Inhibition of Acetylcholinesterase Modulates NMDA Receptor Antagonist Mediated Alterations in the Developing Brain



Abstract
:1. Introduction
2. Results and Discussion
2.1. Results
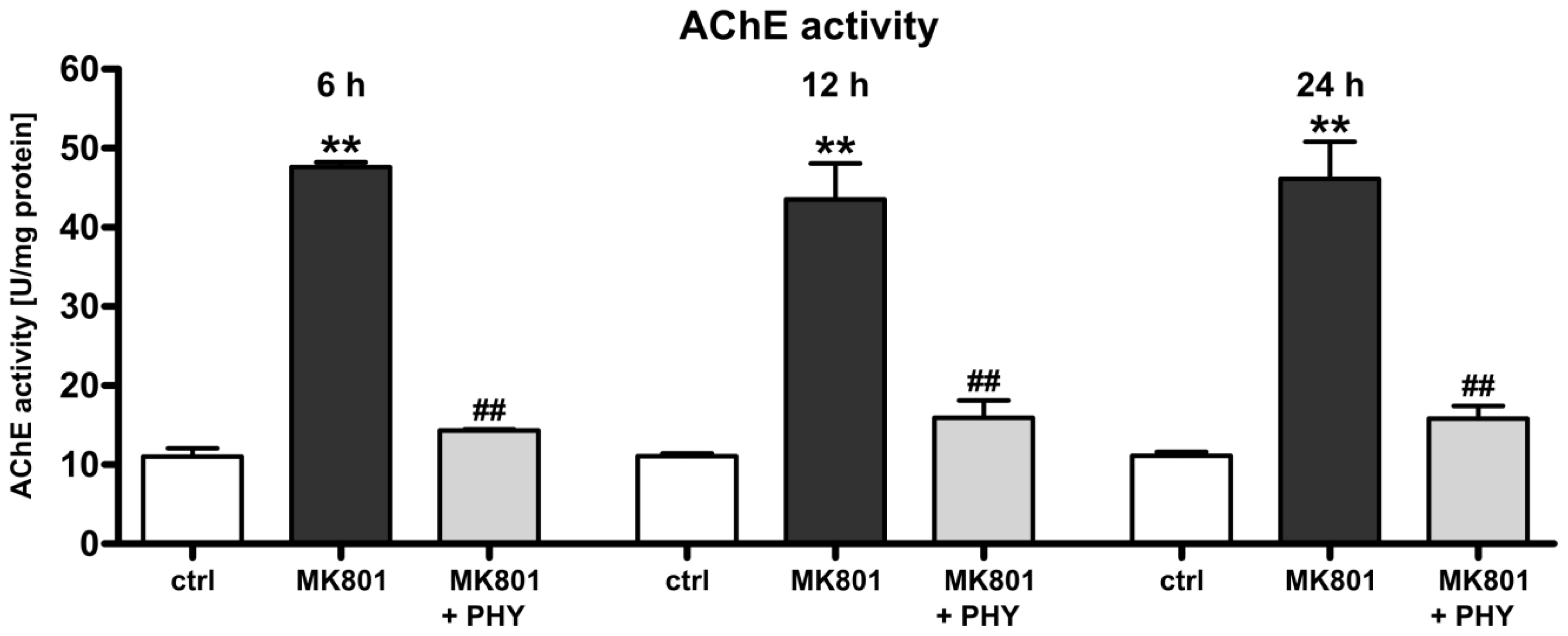
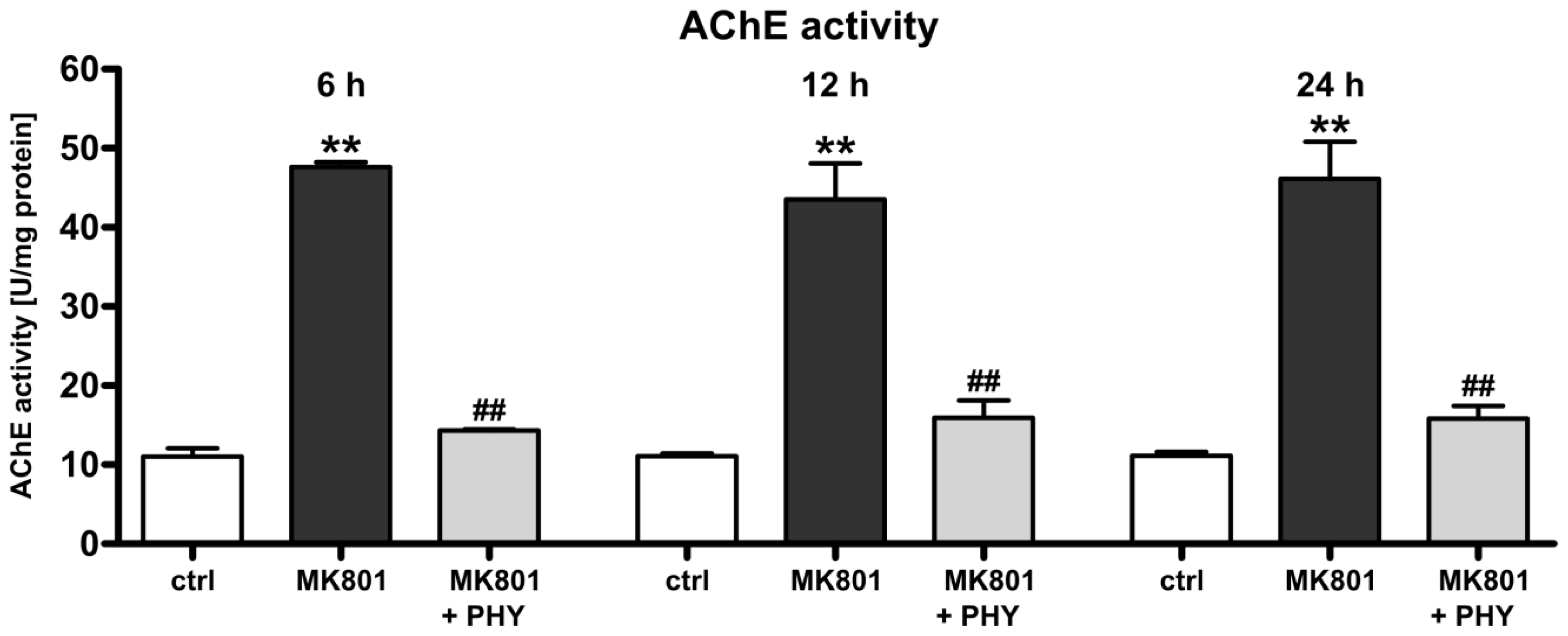
