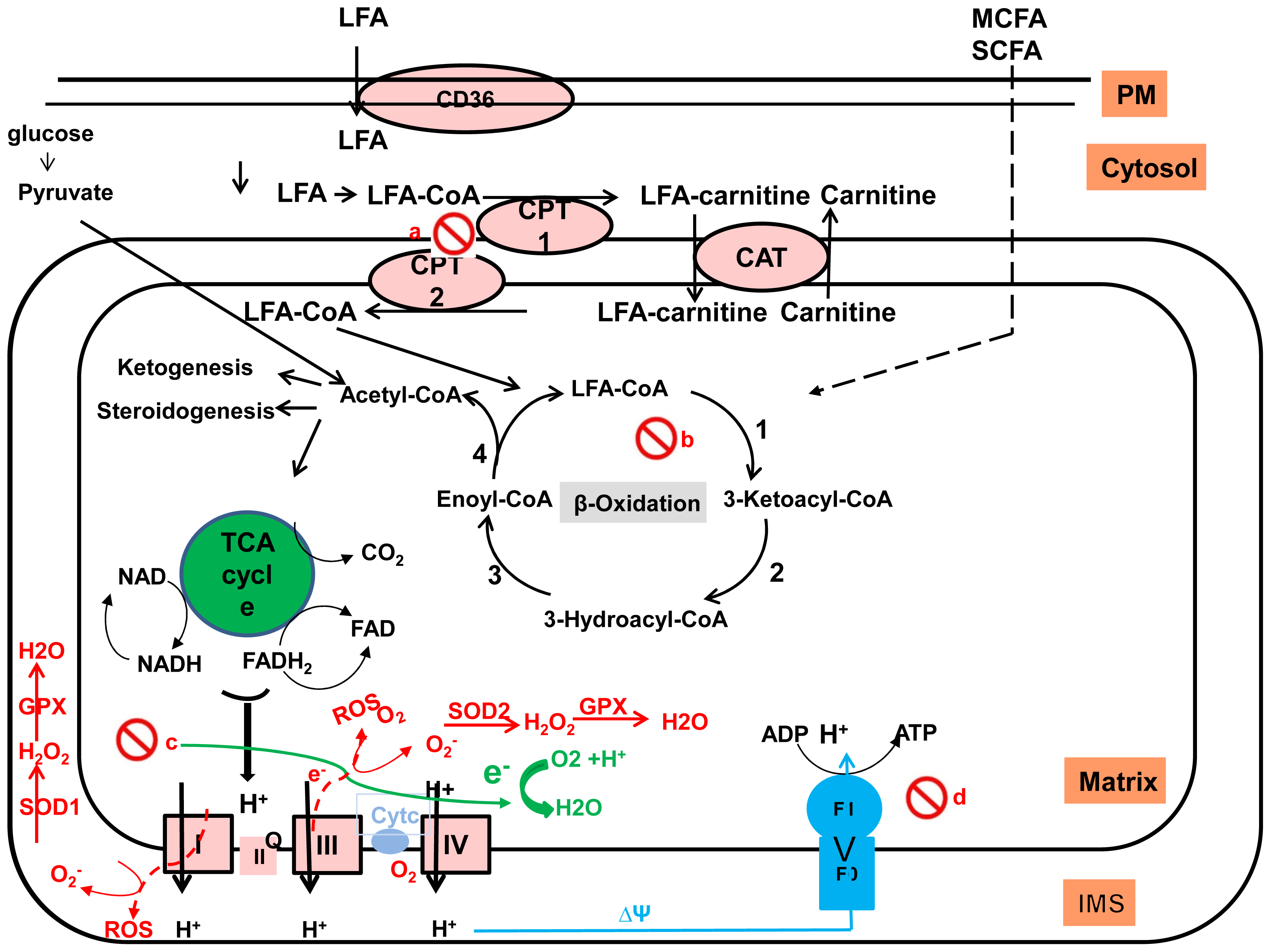
2.3.2. Fatty Acid Oxidation
2.4. Alterations in Lipid Metabolism Leading to NAFLD
3. Role of Mitochondria in NAFLD
3.1. Mitochondrial Structure
3.2. Mitochondrial Function
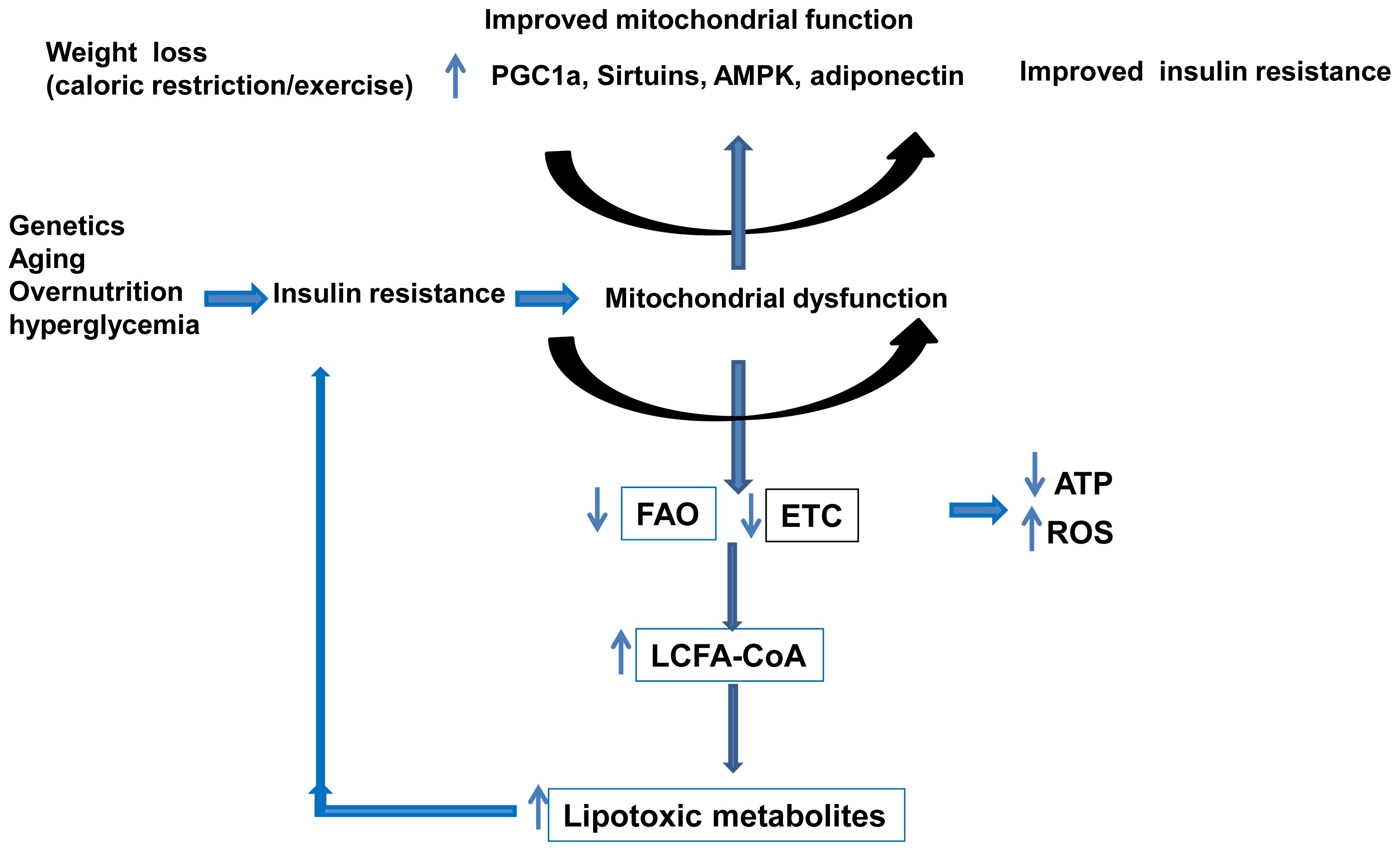
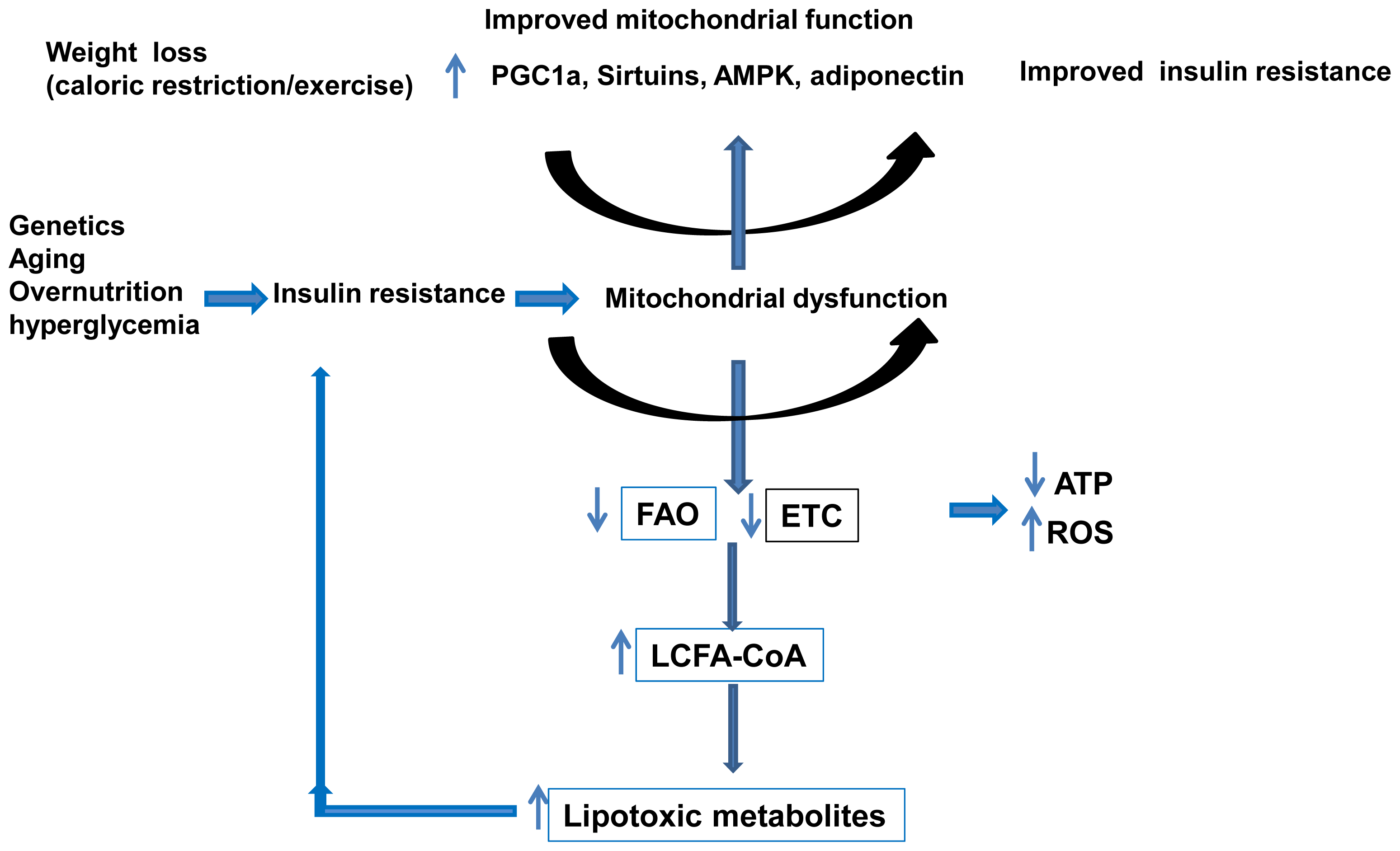
3.3. Mitochondria and NAFLD
4. Mechanisms Regulating Mitochondrial Function
4.1. Role of the Peroxisome Proliferator-Activated Receptor-γ Coactivator 1 (PGC-1)
4.2. Emerging Role of Sirtuins
4.2.1. SIRT1
4.2.2. SIRT3
4.3. Molecular Pathways Regulating Mitochondrial Content
5. Prevention and Treatment of NAFLD
6. Conclusions
Acknowledgments
Conflicts of Interest
- Author ContributionsFatiha Nassir reviewed the literature and wrote the initial draft of the manuscript. Jamal Ibdah contributed to the writing and provided the overall design and editing of the manuscript.