2.1.1. Physostigmine Normalizes N-Methyl-d-aspartate (NMDA) Receptor Antagonist-Induced Increase of Acetylcholinesterase Activity
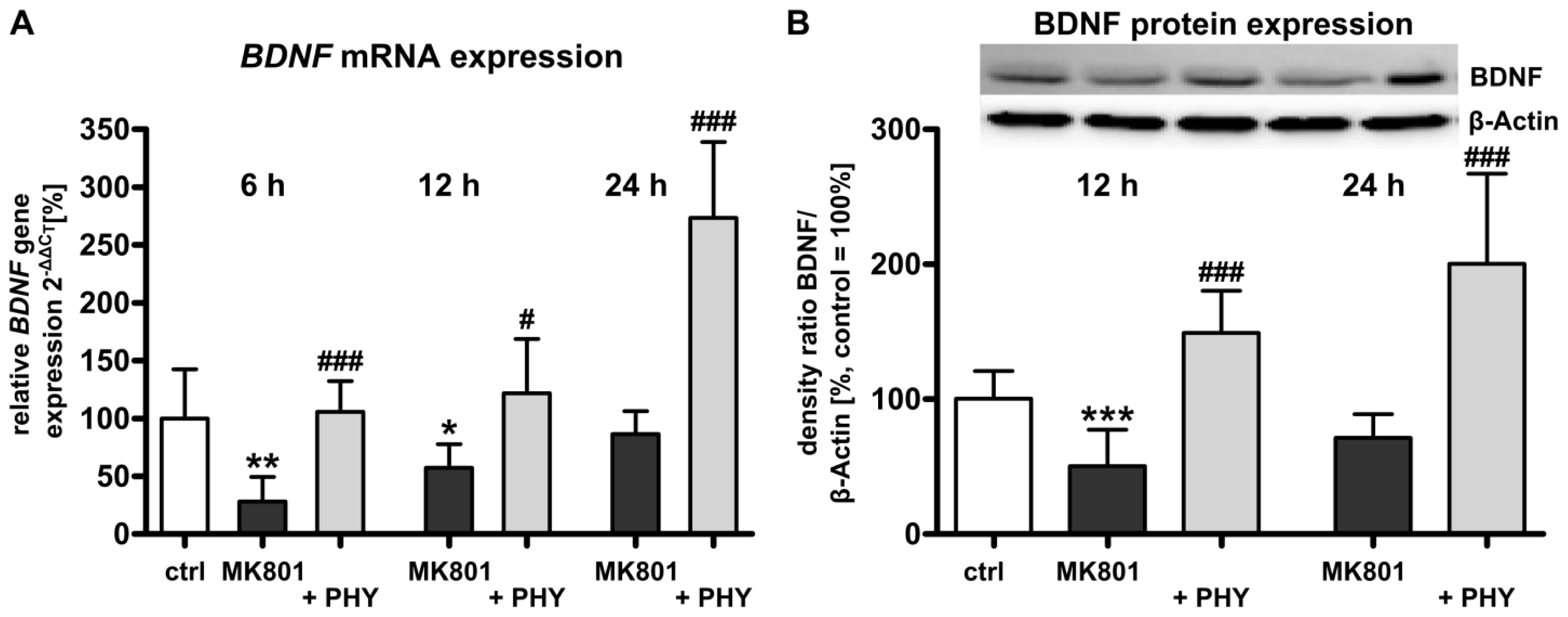
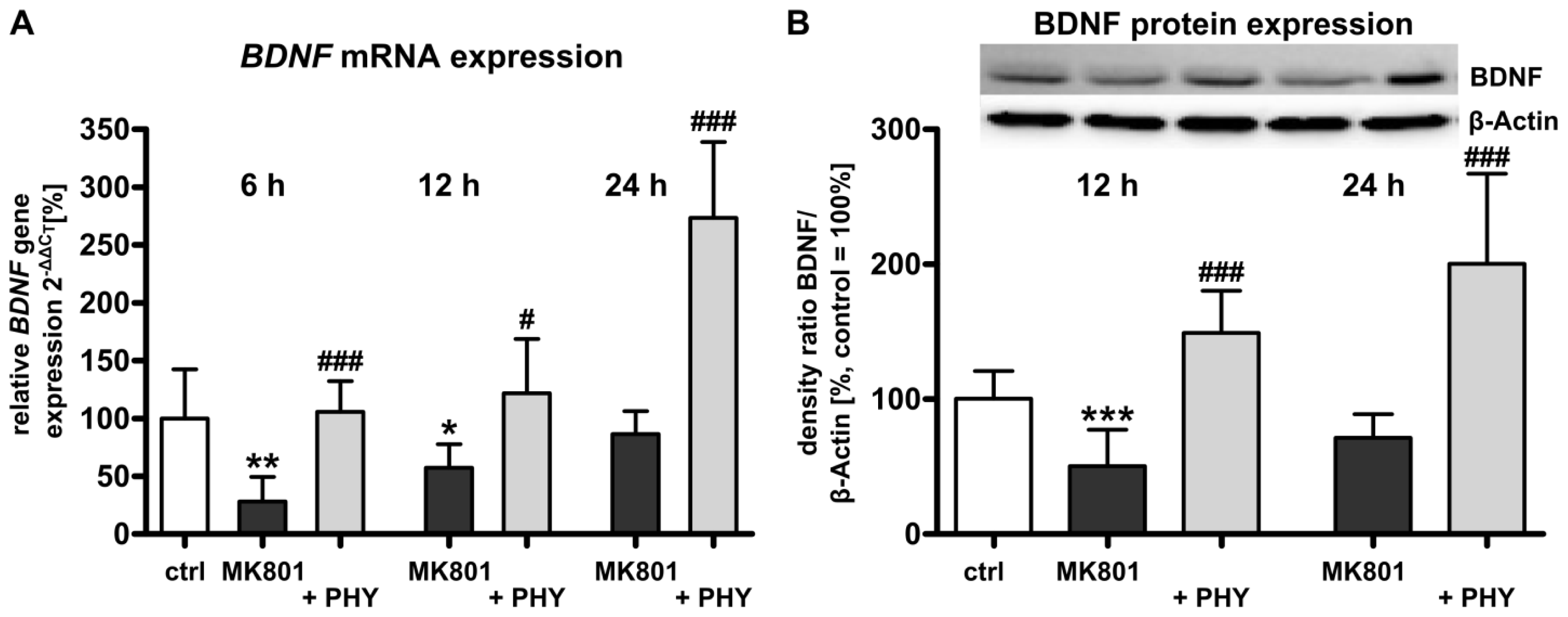
2.1.2. Acetylcholinesterase (AChE) Inhibition Increases Brain-Derived Neurotrophic Factor (BDNF) Expression