References
- Milic, S.; Stimac, D. Nonalcoholic fatty liver disease/steatohepatitis: Epidemiology, pathogenesis, clinical presentation and treatment. Dig. Dis. (Basel. Switz.) 2012, 30, 158–162. [Google Scholar]
- Nassir, F.; Adewole, O.L.; Brunt, E.M.; Abumrad, N.A. CD36 deletion reduces VLDL secretion, modulates liver prostaglandins, and exacerbates hepatic steatosis in ob/ob mice. J. Lipid Res 2013, 54, 2988–2997. [Google Scholar]
- Cohen, J.C.; Horton, J.D.; Hobbs, H.H. Human fatty liver disease: Old questions and new insights. Science 2011, 332, 1519–1523. [Google Scholar]
- Fabbrini, E.; Sullivan, S.; Klein, S. Obesity and nonalcoholic fatty liver disease: Biochemical, metabolic, and clinical implications. Hepatology (Baltim., Md.) 2010, 51, 679–689. [Google Scholar]
- Grattagliano, I.; Russmann, S.; Diogo, C.; Bonfrate, L.; Oliveira, P.J.; Wang, D.Q.; Portincasa, P. Mitochondria in chronic liver disease. Curr. Drug Targets 2011, 12, 879–893. [Google Scholar]
- Szczepaniak, L.S.; Nurenberg, P.; Leonard, D.; Browning, J.D.; Reingold, J.S.; Grundy, S.; Hobbs, H.H.; Dobbins, R.L. Magnetic resonance spectroscopy to measure hepatic triglyceride content: Prevalence of hepatic steatosis in the general population. Am. J. Physiol. Endocrinol. Metab 2005, 288, E462–E468. [Google Scholar]
- Scaglioni, F.; Ciccia, S.; Marino, M.; Bedogni, G.; Bellentani, S. ASH and NASH. Dig. Dis. (Basel, Switz.) 2011, 29, 202–210. [Google Scholar]
- Chalasani, N.; Younossi, Z.; Lavine, J.E.; Diehl, A.M.; Brunt, E.M.; Cusi, K.; Charlton, M.; Sanyal, A.J. The diagnosis and management of non-alcoholic fatty liver disease: Practice Guideline by the American Association for the Study of Liver Diseases, American College of Gastroenterology, and the American Gastroenterological Association. Hepatology (Baltim., Md.) 2012, 55, 2005–2023. [Google Scholar]
- Adams, L.A.; Lymp, J.F.; St Sauver, J.; Sanderson, S.O.; Lindor, K.D.; Feldstein, A.; Angulo, P. The natural history of nonalcoholic fatty liver disease: A population-based cohort study. Gastroenterology 2005, 129, 113–121. [Google Scholar]
- Vernon, G.; Baranova, A.; Younossi, Z.M. Systematic review: The epidemiology and natural history of non-alcoholic fatty liver disease and non-alcoholic steatohepatitis in adults. Aliment. Pharmacol. Therap 2011, 34, 274–285. [Google Scholar]
- Tetri, L.H.; Basaranoglu, M.; Brunt, E.M.; Yerian, L.M.; Neuschwander-Tetri, B.A. Severe NAFLD with hepatic necroinflammatory changes in mice fed trans fats and a high-fructose corn syrup equivalent. Am. J. Physiol. Gastrointest. Liver Physiol 2008, 295, G987–G995. [Google Scholar]
- Alisi, A.; Manco, M.; Panera, N.; Nobili, V. Association between type two diabetes and non-alcoholic fatty liver disease in youth. Ann. Hepatol 2009, 8 Suppl 1, S44–S50. [Google Scholar]
- Parikh, R.M.; Mohan, V. Changing definitions of metabolic syndrome. Indian J. Endocrinol. Metab 2012, 16, 7–12. [Google Scholar]
- Rahman, R.; Hammoud, G.M.; Almashhrawi, A.A.; Ahmed, K.T.; Ibdah, J.A. Primary hepatocellular carcinoma and metabolic syndrome: An update. World J. Gastrointest. Oncol 2013, 5, 186–194. [Google Scholar]
- Clark, J.M.; Brancati, F.L.; Diehl, A.M. Nonalcoholic fatty liver disease. Gastroenterology 2002, 122, 1649–1657. [Google Scholar]
- Machado, M.; Marques-Vidal, P.; Cortez-Pinto, H. Hepatic histology in obese patients undergoing bariatric surgery. J. Hepatol 2006, 45, 600–606. [Google Scholar]
- Loria, P.; Lonardo, A.; Anania, F. Liver and diabetes: A vicious circle. Hepatol. Res 2013, 43, 51–64. [Google Scholar]
- Takamura, T.; Misu, H.; Ota, T.; Kaneko, S. Fatty liver as a consequence and cause of insulin resistance: Lessons from type 2 diabetic liver. Endocr. J 2012, 59, 745–763. [Google Scholar]
- Younossi, Z.M.; Gramlich, T.; Matteoni, C.A.; Boparai, N.; McCullough, A.J. Nonalcoholic fatty liver disease in patients with type 2 diabetes. Clin. Gastroenterol. Hepatol 2004, 2, 262–265. [Google Scholar]
- Abdelmalek, M.; Ludwig, J.; Lindor, K.D. Two cases from the spectrum of nonalcoholic steatohepatitis. J. Clin. Gastroenterol 1995, 20, 127–130. [Google Scholar]
- Bacon, B.R.; Farahvash, M.J.; Janney, C.G.; Neuschwander-Tetri, B.A. Nonalcoholic steatohepatitis: An expanded clinical entity. Gastroenterology 1994, 107, 1103–1109. [Google Scholar]
- Teli, M.R.; James, O.F.; Burt, A.D.; Bennett, M.K.; Day, C.P. The natural history of nonalcoholic fatty liver: A follow-up study. Hepatology (Baltim., Md.) 1995, 22, 1714–1719. [Google Scholar]
- Guerrero, R.; Vega, G.L.; Grundy, S.M.; Browning, J.D. Ethnic differences in hepatic steatosis: An insulin resistance paradox? Hepatology (Baltim., Md.) 2009, 49, 791–801. [Google Scholar]
- Lewis, J.R.; Mohanty, S.R. Nonalcoholic fatty liver disease: A review and update. Dig. Dis. Sci 2010, 55, 560–578. [Google Scholar]
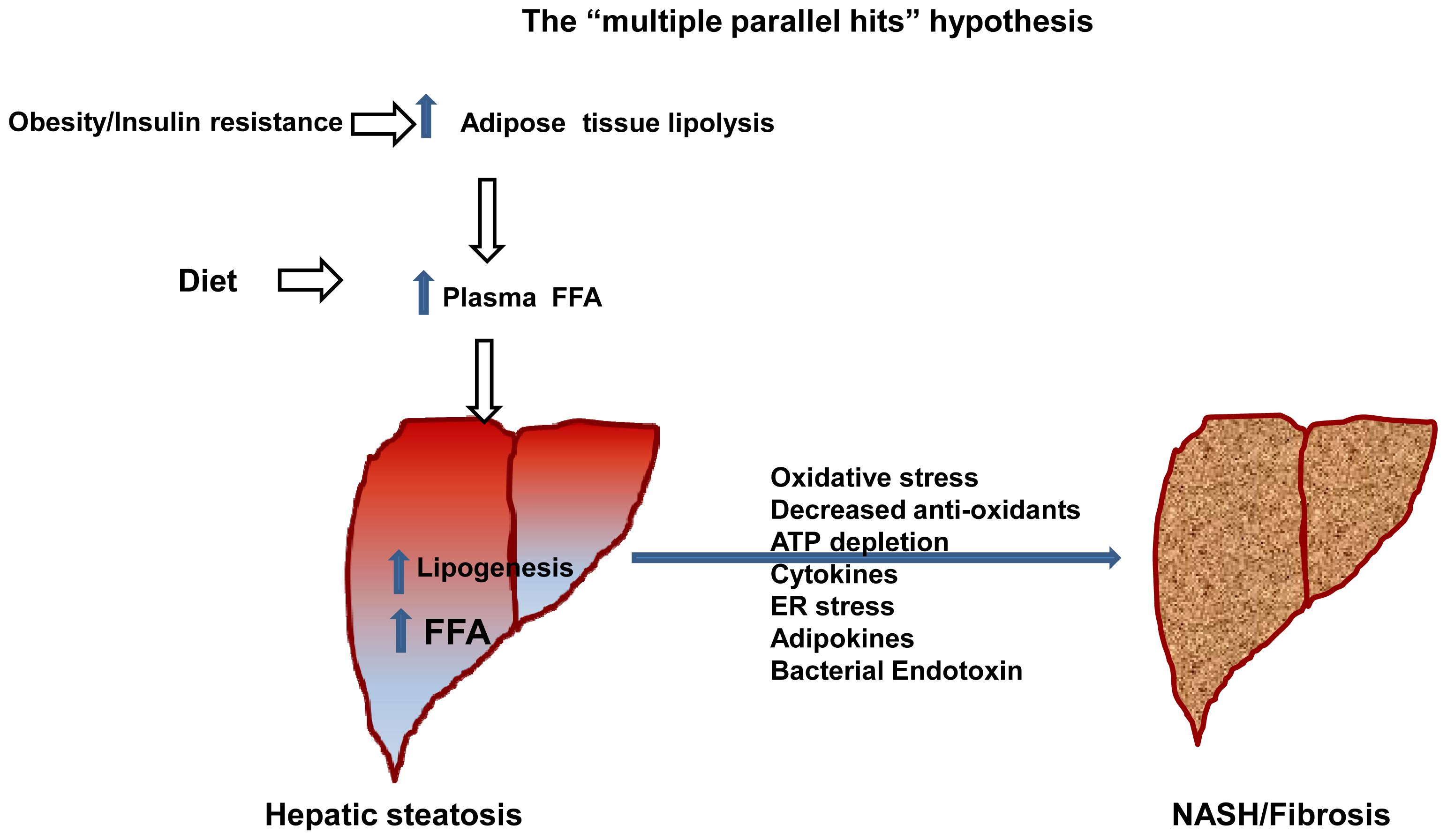
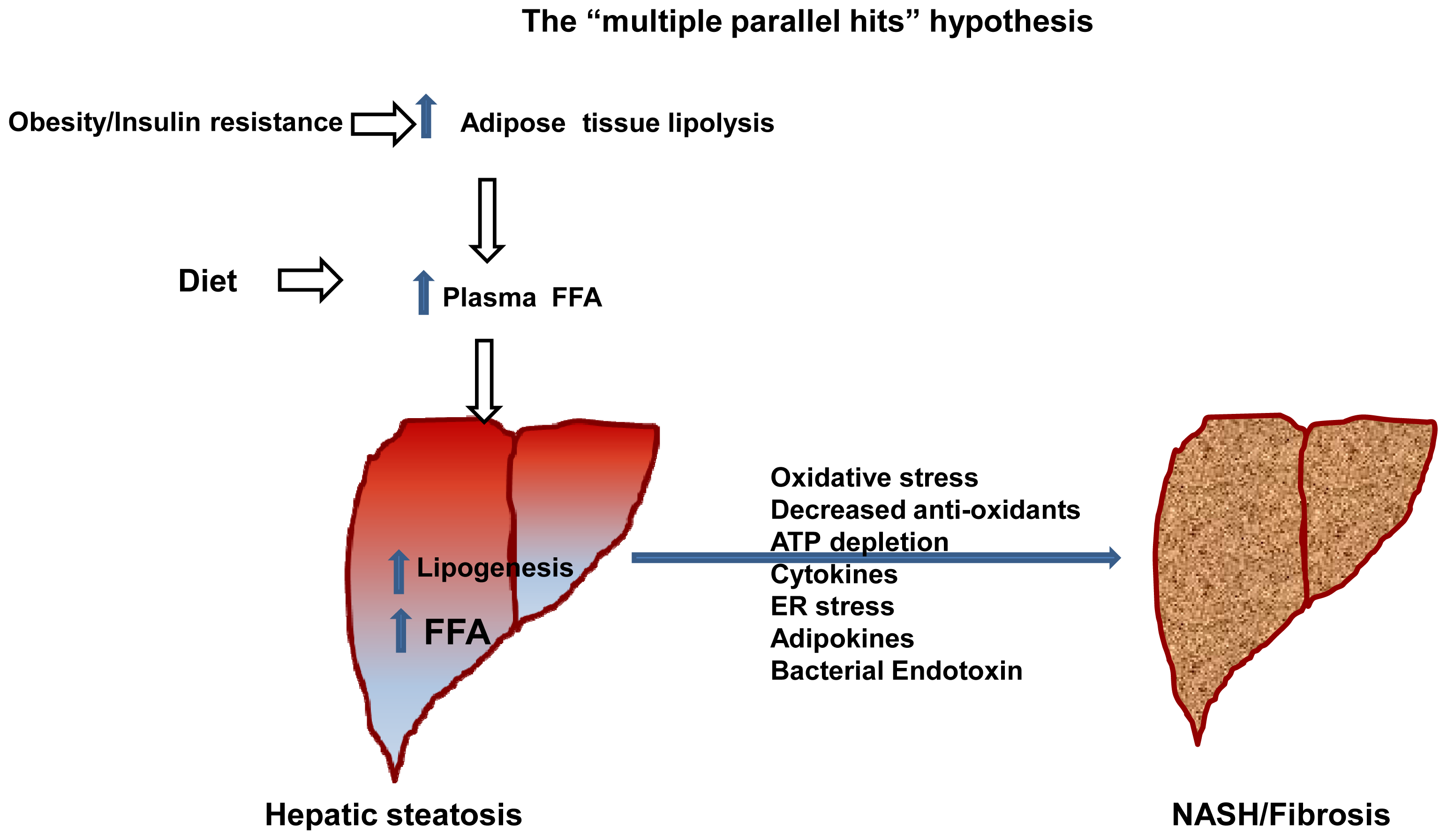
- Day, C.P.; James, O.F. Steatohepatitis: A tale of two “hits”? Gastroenterology 1998, 114, 842–845. [Google Scholar]
- Tilg, H.; Moschen, A.R. Evolution of inflammation in nonalcoholic fatty liver disease: The multiple parallel hits hypothesis. Hepatology (Baltim., Md.) 2010, 52, 1836–1846. [Google Scholar]
- Lonardo, A.; Bellentani, S.; Ratziu, V.; Loria, P. Insulin resistance in nonalcoholic steatohepatitis: Necessary but not sufficient—Death of a dogma from analysis of therapeutic studies? Expert Rev. Gastroenterol. Hepatol 2011, 5, 279–289. [Google Scholar]
- Yilmaz, Y. Review article: Is non-alcoholic fatty liver disease a spectrum, or are steatosis and non-alcoholic steatohepatitis distinct conditions? Aliment. Pharmacol. Therap 2012, 36, 815–823. [Google Scholar]
- Amaro, A.; Fabbrini, E.; Kars, M.; Yue, P.; Schechtman, K.; Schonfeld, G.; Klein, S. Dissociation between intrahepatic triglyceride content and insulin resistance in familial hypobetalipoproteinemia. Gastroenterology 2010, 139, 149–153. [Google Scholar]
- Kantartzis, K.; Machicao, F.; Machann, J.; Schick, F.; Fritsche, A.; Haring, H.U.; Stefan, N. The DGAT2 gene is a candidate for the dissociation between fatty liver and insulin resistance in humans. Clin. Sci. (Lond., Engl.) 2009, 116, 531–537. [Google Scholar]
- Kantartzis, K.; Peter, A.; Machicao, F.; Machann, J.; Wagner, S.; Konigsrainer, I.; Konigsrainer, A.; Schick, F.; Fritsche, A.; Haring, H.U.; et al. Dissociation between fatty liver and insulin resistance in humans carrying a variant of the patatin-like phospholipase 3 gene. Diabetes 2009, 58, 2616–2623. [Google Scholar]
- Stefan, N.; Staiger, H.; Haring, H.U. Dissociation between fatty liver and insulin resistance: The role of adipose triacylglycerol lipase. Diabetologia 2011, 54, 7–9. [Google Scholar]
- Brumbaugh, D.E.; Friedman, J.E. Developmental origins of nonalcoholic fatty liver disease. Pediatr. Res 2014, 75, 140–147. [Google Scholar]
- Nestel, P.J.; Havel, R.J.; Bezman, A. Sites of initial removal of chylomicron triglyceride fatty acids from the blood. J. Clin. Investig 1962, 41, 1915–1921. [Google Scholar]
- Donnelly, K.L.; Smith, C.I.; Schwarzenberg, S.J.; Jessurun, J.; Boldt, M.D.; Parks, E.J. Sources of fatty acids stored in liver and secreted via lipoproteins in patients with nonalcoholic fatty liver disease. J. Clin. Investig 2005, 115, 1343–1351. [Google Scholar]
- Lafontan, M.; Langin, D. Lipolysis and lipid mobilization in human adipose tissue. Prog. Lipid Res 2009, 48, 275–297. [Google Scholar]
- Ferre, P.; Foufelle, F. Hepatic steatosis: A role for de novo lipogenesis and the transcription factor SREBP-1c. Diabetes Obes. Metab 2010, 12 Suppl 2, 83–92. [Google Scholar]
- Horton, J.D.; Goldstein, J.L.; Brown, M.S. SREBPs: Transcriptional mediators of lipid homeostasis. Cold Spring Harb. Symp. Quant. Biol 2002, 67, 491–498. [Google Scholar]
- Kazantzis, M.; Stahl, A. Fatty acid transport proteins, implications in physiology and disease. Biochim. Biophys. Acta 2012, 1821, 852–857. [Google Scholar]
- Doege, H.; Baillie, R.A.; Ortegon, A.M.; Tsang, B.; Wu, Q.; Punreddy, S.; Hirsch, D.; Watson, N.; Gimeno, R.E.; Stahl, A. Targeted deletion of FATP5 reveals multiple functions in liver metabolism: Alterations in hepatic lipid homeostasis. Gastroenterology 2006, 130, 1245–1258. [Google Scholar]
- Doege, H.; Grimm, D.; Falcon, A.; Tsang, B.; Storm, T.A.; Xu, H.; Ortegon, A.M.; Kazantzis, M.; Kay, M.A.; Stahl, A. Silencing of hepatic fatty acid transporter protein 5 in vivo reverses diet-induced non-alcoholic fatty liver disease and improves hyperglycemia. J. Biol. Chem 2008, 283, 22186–22192. [Google Scholar]
- Falcon, A.; Doege, H.; Fluitt, A.; Tsang, B.; Watson, N.; Kay, M.A.; Stahl, A. FATP2 is a hepatic fatty acid transporter and peroxisomal very long-chain acyl-CoA synthetase. Am. J. Physiol. Endocrinol. Metab 2010, 299, E384–E393. [Google Scholar]
- Fernandez, M.A.; Albor, C.; Ingelmo-Torres, M.; Nixon, S.J.; Ferguson, C.; Kurzchalia, T.; Tebar, F.; Enrich, C.; Parton, R.G.; Pol, A. Caveolin-1 is essential for liver regeneration. Science 2006, 313, 1628–1632. [Google Scholar]
- Su, X.; Abumrad, N.A. Cellular fatty acid uptake: A pathway under construction. Trends Endocrinol. Metab 2009, 20, 72–77. [Google Scholar]
- Koonen, D.P.Y.; Jacobs, R.L.; Febbraio, M.; Young, M.E.; Soltys, C.-L.M.; Ong, H.; Vance, D.E.; Dyck, J.R.B. Increased hepatic CD36 expression contributes to dyslipidemia associated with diet-induced obesity. Diabetes 2007, 56, 2863–2871. [Google Scholar]
- Miquilena-Colina, M.E.; Lima-Cabello, E.; Sanchez-Campos, S.; Garcia-Mediavilla, M.V.; Fernandez-Bermejo, M.; Lozano-Rodriguez, T.; Vargas-Castrillon, J.; Buque, X.; Ochoa, B.; Aspichueta, P.; et al. Hepatic fatty acid translocase CD36 upregulation is associated with insulin resistance, hyperinsulinaemia and increased steatosis in non-alcoholic steatohepatitis and chronic hepatitis, C. Gut 2011, 60, 1394–1402. [Google Scholar]
- Zhou, D.; Samovski, D.; Okunade, A.L.; Stahl, P.D.; Abumrad, N.A.; Su, X. CD36 level and trafficking are determinants of lipolysis in adipocytes. FASEB J 2012, 26, 4733–4742. [Google Scholar]
- Fabbrini, E.; Magkos, F.; Mohammed, B.S.; Pietka, T.; Abumrad, N.A.; Patterson, B.W.; Okunade, A.; Klein, S. Intrahepatic fat, not visceral fat, is linked with metabolic complications of obesity. Proc. Natl. Acad. Sci. USA 2009, 106, 15430–15435. [Google Scholar]
- Stremmel, W.; Pohl, L.; Ring, A.; Herrmann, T. A new concept of cellular uptake and intracellular trafficking of long-chain fatty acids. Lipids 2001, 36, 981–989. [Google Scholar]
- Storch, J.; Corsico, B. The emerging functions and mechanisms of mammalian fatty acid-binding proteins. Annu. Rev. Nutr 2008, 28, 73–95. [Google Scholar]
- Newberry, E.P.; Xie, Y.; Kennedy, S.; Han, X.; Buhman, K.K.; Luo, J.; Gross, R.W.; Davidson, N.O. Decreased hepatic triglyceride accumulation and altered fatty acid uptake in mice with deletion of the liver fatty acid-binding protein gene. J. Biol. Chem 2003, 278, 51664–51672. [Google Scholar]
- Wolins, N.E.; Brasaemle, D.L.; Bickel, P.E. A proposed model of fat packaging by exchangeable lipid droplet proteins. FEBS Lett 2006, 580, 5484–5491. [Google Scholar]
- Schonfeld, G. Familial hypobetalipoproteinemia: A review. J. Lipid Res 2003, 44, 878–883. [Google Scholar]
- Sen, D.; Dagdelen, S.; Erbas, T. Hepatosteatosis with hypobetalipoproteinemia. J. Natl. Med. Assoc 2007, 99, 284–286. [Google Scholar]
- Ginsberg, H.N.; Fisher, E.A. The ever-expanding role of degradation in the regulation of apolipoprotein B metabolism. J. Lipid Res 2009, 50 Suppl, S162–S166. [Google Scholar]
- Tarugi, P.; Lonardo, A.; Ballarini, G.; Grisendi, A.; Pulvirenti, M.; Bagni, A.; Calandra, S. Fatty liver in heterozygous hypobetalipoproteinemia caused by a novel truncated form of apolipoprotein, B. Gastroenterology 1996, 111, 1125–1133. [Google Scholar]
- Welty, F.K. Hypobetalipoproteinemia and abetalipoproteinemia. Curr. Opin. Lipidol 2014, 25, 161–168. [Google Scholar]
- Wolfrum, C.; Stoffel, M. Coactivation of Foxa2 through Pgc-1β promotes liver fatty acid oxidation and triglyceride/VLDL secretion. Cell Metab 2006, 3, 99–110. [Google Scholar]
- Choi, S.H.; Ginsberg, H.N. Increased very low density lipoprotein (VLDL) secretion, hepatic steatosis, and insulin resistance. Trends Endocrinol. Metab 2011, 22, 353–363. [Google Scholar]
- Van der Poorten, D.; Samer, C.F.; Ramezani-Moghadam, M.; Coulter, S.; Kacevska, M.; Schrijnders, D.; Wu, L.E.; McLeod, D.; Bugianesi, E.; Komuta, M.; et al. Hepatic fat loss in advanced nonalcoholic steatohepatitis: Are alterations in serum adiponectin the cause? Hepatology (Baltim., Md.) 2013, 57, 2180–2188. [Google Scholar]
- Begriche, K.; Massart, J.; Robin, M.A.; Bonnet, F.; Fromenty, B. Mitochondrial adaptations and dysfunctions in nonalcoholic fatty liver disease. Hepatology (Baltim., Md.) 2013, 58, 1497–1507. [Google Scholar]
- Eaton, S.; Bartlett, K.; Pourfarzam, M. Mammalian mitochondrial β-oxidation. Biochem. J 1996, 320, 345–357. [Google Scholar]
- McGarry, J.D.; Foster, D.W. Regulation of hepatic fatty acid oxidation and ketone body production. Annu. Rev. Biochem 1980, 49, 395–420. [Google Scholar]
- Sidossis, L.S.; Stuart, C.A.; Shulman, G.I.; Lopaschuk, G.D.; Wolfe, R.R. Glucose plus insulin regulate fat oxidation by controlling the rate of fatty acid entry into the mitochondria. J. Clin. Investig 1996, 98, 2244–2250. [Google Scholar]
- Longuet, C.; Sinclair, E.M.; Maida, A.; Baggio, L.L.; Maziarz, M.; Charron, M.J.; Drucker, D.J. The glucagon receptor is required for the adaptive metabolic response to fasting. Cell Metab 2008, 8, 359–371. [Google Scholar]