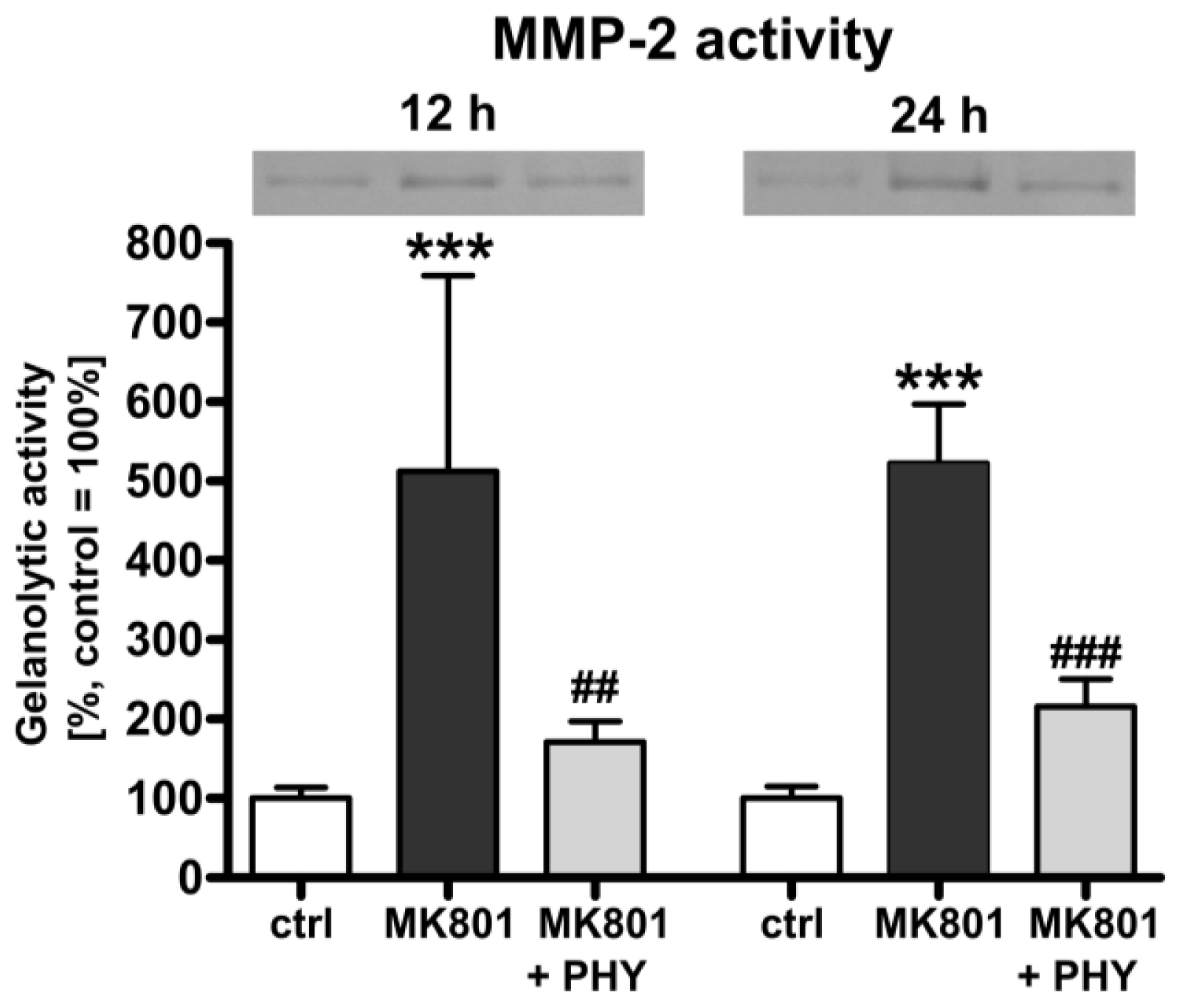
2.1.3. Physostigmine Modulates MK801 (Dizocilpine)-Induced Increase of Matrix Metalloproteinase (MMP)-2 Activity
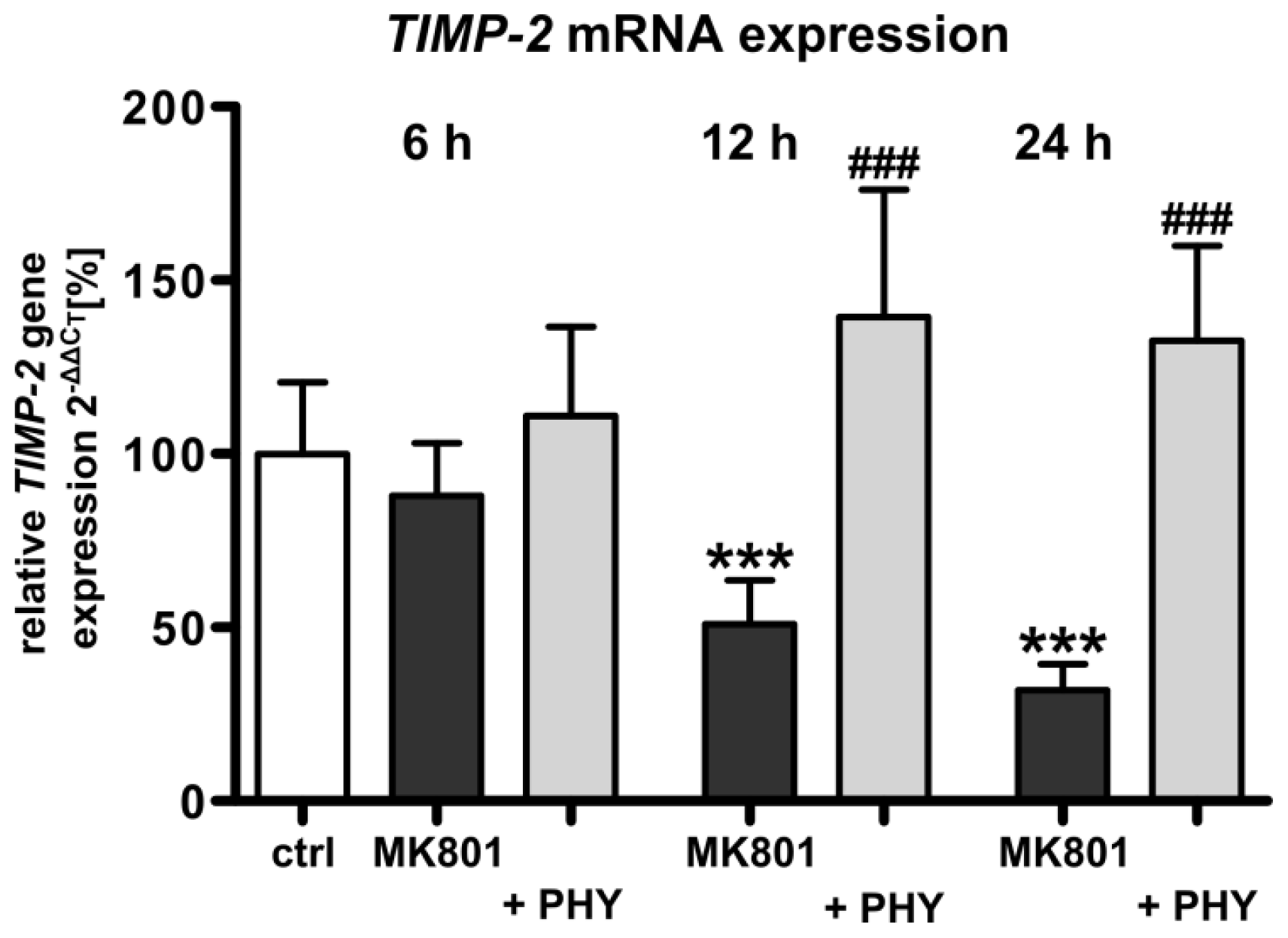
2.1.4. NMDA Receptor Antagonist Mediated Reduction of Tissue Inhibitor of Metalloproteinase-2 (TIMP-2) Expression Is Increased by Physostigmine Co-Treatment
2.2. Discussion
3. Experimental Section
3.1. Animals and Experimental Procedure
3.1.1. Drug Treatment
3.1.2. Tissue Sampling
3.2. AChE Activity Assay
3.3. Semiquantitative Real-Time PCR
3.4. Immunoblotting
3.5. Gelatin Zymography
3.6. Statistical Analysis
4. Conclusions
Acknowledgments
Conflicts of Interest
- Author ContributionsC.D.S., M.Si. and S.E. conceived and designed the study; I.B., M.Se., J.H., C.H., F.N., B.R., S.E., U.F.-M. and M.Si. performed the experiments and analysed the data; I.B. and M.Se. wrote the manuscript.