- Mandard, S.; Muller, M.; Kersten, S. Peroxisome proliferator-activated receptor α target genes. Cell. Mol. Life Sci 2004, 61, 393–416. [Google Scholar]
- Potthoff, M.J.; Inagaki, T.; Satapati, S.; Ding, X.; He, T.; Goetz, R.; Mohammadi, M.; Finck, B.N.; Mangelsdorf, D.J.; Kliewer, S.A.; et al. FGF21 induces PGC-1α and regulates carbohydrate and fatty acid metabolism during the adaptive starvation response. Proc. Natl. Acad. Sci. USA 2009, 106, 10853–10858. [Google Scholar]
- Pellicoro, A.; Ramachandran, P.; Iredale, J.P.; Fallowfield, J.A. Liver fibrosis and repair: Immune regulation of wound healing in a solid organ. Nat. Rev. Immunol 2014, 14, 181–194. [Google Scholar]
- Wang, K. Molecular mechanisms of hepatic apoptosis. Cell Death Dis 2014, 5, e996. [Google Scholar]
- Malaguarnera, M.; di Rosa, M.; Nicoletti, F.; Malaguarnera, L. Molecular mechanisms involved in NAFLD progression. J. Mol. Med. (Berl., Ger.) 2009, 87, 679–695. [Google Scholar]
- Musso, G.; Gambino, R.; Cassader, M. Obesity, diabetes, and gut microbiota: The hygiene hypothesis expanded? Diabetes Care 2010, 33, 2277–2284. [Google Scholar]
- Calvo, S.E.; Mootha, V.K. The mitochondrial proteome and human disease. Annu. Rev. Genomics Hum. Genet 2010, 11, 25–44. [Google Scholar]
- Pessayre, D.; Mansouri, A.; Fromenty, B. Nonalcoholic steatosis and steatohepatitis. V. Mitochondrial dysfunction in steatohepatitis. Am. J. Physiol. Gastrointest. Liver Physiol 2002, 282, G193–G199. [Google Scholar]
- Boveris, A.; Chance, B. The mitochondrial generation of hydrogen peroxide. General properties and effect of hyperbaric oxygen. Biochem. J 1973, 134, 707–716. [Google Scholar]
- Begriche, K.; Igoudjil, A.; Pessayre, D.; Fromenty, B. Mitochondrial dysfunction in NASH: Causes, consequences and possible means to prevent it. Mitochondrion 2006, 6, 1–28. [Google Scholar]
- Mansouri, A.; Gaou, I.; de Kerguenec, C.; Amsellem, S.; Haouzi, D.; Berson, A.; Moreau, A.; Feldmann, G.; Letteron, P.; Pessayre, D.; et al. An alcoholic binge causes massive degradation of hepatic mitochondrial DNA in mice. Gastroenterology 1999, 117, 181–190. [Google Scholar]
- Caldwell, S.H.; Chang, C.Y.; Nakamoto, R.K.; Krugner-Higby, L. Mitochondria in nonalcoholic fatty liver disease. Clin. Liver Dis 2004, 8, 595–617. [Google Scholar]
- Sanyal, A.J.; Campbell-Sargent, C.; Mirshahi, F.; Rizzo, W.B.; Contos, M.J.; Sterling, R.K.; Luketic, V.A.; Shiffman, M.L.; Clore, J.N. Nonalcoholic steatohepatitis: Association of insulin resistance and mitochondrial abnormalities. Gastroenterology 2001, 120, 1183–1192. [Google Scholar]
- Lammens, M.; Laak, H. Contribution of histopathological examination to the diagnosis of OXPHOS disorders. In Oxidative Phosphorylation in Health and Disease; Springer: New York, NY, USA, 2005; pp. 53–78. [Google Scholar]
- Rolo, A.P.; Teodoro, J.S.; Palmeira, C.M. Role of oxidative stress in the pathogenesis of nonalcoholic steatohepatitis. Free Radic. Biol. Med 2012, 52, 59–69. [Google Scholar]
- Ibdah, J.A.; Paul, H.; Zhao, Y.; Binford, S.; Salleng, K.; Cline, M.; Matern, D.; Bennett, M.J.; Rinaldo, P.; Strauss, A.W. Lack of mitochondrial trifunctional protein in mice causes neonatal hypoglycemia and sudden death. J. Clin. Investig 2001, 107, 1403–1409. [Google Scholar]
- Wanders, R.J.; Ijist, L.; Poggi, F.; Bonnefont, J.P.; Munnich, A.; Brivet, M.; Rabier, D.; Saudubray, J.M. Human trifunctional protein deficiency: A new disorder of mitochondrial fatty acid beta-oxidation. Biochem. Biophys. Res. Commun 1992, 188, 1139–1145. [Google Scholar]
- Rector, R.S.; Payne, R.M.; Ibdah, J.A. Mitochondrial trifunctional protein defects: Clinical implications and therapeutic approaches. Adv. Drug Deliv. Rev 2008, 60, 1488–1496. [Google Scholar]
- Ibdah, J.A.; Perlegas, P.; Zhao, Y.; Angdisen, J.; Borgerink, H.; Shadoan, M.K.; Wagner, J.D.; Matern, D.; Rinaldo, P.; Cline, J.M. Mice heterozygous for a defect in mitochondrial trifunctional protein develop hepatic steatosis and insulin resistance. Gastroenterology 2005, 128, 1381–1390. [Google Scholar]
- Rector, R.S.; Morris, E.M.; Ridenhour, S.; Meers, G.M.; Hsu, F.F.; Turk, J.; Ibdah, J.A. Selective hepatic insulin resistance in a murine model heterozygous for a mitochondrial trifunctional protein defect. Hepatology 2013, 57, 2213–2223. [Google Scholar]
- Zhou, M.; Xu, A.; Tam, P.K.; Lam, K.S.; Chan, L.; Hoo, R.L.; Liu, J.; Chow, K.H.; Wang, Y. Mitochondrial dysfunction contributes to the increased vulnerabilities of adiponectin knockout mice to liver injury. Hepatology 2008, 48, 1087–1096. [Google Scholar]
- Thyfault, J.P.; Rector, R.S.; Uptergrove, G.M.; Borengasser, S.J.; Morris, E.M.; Wei, Y.; Laye, M.J.; Burant, C.F.; Qi, N.R.; Ridenhour, S.E.; et al. Rats selectively bred for low aerobic capacity have reduced hepatic mitochondrial oxidative capacity and susceptibility to hepatic steatosis and injury. J. Physiol 2009, 587, 1805–1816. [Google Scholar]
- Rector, R.S.; Thyfault, J.P.; Uptergrove, G.M.; Morris, E.M.; Naples, S.P.; Borengasser, S.J.; Mikus, C.R.; Laye, M.J.; Laughlin, M.H.; Booth, F.W.; et al. Mitochondrial dysfunction precedes insulin resistance and hepatic steatosis and contributes to the natural history of non-alcoholic fatty liver disease in an obese rodent model. J. Hepatol 2010, 52, 727–736. [Google Scholar]
- Kawano, K.; Hirashima, T.; Mori, S.; Saitoh, Y.; Kurosumi, M.; Natori, T. Spontaneous long-term hyperglycemic rat with diabetic complications. Otsuka long-evans tokushima fatty (OLETF) strain. Diabetes 1992, 41, 1422–1428. [Google Scholar]
- Jeong, S.K.; Kim, Y.K.; Park, J.W.; Shin, Y.J.; Kim, D.S. Impact of visceral fat on the metabolic syndrome and nonalcoholic fatty liver disease. J. Korean Med. Sci 2008, 23, 789–795. [Google Scholar]
- Wang, Y.; Zhou, M.; Lam, K.S.; Xu, A. Protective roles of adiponectin in obesity-related fatty liver diseases: Mechanisms and therapeutic implications. Arq. Bras. Endocrinol. Metabol 2009, 53, 201–212. [Google Scholar]
- Bajaj, M.; Suraamornkul, S.; Piper, P.; Hardies, L.J.; Glass, L.; Cersosimo, E.; Pratipanawatr, T.; Miyazaki, Y.; DeFronzo, R.A. Decreased plasma adiponectin concentrations are closely related to hepatic fat content and hepatic insulin resistance in pioglitazone-treated type 2 diabetic patients. J. Clin. Endocrinol. Metab 2004, 89, 200–206. [Google Scholar]
- Hui, J.M.; Hodge, A.; Farrell, G.C.; Kench, J.G.; Kriketos, A.; George, J. Beyond insulin resistance in NASH: TNF-α or adiponectin? Hepatology (Baltim. Md.) 2004, 40, 46–54. [Google Scholar]
- Jung, U.J.; Choi, M.S. Obesity and its metabolic complications: The role of adipokines and the relationship between obesity, inflammation, insulin resistance, dyslipidemia and nonalcoholic fatty liver disease. Int. J. Mol. Sci 2014, 15, 6184–6223. [Google Scholar]
- Negre-Salvayre, A.; Hirtz, C.; Carrera, G.; Cazenave, R.; Troly, M.; Salvayre, R.; Penicaud, L.; Casteilla, L. A role for uncoupling protein-2 as a regulator of mitochondrial hydrogen peroxide generation. FASEB J 1997, 11, 809–815. [Google Scholar]
- Chow, L.; From, A.; Seaquist, E. Skeletal muscle insulin resistance: The interplay of local lipid excess and mitochondrial dysfunction. Metab. Clin. Exp 2010, 59, 70–85. [Google Scholar]
- Morino, K.; Petersen, K.F.; Dufour, S.; Befroy, D.; Frattini, J.; Shatzkes, N.; Neschen, S.; White, M.F.; Bilz, S.; Sono, S.; et al. Reduced mitochondrial density and increased IRS-1 serine phosphorylation in muscle of insulin-resistant offspring of type 2 diabetic parents. J. Clin. Investig 2005, 115, 3587–3593. [Google Scholar]
- Petersen, K.F.; Dufour, S.; Befroy, D.; Garcia, R.; Shulman, G.I. Impaired mitochondrial activity in the insulin-resistant offspring of patients with type 2 diabetes. N. Engl. J. Med 2004, 350, 664–671. [Google Scholar]
- Morino, K.; Petersen, K.F.; Sono, S.; Choi, C.S.; Samuel, V.T.; Lin, A.; Gallo, A.; Zhao, H.; Kashiwagi, A.; Goldberg, I.J.; et al. Regulation of mitochondrial biogenesis by lipoprotein lipase in muscle of insulin-resistant offspring of parents with type 2 diabetes. Diabetes 2012, 61, 877–887. [Google Scholar]
- Petersen, K.F.; Dufour, S.; Morino, K.; Yoo, P.S.; Cline, G.W.; Shulman, G.I. Reversal of muscle insulin resistance by weight reduction in young, lean, insulin-resistant offspring of parents with type 2 diabetes. Proc. Natl. Acad. Sci. USA 2012, 109, 8236–8240. [Google Scholar]
- Andersson, U.; Scarpulla, R.C. Pgc-1-related coactivator, a novel, serum-inducible coactivator of nuclear respiratory factor 1-dependent transcription in mammalian cells. Mol. Cell. Biol 2001, 21, 3738–3749. [Google Scholar]
- Vercauteren, K.; Gleyzer, N.; Scarpulla, R.C. Short hairpin RNA-mediated silencing of PRC (PGC-1-related coactivator) results in a severe respiratory chain deficiency associated with the proliferation of aberrant mitochondria. J. Biol. Chem 2009, 284, 2307–2319. [Google Scholar]
- Handschin, C.; Spiegelman, B.M. Peroxisome proliferator-activated receptor gamma coactivator 1 coactivators, energy homeostasis, and metabolism. Endocr. Rev 2006, 27, 728–735. [Google Scholar]
- Lin, J.; Puigserver, P.; Donovan, J.; Tarr, P.; Spiegelman, B.M. Peroxisome proliferator-activated receptor γ coactivator 1β (PGC-1β ), a novel PGC-1-related transcription coactivator associated with host cell factor. J. Biol. Chem 2002, 277, 1645–1648. [Google Scholar]
- Puigserver, P.; Wu, Z.; Park, C.W.; Graves, R.; Wright, M.; Spiegelman, B.M. A cold-inducible coactivator of nuclear receptors linked to adaptive thermogenesis. Cell 1998, 92, 829–839. [Google Scholar]
- Lai, L.; Leone, T.C.; Zechner, C.; Schaeffer, P.J.; Kelly, S.M.; Flanagan, D.P.; Medeiros, D.M.; Kovacs, A.; Kelly, D.P. Transcriptional coactivators PGC-1α and PGC-lβ control overlapping programs required for perinatal maturation of the heart. Genes Dev 2008, 22, 1948–1961. [Google Scholar]
- Lelliott, C.J.; Medina-Gomez, G.; Petrovic, N.; Kis, A.; Feldmann, H.M.; Bjursell, M.; Parker, N.; Curtis, K.; Campbell, M.; Hu, P.; et al. Ablation of PGC-1β results in defective mitochondrial activity, thermogenesis, hepatic function, and cardiac performance. PLoS Biol 2006, 4, e369. [Google Scholar]
- Leone, T.C.; Lehman, J.J.; Finck, B.N.; Schaeffer, P.J.; Wende, A.R.; Boudina, S.; Courtois, M.; Wozniak, D.F.; Sambandam, N.; Bernal-Mizrachi, C.; et al. PGC-1α deficiency causes multi-system energy metabolic derangements: Muscle dysfunction, abnormal weight control and hepatic steatosis. PLoS Biol 2005, 3, e101. [Google Scholar]
- Sonoda, J.; Mehl, I.R.; Chong, L.W.; Nofsinger, R.R.; Evans, R.M. PGC-1β controls mitochondrial metabolism to modulate circadian activity, adaptive thermogenesis, and hepatic steatosis. Proc. Natl. Acad. Sci. USA 2007, 104, 5223–5228. [Google Scholar]
- Vianna, C.R.; Huntgeburth, M.; Coppari, R.; Choi, C.S.; Lin, J.; Krauss, S.; Barbatelli, G.; Tzameli, I.; Kim, Y.B.; Cinti, S.; et al. Hypomorphic mutation of PGC-1β causes mitochondrial dysfunction and liver insulin resistance. Cell Metab 2006, 4, 453–464. [Google Scholar]
- Horton, J.D.; Shah, N.A.; Warrington, J.A.; Anderson, N.N.; Park, S.W.; Brown, M.S.; Goldstein, J.L. Combined analysis of oligonucleotide microarray data from transgenic and knockout mice identifies direct SREBP target genes. Proc. Natl. Acad. Sci. USA 2003, 100, 12027–12032. [Google Scholar]
- Lin, J.; Yang, R.; Tarr, P.T.; Wu, P.-H.; Handschin, C.; Li, S.; Yang, W.; Pei, L.; Uldry, M.; Tontonoz, P.; et al. Hyperlipidemic effects of dietary saturated fats mediated through PGC-1β coactivation of SREBP. Cell 2005, 120, 261–273. [Google Scholar]
- Bellafante, E.; Murzilli, S.; Salvatore, L.; Latorre, D.; Villani, G.; Moschetta, A. Hepatic-specific activation of peroxisome proliferator-activated receptor γ coactivator-1β protects against steatohepatitis. Hepatology (Baltim. Md.) 2013, 57, 1343–1356. [Google Scholar]
- Morris, E.M.; Meers, G.M.; Booth, F.W.; Fritsche, K.L.; Hardin, C.D.; Thyfault, J.P.; Ibdah, J.A. PGC-1α overexpression results in increased hepatic fatty acid oxidation with reduced triacylglycerol accumulation and secretion. Am. J. Physiol. Gastrointest. Liver Physiol 2012, 303, G979–G992. [Google Scholar]
- Laye, M.J.; Rector, R.S.; Borengasser, S.J.; Naples, S.P.; Uptergrove, G.M.; Ibdah, J.A.; Booth, F.W.; Thyfault, J.P. Cessation of daily wheel running differentially alters fat oxidation capacity in liver, muscle, and adipose tissue. J. Appl. Physiol. (Bethesda. Md.) 2009, 106, 161–168. [Google Scholar]
- Hirschey, M.D.; Shimazu, T.; Jing, E.; Grueter, C.A.; Collins, A.M.; Aouizerat, B.; Stancakova, A.; Goetzman, E.; Lam, M.M.; Schwer, B.; et al. SIRT3 deficiency and mitochondrial protein hyperacetylation accelerate the development of the metabolic syndrome. Mol. Cell 2011, 44, 177–190. [Google Scholar]
- Puigserver, P. Tissue-specific regulation of metabolic pathways through the transcriptional coactivator PGC1-α. Int. J. Obes. Relat. Metab. Disord 2005, 29, S5–S9. [Google Scholar]
- Wu, Z.; Puigserver, P.; Andersson, U.; Zhang, C.; Adelmant, G.; Mootha, V.; Troy, A.; Cinti, S.; Lowell, B.; Scarpulla, R.C.; et al. Mechanisms controlling mitochondrial biogenesis and respiration through the thermogenic coactivator PGC-1. Cell 1999, 98, 115–124. [Google Scholar]
- Feige, J.N.; Auwerx, J. Transcriptional coregulators in the control of energy homeostasis. Trends Cell Biol 2007, 17, 292–301. [Google Scholar]
- Wright, D.C.; Geiger, P.C.; Han, D.H.; Jones, T.E.; Holloszy, J.O. Calcium induces increases in peroxisome proliferator-activated receptor γ coactivator-1α and mitochondrial biogenesis by a pathway leading to p38 mitogen-activated protein kinase activation. J. Biol. Chem 2007, 282, 18793–9. [Google Scholar]