References
- Ikonomidou, C.; Bosch, F.; Miksa, M.; Bittigau, P.; Vöckler, J.; Dikranian, K.; Tenkova, T.I.; Stefovska, V.; Turski, L.; Olney, J.W. Blockade of NMDA receptors and apoptotic neurodegeneration in the developing brain. Science 1999, 283, 70–74. [Google Scholar]
- Ikonomidou, C.; Stefovska, V.; Turski, L. Neuronal death enhanced by N-methyl-d-aspartate antagonists. Proc. Natl. Acad. Sci. USA 2000, 97, 12885–12890. [Google Scholar]
- Jevtovic-Todorovic, V.; Hartman, R.E.; Izumi, Y.; Benshoff, N.D.; Dikranian, K.; Zorumski, C.F.; Olney, J.W.; Wozniak, D.F. Early exposure to common anesthetic agents causes widespread neurodegeneration in the developing rat brain and persistent learning deficits. J. Neurosci. 2003, 23, 876–882. [Google Scholar]
- Dobbing, J.; Sands, J. Comparative aspects of the brain growth spurt. Early Hum. Dev. 1979, 3, 79–83. [Google Scholar]
- Reynolds, J.D.; Brien, J.F. Ethanol neurobehavioural teratogenesis and the role of l-glutamate in the fetal hippocampus. Can. J. Physiol. Pharmacol. 1995, 73, 1209–1223. [Google Scholar]
- Dzietko, M.; Felderhoff-Mueser, U.; Sifringer, M.; Krutz, B.; Bittigau, P.; Thor, F.; Heumann, R.; Bührer, C.; Ikonomidou, C.; Hansen, H.H. Erythropoietin protects the developing brain against N-methyl-d-aspartate receptor antagonist neurotoxicity. Neurobiol. Dis. 2004, 15, 177–187. [Google Scholar]
- Haberny, K.A.; Paule, M.G.; Scallet, A.C.; Sistare, F.D.; Lester, D.S.; Hanig, J.P.; Slikker, W., Jr. Ontogeny of the N-methyl-d-aspartate (NMDA) receptor system and susceptibility to neurotoxicity. Toxicol. Sci. 2002, 68, 9–17. [Google Scholar]
- Hansen, H.H.; Briem, T.; Dzietko, M.; Sifringer, M.; Voss, A.; Rzeski, W.; Zdzisinska, B.; Thor, F.; Heumann, R.; Stepulak, A.; et al. Mechanisms leading to disseminated apoptosis following NMDA receptor blockade in the developing rat brain. Neurobiol. Dis. 2004, 16, 440–453. [Google Scholar]
- Hardingham, G.E.; Bading, H. Coupling of extrasynaptic NMDA receptors to a CREB shut-off pathway is developmentally regulated. Biochim. Biophys. Acta 2002, 1600, 148–153. [Google Scholar]
- Kaindl, A.M.; Koppelstaetter, A.; Nebrich, G.; Stuwe, J.; Sifringer, M.; Zabel, C.; Klose, J.; Ikonomidou, C. Brief alteration of NMDA or GABAA receptor-mediated neurotransmission has long term effects on the developing cerebral cortex. Mol. Cell. Proteomics 2008, 7, 2293–2310. [Google Scholar]
- Stefovska, V.G.; Uckermann, O.; Czuczwar, M.; Smitka, M.; Czuczwar, P.; Kis, J.; Kaindl, A.M.; Turski, L.; Turski, W.A.; Ikonomidou, C. Sedative and anticonvulsant drugs suppress postnatal neurogenesis. Ann. Neurol. 2008, 64, 434–445. [Google Scholar]
- Gomes, L.M.; Garcia, J.B.; Ribamar, J.S., Jr; Nascimento, A.G. Neurotoxicity of subarachnoid preservative-free S(+)-ketamine in dogs. Pain Physician 2011, 14, 83–90. [Google Scholar]
- Liu, F.; Paule, M.G.; Ali, S.; Wang, C. Ketamine-induced neurotoxicity and changes in gene expression in the developing rat brain. Curr. Neuropharmacol. 2011, 9, 256–261. [Google Scholar]
- Paule, M.G.; Li, M.; Allen, R.R.; Liu, F.; Zou, X.; Hotchkiss, C.; Hanig, J.P.; Patterson, T.A.; Slikker, W., Jr; Wang, C. Ketamine anesthesia during the first week of life can cause long-lasting cognitive deficits in rhesus monkeys. Neurotoxicol. Teratol. 2011, 33, 220–230. [Google Scholar]
- Shi, Q.; Guo, L.; Patterson, T.A.; Dial, S.; Li, Q.; Sadovova, N.; Zhang, X.; Hanig, J.P.; Paule, M.G.; Slikker, W., Jr; et al. Gene expression profiling in the developing rat brain exposed to ketamine. Neuroscience 2010, 166, 852–863. [Google Scholar]
- Zou, X.; Patterson, T.A.; Divine, R.L.; Sadovova, N.; Zhang, X.; Hanig, J.P.; Paule, M.G.; Slikker, W., Jr; Wang, C. Prolonged exposure to ketamine increases neurodegeneration in the developing monkey brain. Int. J. Dev. Neurosci. 2009, 27, 727–731. [Google Scholar]
- Zou, X.; Patterson, T.A.; Sadovova, N.; Twaddle, N.C.; Doerge, D.R.; Zhang, X.; Fu, X.; Hanig, J.P.; Paule, M.G.; Slikker, W.; et al. Potential neurotoxicity of ketamine in the developing rat brain. Toxicol. Sci. 2009, 108, 149–158. [Google Scholar]
- Soreq, H.; Seidman, S. Acetylcholinesterase-new roles for an old actor. Nat. Rev. Neurosci. 2001, 2, 294–302. [Google Scholar]
- Bigbee, J.W.; Sharma, K.V. The adhesive role of acetylcholinesterase (AChE): Detection of AChE binding proteins in developing rat spinal cord. Neurochem. Res. 2004, 29, 2043–2050. [Google Scholar]
- Bigbee, J.W.; Sharma, K.V.; Chan, E.L.; Bogler, O. Evidence for the direct role of acetylcholinesterase in neurite outgrowth in primary dorsal root ganglion neurons. Brain Res. 2000, 861, 354–362. [Google Scholar]
- Byers, D.M.; Irwin, L.N.; Moss, D.E.; Sumaya, I.C.; Hohmann, C.F. Prenatal exposure to the acetylcholinesterase inhibitor methanesulfonyl fluoride alters forebrain morphology and gene expression. Dev. Brain Res. 2005, 158, 13–22. [Google Scholar]
- Dori, A.; Cohen, J.; Silverman, W.F.; Pollack, Y.; Soreq, H. Functional manipulations of acetylcholinesterase splice variants highlight alternative splicing contributions to murine neocortical development. Cereb. Cortex. 2005, 15, 419–430. [Google Scholar]
- Slotkin, T.A.; Ryde, I.T.; Wrench, N.; Card, J.A.; Seidler, F.J. Nonenzymatic role of acetylcholinesterase in neuritic sprouting: Regional changes in acetylcholinesterase and choline acetyltransferase after neonatal 6-hydroxydopamine lesions. Neurotoxicol. Teratol. 2009, 31, 183–186. [Google Scholar]
- Csernansky, J.G.; Martin, M.; Shah, R.; Bertchume, A.; Colvin, J.; Dong, H. Cholinesterase inhibitors ameliorate behavioral deficits induced by MK-801 in mice. Neuropsychopharmacology 2005, 30, 2135–2143. [Google Scholar]
- Su, Y.A.; Huang, R.H.; Wang, X.D.; Li, J.T.; Si, T.M. Impaired working memory by repeated neonatal MK-801 treatment is ameliorated by galantamine in adult rats. Eur. J. Pharmacol. 2014, 725, 32–39. [Google Scholar]
- Sifringer, M.; Bendix, I.; von Haefen, C.; Endesfelder, S.; Kalb, A.; Bührer, C.; Felderhoff-Mueser, U.; Spies, C.D. Oxygen toxicity is reduced by acetylcholinesterase inhibition in the developing rat brain. Dev. Neurosci. 2013, 35, 255–264. [Google Scholar]
- Leyhe, T.; Stransky, E.; Eschweiler, G.W.; Buchkremer, G.; Laske, C. Increase of BDNF serum concentration during donepezil treatment of patients with early alzheimer’s disease. Eur. Arch. Psychiatry Clin. Neurosci. 2008, 258, 124–128. [Google Scholar]
- Wang, Z.F.; Tang, L.L.; Yan, H.; Wang, Y.J.; Tang, X.C. Effects of huperzine A on memory deficits and neurotrophic factors production after transient cerebral ischemia and reperfusion in mice. Pharmacol. Biochem. Behav. 2006, 83, 603–611. [Google Scholar]