- Canto, C.; Gerhart-Hines, Z.; Feige, J.N.; Lagouge, M.; Noriega, L.; Milne, J.C.; Elliott, P.J.; Puigserver, P.; Auwerx, J. AMPK regulates energy expenditure by modulating NAD+ metabolism and SIRT1 activity. Nature 2009, 458, 1056–1060. [Google Scholar]
- Haigis, M.C.; Guarente, L.P. Mammalian sirtuins—Emerging roles in physiology, aging, and calorie restriction. Genes Dev 2006, 20, 2913–1921. [Google Scholar]
- Haigis, M.C.; Sinclair, D.A. Mammalian sirtuins: Biological insights and disease relevance. Annu. Rev. Pathol 2010, 5, 253–295. [Google Scholar]
- Nogueiras, R.; Habegger, K.M.; Chaudhary, N.; Finan, B.; Banks, A.S.; Dietrich, M.O.; Horvath, T.L.; Sinclair, D.A.; Pfluger, P.T.; Tschop, M.H. Sirtuin 1 and sirtuin 3, physiological modulators of metabolism. Physiol. Rev 2012, 92, 1479–1514. [Google Scholar]
- Guarente, L. Calorie restriction and sirtuins revisited. Genes Dev 2013, 27, 2072–2085. [Google Scholar]
- Banks, A.S.; Kon, N.; Knight, C.; Matsumoto, M.; Gutierrez-Juarez, R.; Rossetti, L.; Gu, W.; Accili, D. SirT1 gain of function increases energy efficiency and prevents diabetes in mice. Cell Metab 2008, 8, 333–341. [Google Scholar]
- Kitada, M.; Kume, S.; Kanasaki, K.; Takeda-Watanabe, A.; Koya, D. Sirtuins as possible drug targets in type 2 diabetes. Curr. Drug Targets 2013, 14, 622–636. [Google Scholar]
- Garcia-Rodriguez, J.L.; Barbier-Torres, L.; Fernandez-Alvarez, S.; Juan, V.G.; Monte, M.J.; Halilbasic, E.; Herranz, D.; Alvarez, L.; Aspichueta, P.; Marin, J.J.; et al. SIRT1 controls liver regeneration by regulating BA metabolism through FXR and mTOR signaling. Hepatology (Baltim. Md.) 2014, 59, 1972–1983. [Google Scholar]
- Ruderman, N.B.; Xu, X.J.; Nelson, L.; Cacicedo, J.M.; Saha, A.K.; Lan, F.; Ido, Y. AMPK and SirT1, a long-standing partnership? Am. J. Physiol. Endocrinol. Metab 2010, 298, E751–E760. [Google Scholar]
- Chen, L.L.; Deng, X.Q.; Li, N.X. Effects of calorie restriction on SIRT1 expression in liver of nonalcoholic fatty liver disease: Experiment with rats. Zhonghua Yi Xue Za Zhi 2007, 87, 1434–1437. [Google Scholar]
- Deng, X.Q.; Chen, L.L.; Li, N.X. The expression of SirT1 in nonalcoholic fatty liver disease induced by high-fat diet in rats. Liver Int 2007, 27, 708–715. [Google Scholar]
- Colak, Y.; Ozturk, O.; Senates, E.; Tuncer, I.; Yorulmaz, E.; Adali, G.; Doganay, L.; Enc, F.Y. SirT1 as a potential therapeutic target for treatment of nonalcoholic fatty liver disease. Med. Sci. Monit 2011, 17, HY5–HY9. [Google Scholar]
- Li, Y.; Xu, S.; Giles, A.; Nakamura, K.; Lee, J.W.; Hou, X.; Donmez, G.; Li, J.; Luo, Z.; Walsh, K.; et al. Hepatic overexpression of SirT1 in mice attenuates endoplasmic reticulum stress and insulin resistance in the liver. FASEB J 2011, 25, 1664–1679. [Google Scholar]
- Li, Y.; Wong, K.; Giles, A.; Jiang, J.; Lee, J.W.; Adams, A.C.; Kharitonenkov, A.; Yang, Q.; Gao, B.; Guarente, L.; et al. Hepatic SirT1 attenuates hepatic steatosis and controls energy balance in mice by inducing fibroblast growth factor 21. Gastroenterology 2014, 146, 539–549. [Google Scholar]
- Purushotham, A.; Schug, T.T.; Xu, Q.; Surapureddi, S.; Guo, X.; Li, X. Hepatocyte-specific deletion of SirT1 alters fatty acid metabolism and results in hepatic steatosis and inflammation. Cell Metab 2009, 9, 327–338. [Google Scholar]
- Shin, S.Y.; Kim, T.H.; Wu, H.; Choi, Y.H.; Kim, S.G. SirT1 activation by methylene blue, a repurposed drug, leads to AMPK-mediated inhibition of steatosis and steatohepatitis. Eur. J. Pharmacol 2014, 727C, 115–124. [Google Scholar]
- Lombard, D.B.; Alt, F.W.; Cheng, H.L.; Bunkenborg, J.; Streeper, R.S.; Mostoslavsky, R.; Kim, J.; Yancopoulos, G.; Valenzuela, D.; Murphy, A.; et al. Mammalian SirT2 homolog SirT3 regulates global mitochondrial lysine acetylation. Mol. Cell. Biol 2007, 27, 8807–8814. [Google Scholar]
- Hebert, A.S.; Dittenhafer-Reed, K.E.; Yu, W.; Bailey, D.J.; Selen, E.S.; Boersma, M.D.; Carson, J.J.; Tonelli, M.; Balloon, A.J.; Higbee, A.J.; et al. Calorie restriction and SirT3 trigger global reprogramming of the mitochondrial protein acetylome. Mol. Cell 2013, 49, 186–199. [Google Scholar]
- Hirschey, M.D.; Shimazu, T.; Goetzman, E.; Jing, E.; Schwer, B.; Lombard, D.B.; Grueter, C.A.; Harris, C.; Biddinger, S.; Ilkayeva, O.R.; et al. SirT3 regulates mitochondrial fatty-acid oxidation by reversible enzyme deacetylation. Nature 2010, 464, 121–125. [Google Scholar]
- Shimazu, T.; Hirschey, M.D.; Hua, L.; Dittenhafer-Reed, K.E.; Schwer, B.; Lombard, D.B.; Li, Y.; Bunkenborg, J.; Alt, F.W.; Denu, J.M.; et al. SirT3 deacetylates mitochondrial 3-hydroxy-3- methylglutaryl CoA synthase 2 and regulates ketone body production. Cell Metab 2010, 12, 654–661. [Google Scholar]
- Fujino, T.; Kondo, J.; Ishikawa, M.; Morikawa, K.; Yamamoto, T.T. Acetyl-CoA synthetase 2, a mitochondrial matrix enzyme involved in the oxidation of acetate. J. Biol. Chem 2001, 276, 11420–11426. [Google Scholar]
- Ahn, B.-H.; Kim, H.-S.; Song, S.; Lee, I.H.; Liu, J.; Vassilopoulos, A.; Deng, C.-X.; Finkel, T. A role for the mitochondrial deacetylase SirT3 in regulating energy homeostasis. Proc. Natl. Acad. Sci. USA 2008, 105, 14447–14452. [Google Scholar]
- Cimen, H.; Han, M.-J.; Yang, Y.; Tong, Q.; Koc, H.; Koc, E.C. Regulation of succinate dehydrogenase activity by SirT3 in mammalian mitochondria. Biochemistry 2009, 49, 304–311. [Google Scholar]
- Finley, L.W.; Haas, W.; Desquiret-Dumas, V.; Wallace, D.C.; Procaccio, V.; Gygi, S.P.; Haigis, M.C. Succinate dehydrogenase is a direct target of sirtuin 3 deacetylase activity. PLoS One 2011, 6, e23295. [Google Scholar]
- Zhang, D.; Liu, Z.X.; Choi, C.S.; Tian, L.; Kibbey, R.; Dong, J.; Cline, G.W.; Wood, P.A.; Shulman, G.I. Mitochondrial dysfunction due to long-chain acyl-CoA dehydrogenase deficiency causes hepatic steatosis and hepatic insulin resistance. Proc. Natl. Acad. Sci. USA 2007, 104, 17075–17080. [Google Scholar]
- Kurtz, D.M.; Rinaldo, P.; Rhead, W.J.; Tian, L.; Millington, D.S.; Vockley, J.; Hamm, D.A.; Brix, A.E.; Lindsey, J.R.; Pinkert, C.A.; et al. Targeted disruption of mouse long-chain acyl-CoA dehydrogenase gene reveals crucial roles for fatty acid oxidation. Proc. Natl. Acad. Sci. USA 1998, 95, 15592–15597. [Google Scholar]
- Qiu, X.; Brown, K.; Hirschey, M.D.; Verdin, E.; Chen, D. Calorie restriction reduces oxidative stress by SirT3-mediated SOD2 activation. Cell Metab 2010, 12, 662–667. [Google Scholar]
- Hirschey, M.D.; Shimazu, T.; Huang, J.Y.; Schwer, B.; Verdin, E. SirT3 regulates mitochondrial protein acetylation and intermediary metabolism. Cold Spring Harb. Symp. Quant. Biol 2011, 76, 267–277. [Google Scholar]
- Petro, A.E.; Cotter, J.; Cooper, D.A.; Peters, J.C.; Surwit, S.J.; Surwit, R.S. Fat, carbohydrate, and calories in the development of diabetes and obesity in the C57BL/6J mouse. Metab. Clin. Exp 2004, 53, 454–457. [Google Scholar]
- Rossmeisl, M.; Rim, J.S.; Koza, R.A.; Kozak, L.P. Variation in type 2 diabetes—Related traits in mouse strains susceptible to diet-induced obesity. Diabetes 2003, 52, 1958–1966. [Google Scholar]
- Surwit, R.S.; Feinglos, M.N.; Rodin, J.; Sutherland, A.; Petro, A.E.; Opara, E.C.; Kuhn, C.M.; Rebuffe-Scrive, M. Differential effects of fat and sucrose on the development of obesity and diabetes in C57BL/6J and A/J mice. Metab. Clin. Exp 1995, 44, 645–651. [Google Scholar]
- Kendrick, A.A.; Choudhury, M.; Rahman, S.M.; McCurdy, C.E.; Friederich, M.; van Hove, J.L.; Watson, P.A.; Birdsey, N.; Bao, J.; Gius, D.; et al. Fatty liver is associated with reduced SirT3 activity and mitochondrial protein hyperacetylation. Biochem. J 2011, 433, 505–514. [Google Scholar]
- Crunkhorn, S.; Dearie, F.; Mantzoros, C.; Gami, H.; da Silva, W.S.; Espinoza, D.; Faucette, R.; Barry, K.; Bianco, A.C.; Patti, M.E. Peroxisome proliferator activator receptor gamma coactivator-1 expression is reduced in obesity: Potential pathogenic role of saturated fatty acids and p38 mitogen-activated protein kinase activation. J. Biol. Chem 2007, 282, 15439–15450. [Google Scholar]
- Ji, H.; Friedman, M.I. Reduced capacity for fatty acid oxidation in rats with inherited susceptibility to diet-induced obesity. Metab. Clin. Exp 2007, 56, 1124–1130. [Google Scholar]
- Jing, E.; Emanuelli, B.; Hirschey, M.D.; Boucher, J.; Lee, K.Y.; Lombard, D.; Verdin, E.M.; Kahn, C.R. Sirtuin-3 (SirT3) regulates skeletal muscle metabolism and insulin signaling via altered mitochondrial oxidation and reactive oxygen species production. Proc. Natl. Acad. Sci. USA 2011, 108, 14608–14613. [Google Scholar]
- Bao, J.; Scott, I.; Lu, Z.; Pang, L.; Dimond, C.C.; Gius, D.; Sack, M.N. SirT3 is regulated by nutrient excess and modulates hepatic susceptibility to lipotoxicity. Free Radic. Biol. Med 2010, 49, 1230–1237. [Google Scholar]
- Mailloux, R.J.; Beriault, R.; Lemire, J.; Singh, R.; Chenier, D.R.; Hamel, R.D.; Appanna, V.D. The tricarboxylic acid cycle, an ancient metabolic network with a novel twist. PLoS One 2007, 2, e690. [Google Scholar]
- Someya, S.; Yu, W.; Hallows, W.C.; Xu, J.; Vann, J.M.; Leeuwenburgh, C.; Tanokura, M.; Denu, J.M.; Prolla, T.A. SirT3 mediates reduction of oxidative damage and prevention of age-related hearing loss under caloric restriction. Cell 2010, 143, 802–812. [Google Scholar]
- Yu, H.S.; Oyama, T.; Isse, T.; Kitagawa, K.; Pham, T.T.; Tanaka, M.; Kawamoto, T. Formation of acetaldehyde-derived DNA adducts due to alcohol exposure. Chem. Biol. Interact 2010, 188, 367–375. [Google Scholar]
- Spitz, D.R.; Oberley, L.W. An assay for superoxide dismutase activity in mammalian tissue homogenates. Anal. Biochem 1989, 179, 8–18. [Google Scholar]
- Tao, R.; Coleman, M.C.; Pennington, J.D.; Ozden, O.; Park, S.-H.; Jiang, H.; Kim, H.-S.; Flynn, C.R.; Hill, S.; Hayes McDonald, W.; et al. SirT3-mediated deacetylation of evolutionarily conserved lysine 122 regulates MnSOD activity in response to stress. Mol. Cell 2010, 40, 893–904. [Google Scholar]
- He, J.; Hu, B.; Shi, X.; Weidert, E.R.; Lu, P.; Xu, M.; Huang, M.; Kelley, E.E.; Xie, W. Activation of the aryl hydrocarbon receptor sensitizes mice to nonalcoholic steatohepatitis by deactivating mitochondrial sirtuin deacetylase SirT3. Mol. Cell. Biol 2013, 33, 2047–2055. [Google Scholar]
- Amir, M.; Czaja, M.J. Autophagy in nonalcoholic steatohepatitis. Expert Rev. Gastroenterol. Hepatol 2011, 5, 159–166. [Google Scholar]
- Singh, R.; Kaushik, S.; Wang, Y.; Xiang, Y.; Novak, I.; Komatsu, M.; Tanaka, K.; Cuervo, A.M.; Czaja, M.J. Autophagy regulates lipid metabolism. Nature 2009, 458, 1131–1135. [Google Scholar]
- Yang, L.; Li, P.; Fu, S.; Calay, E.S.; Hotamisligil, G.S. Defective hepatic autophagy in obesity promotes ER stress and causes insulin resistance. Cell Metab 2010, 11, 467–478. [Google Scholar]
- Scarpulla, R.C.; Vega, R.B.; Kelly, D.P. Transcriptional integration of mitochondrial biogenesis. Trends Endocrinol. Metab 2012, 23, 459–466. [Google Scholar]
- Aatsinki, S.M.; Buler, M.; Salomaki, H.; Koulu, M.; Pavek, P.; Hakkola, J. Metformin induces PGC-1α expression and selectively affects hepatic PGC-1α functions. Br. J. Pharmacol 2014, 171, 2351–2363. [Google Scholar]
- Srivastava, S.; Diaz, F.; Iommarini, L.; Aure, K.; Lombes, A.; Moraes, C.T. PGC-1α/β induced expression partially compensates for respiratory chain defects in cells from patients with mitochondrial disorders. Hum. Mol. Genet 2009, 18, 1805–1812. [Google Scholar]
- Kong, X.; Wang, R.; Xue, Y.; Liu, X.; Zhang, H.; Chen, Y.; Fang, F.; Chang, Y. Sirtuin 3, a new target of PGC-1α, plays an important role in the suppression of ROS and mitochondrial biogenesis. PLoS One 2010, 5, e11707. [Google Scholar]
- St-Pierre, J.; Drori, S.; Uldry, M.; Silvaggi, J.M.; Rhee, J.; Jager, S.; Handschin, C.; Zheng, K.; Lin, J.; Yang, W.; et al. Suppression of reactive oxygen species and neurodegeneration by the PGC-1 transcriptional coactivators. Cell 2006, 127, 397–408. [Google Scholar]
- Giralt, A.; Hondares, E.; Villena, J.A.; Ribas, F.; Díaz-Delfín, J.; Giralt, M.; Iglesias, R.; Villarroya, F. Peroxisome proliferator-activated receptor-γ coactivator-1α controls transcription of the SirT3 Gene, an essential component of the thermogenic brown adipocyte phenotype. J. Biol. Chem 2011, 286, 16958–16966. [Google Scholar]
- Kubli, D.A.; Gustafsson, Å.B. Mitochondria and mitophagy: The yin and yang of cell death control. Circ. Res 2012, 111, 1208–1221. [Google Scholar]
- Babbar, M.; Sheikh, M.S. Metabolic stress and disorders related to alterations in mitochondrial fission or fusion. Mol. Cell. Pharmacol 2013, 5, 109–133. [Google Scholar]
- Palikaras, K.; Tavernarakis, N. Mitochondrial homeostasis: The interplay between mitophagy and mitochondrial biogenesis. Exp. Gerontol 2014. [Google Scholar] [CrossRef]
- Van der Bliek, A.M.; Shen, Q.; Kawajiri, S. Mechanisms of mitochondrial fission and fusion. Cold Spring Harb. Perspect. Biol 2013. [Google Scholar] [CrossRef]
- Geisler, S.; Holmstrom, K.M.; Treis, A.; Skujat, D.; Weber, S.S.; Fiesel, F.C.; Kahle, P.J.; Springer, W. The PINK1/Parkin-mediated mitophagy is compromised by PD-associated mutations. Autophagy 2010, 6, 871–878. [Google Scholar]
- Geisler, S.; Holmstrom, K.M.; Skujat, D.; Fiesel, F.C.; Rothfuss, O.C.; Kahle, P.J.; Springer, W. PINK1/Parkin-mediated mitophagy is dependent on VDAC1 and p62/SQSTM1. Nat. Cell Biol 2010, 12, 119–131. [Google Scholar]
- Jin, S.M.; Youle, R.J. PINK1- and Parkin-mediated mitophagy at a glance. J. Cell Sci 2012, 125, 795–799. [Google Scholar]
- Chan, N.C.; Salazar, A.M.; Pham, A.H.; Sweredoski, M.J.; Kolawa, N.J.; Graham, R.L.J.; Hess, S.; Chan, D.C. Broad activation of the ubiquitin–proteasome system by Parkin is critical for mitophagy. Hum. Mol. Genet 2011, 20, 1726–1737. [Google Scholar]
- Poole, A.C.; Thomas, R.E.; Yu, S.; Vincow, E.S.; Pallanck, L. The mitochondrial fusion-promoting factor mitofusin is a substrate of the PINK1/Parkin pathway. PLoS One 2010, 5, e10054. [Google Scholar]
- Gegg, M.E.; Cooper, J.M.; Chau, K.-Y.; Rojo, M.; Schapira, A.H.V.; Taanman, J.-W. Mitofusin 1 and mitofusin 2 are ubiquitinated in a PINK1/Parkin-dependent manner upon induction of mitophagy. Hum. Mol. Genet 2010, 19, 4861–4870. [Google Scholar]
- Bhat, G.; Baba, C.S.; Pandey, A.; Kumari, N.; Choudhuri, G. Life style modification improves insulin resistance and liver histology in patients with non-alcoholic fatty liver disease. World J. Hepatol 2012, 4, 209–217. [Google Scholar]
- Browning, J.D.; Baker, J.A.; Rogers, T.; Davis, J.; Satapati, S.; Burgess, S.C. Short-term weight loss and hepatic triglyceride reduction: Evidence of a metabolic advantage with dietary carbohydrate restriction. Am. J. Clin. Nutr 2011, 93, 1048–1052. [Google Scholar]
- Elias, M.C.; Parise, E.R.; de Carvalho, L.; Szejnfeld, D.; Netto, J.P. Effect of 6-month nutritional intervention on non-alcoholic fatty liver disease. Nutrition 2010, 26, 1094–1099. [Google Scholar]
- Goncalves, I.O.; Oliveira, P.J.; Ascensao, A.; Magalhaes, J. Exercise as a therapeutic tool to prevent mitochondrial degeneration in nonalcoholic steatohepatitis. Eur. J. Clin. Investig 2013, 43, 1184–1194. [Google Scholar]
- Goncalves, I.O.; Passos, E.; Rocha-Rodrigues, S.; Diogo, C.V.; Torrella, J.R.; Rizo, D.; Viscor, G.; Santos-Alves, E.; Marques-Aleixo, I.; Oliveira, P.J.; et al. Physical exercise prevents and mitigates non-alcoholic steatohepatitis-induced liver mitochondrial structural and bioenergetics impairments. Mitochondrion 2014, 15, 40–51. [Google Scholar]
- Haus, J.M.; Solomon, T.P.; Kelly, K.R.; Fealy, C.E.; Kullman, E.L.; Scelsi, A.R.; Lu, L.; Pagadala, M.R.; McCullough, A.J.; Flask, C.A.; et al. Improved hepatic lipid composition following short-term exercise in nonalcoholic fatty liver disease. J. Clin. Endocrinol. Metab 2013, 98, E1181–E1188. [Google Scholar]
- He, Y.; Zhang, H.; Fu, F. The effects of swimming exercise on high-fat-diet-induced steatohepatitis. J. Sports Med. Phys. Fit 2008, 48, 259–265. [Google Scholar]
- Larson-Meyer, D.E.; Newcomer, B.R.; Heilbronn, L.K.; Volaufova, J.; Smith, S.R.; Alfonso, A.J.; Lefevre, M.; Rood, J.C.; Williamson, D.A.; Ravussin, E.; et al. Effect of 6-month calorie restriction and exercise on serum and liver lipids and markers of liver function. Obesity (Silver Spring. Md.) 2008, 16, 1355–1362. [Google Scholar]
- Rector, R.S.; Thyfault, J.P.; Morris, R.T.; Laye, M.J.; Borengasser, S.J.; Booth, F.W.; Ibdah, J.A. Daily exercise increases hepatic fatty acid oxidation and prevents steatosis in otsuka long-evans tokushima Fatty rats. Am. J. Physiol. Gastrointest. Liver Physiol 2008, 294, G619–G626. [Google Scholar]
- Pacana, T.; Sanyal, A.J. Vitamin E and nonalcoholic fatty liver disease. Curr. Opin. Clin. Nutr. Metab. Care 2012, 15, 641–648. [Google Scholar]
- Linden, M.A.; Fletcher, J.A.; Morris, E.M.; Meers, G.M.; Kearney, M.L.; Crissey, J.M.; Laughlin, M.H.; Booth, F.W.; Sowers, J.R.; Ibdah, J.A.; et al. Combining metformin and aerobic exercise training in the treatment of type 2 diabetes and NAFLD in OLETF rats. Am. J. Physiol. Endocrinol. Metab 2014, 306, E300–E310. [Google Scholar]
- Chachay, V.S.; Macdonald, G.A.; Martin, J.H.; Whitehead, J.P.; O’Moore-Sullivan, T.M.; Lee, P.; Franklin, M.; Klein, K.; Taylor, P.J.; Ferguson, M.; et al. Resveratrol does not benefit patients with non-alcoholic fatty liver disease. Clin. Gastroenterol. Hepatol 2014. [Google Scholar] [CrossRef] [Green Version]
- Poulsen, M.M.; Larsen, J.O.; Hamilton-Dutoit, S.; Clasen, B.F.; Jessen, N.; Paulsen, S.K.; Kjaer, T.N.; Richelsen, B.; Pedersen, S.B. Resveratrol up-regulates hepatic uncoupling protein 2 and prevents development of nonalcoholic fatty liver disease in rats fed a high-fat diet. Nutr. Res 2012, 32, 701–708. [Google Scholar]
- Poulsen, M.M.; Vestergaard, P.F.; Clasen, B.F.; Radko, Y.; Christensen, L.P.; Stodkilde-Jorgensen, H.; Moller, N.; Jessen, N.; Pedersen, S.B.; Jorgensen, J.O. High-dose resveratrol supplementation in obese men: An investigator-initiated, randomized, placebo-controlled clinical trial of substrate metabolism, insulin sensitivity, and body composition. Diabetes 2013, 62, 1186–1195. [Google Scholar]
- Timmers, S.; Konings, E.; Bilet, L.; Houtkooper, R.H.; van de Weijer, T.; Goossens, G.H.; Hoeks, J.; van der Krieken, S.; Ryu, D.; Kersten, S.; et al. Calorie restriction-like effects of 30 days of resveratrol supplementation on energy metabolism and metabolic profile in obese humans. Cell Metab 2011, 14, 612–622. [Google Scholar]
- Teodoro, J.S.; Duarte, F.V.; Gomes, A.P.; Varela, A.T.; Peixoto, F.M.; Rolo, A.P.; Palmeira, C.M. Berberine reverts hepatic mitochondrial dysfunction in high-fat fed rats: A possible role for SirT3 activation. Mitochondrion 2013, 13, 637–646. [Google Scholar]

{kind=link}
{kind=link}
{kind=link}
| Authors | Journal | Title |
|---|---|---|
| Sanyal et al. [78] | Gastroenterology 2001, 120, 1183–1192 | Nonalcoholic steatohepatitis: association of insulin resistance and mitochondrial abnormalities |
| Ibdah et al. [84] | Gastroenterology 2005, 128, 1381–1390 | Mice heterozygous for a defect in mitochondrial trifunctional protein develop hepatic steatosis and insulin resistance |
| Zhou et al. [86] | Hepatology 2008, 48, 1087–1096 | Mitochondrial dysfunction contributes to the increased vulnerabilities of adiponectin knockout mice to liver injury |
| Thyfault et al. [87] | J. Physiol. 2009, 587, 1805–1816 | Rats selectively bred for low aerobic capacity have reduced hepatic mitochondrial oxidative capacity and susceptibility to hepatic steatosis and injury |
| Rector et al. [88] | J. Hepatol. 2010, 52, 727–736 | Mitochondrial dysfunction precedes insulin resistance and hepatic steatosis and contributes to the natural history of non-alcoholic fatty liver disease in an obese rodent model |
| Rector et al. [85] | Hepatology 2013, 57, 2213–2223 | Selective hepatic insulin resistance in a murine model heterozygous for a mitochondrial trifunctional protein defect |
© 2014 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Nassir, F.; Ibdah, J.A. Role of Mitochondria in Nonalcoholic Fatty Liver Disease. Int. J. Mol. Sci. 2014, 15, 8713-8742. https://doi.org/10.3390/ijms15058713
Nassir F, Ibdah JA. Role of Mitochondria in Nonalcoholic Fatty Liver Disease. International Journal of Molecular Sciences. 2014; 15(5):8713-8742. https://doi.org/10.3390/ijms15058713
Chicago/Turabian StyleNassir, Fatiha, and Jamal A. Ibdah. 2014. "Role of Mitochondria in Nonalcoholic Fatty Liver Disease" International Journal of Molecular Sciences 15, no. 5: 8713-8742. https://doi.org/10.3390/ijms15058713