- Almli, C.R.; Levy, T.J.; Han, B.H.; Shah, A.R.; Gidday, J.M.; Holtzman, D.M. BDNF protects against spatial memory deficits following neonatal hypoxia-ischemia. Exp. Neurol. 2000, 166, 99–114. [Google Scholar]
- Barco, A.; Patterson, S.L.; Alarcon, J.M.; Gromova, P.; Mata-Roig, M.; Morozov, A.; Kandel, E.R. Gene expression profiling of facilitated L-LTP in VP16-CREB mice reveals that BDNF is critical for the maintenance of LTP and its synaptic capture. Neuron 2005, 48, 123–137. [Google Scholar]
- Marini, A.M.; Jiang, X.; Wu, X.; Pan, H.; Guo, Z.; Mattson, M.P.; Blondeau, N.; Novelli, A.; Lipsky, R.H. Preconditioning and neurotrophins: A model for brain adaptation to seizures ischemia and other stressful stimuli. Amino Acids 2007, 32, 299–304. [Google Scholar]
- Poo, M.M. Neurotrophins as synaptic modulators. Nat. Rev. Neurosci. 2001, 2, 24–32. [Google Scholar]
- Nikolaou, K.E.; Malamitsi-Puchner, A.; Boutsikou, T.; Economou, E.; Boutsikou, M.; Puchner, K.P.; Baka, S.; Hassiakos, D. The varying patterns of neurotrophin changes in the perinatal period. Ann. N. Y. Acad. Sci. 2006, 1092, 426–433. [Google Scholar]
- Webster, M.J.; Herman, M.M.; Kleinman, J.E.; Shannon Weickert, C. BDNF and trkB mRNA expression in the hippocampus and temporal cortex during the human lifespan. Gene Expr. Patterns 2006, 6, 941–951. [Google Scholar]
- Asahi, M.; Wang, X.; Mori, T.; Sumii, T.; Jung, J.C.; Moskowitz, M.A.; Fini, M.E.; Lo, E.H. Effects of matrix metalloproteinase-9 gene knock-out on the proteolysis of blood-brain barrier and white matter components after cerebral ischemia. J. Neurosci. 2001, 21, 7724–7732. [Google Scholar]
- Heo, J.H.; Lucero, J.; Abumiya, T.; Koziol, J.A.; Copeland, B.R.; del Zoppo, G.J. Matrix metalloproteinases increase very early during experimental focal cerebral ischemia. J. Cereb. Blood Flow Metab. 1999, 19, 624–633. [Google Scholar]
- Lo, E.H.; Dalkara, T.; Moskowitz, M.A. Mechanisms challenges and opportunities in stroke. Nat. Rev. Neurosci. 2003, 4, 399–415. [Google Scholar]
- Lukashev, M.E.; Werb, Z. ECM signalling: Orchestrating cell behaviour and misbehaviour. Trends Cell Biol. 1998, 8, 437–441. [Google Scholar]
- Montaner, J.; Alvarez-Sabin, J.; Molina, C.; Angles, A.; Abilleira, S.; Arenillas, J.; Gonzalez, M.A.; Monasterio, J. Matrix metalloproteinase expression after human cardioembolic stroke: Temporal profile and relation to neurological impairment. Stroke 2001, 32, 1759–1766. [Google Scholar]
- Morita-Fujimura, Y.; Fujimura, M.; Gasche, Y.; Copin, J.C.; Chan, P.H. Overexpression of copper and zinc superoxide dismutase in transgenic mice prevents the induction and activation of matrix metalloproteinases after cold injury-induced brain trauma. J. Cereb. Blood Flow Metab. 2000, 20, 130–138. [Google Scholar]
- Romanic, A.M.; White, R.F.; Arleth, A.J.; Ohlstein, E.H.; Barone, F.C. Matrix metalloproteinase expression increases after cerebral focal ischemia in rats: Inhibition of matrix metalloproteinase-9 reduces infarct size. Stroke 1998, 29, 1020–1030. [Google Scholar]
- Shigemori, Y.; Katayama, Y.; Mori, T.; Maeda, T.; Kawamata, T. Matrix metalloproteinase-9 is associated with blood-brain barrier opening and brain edema formation after cortical contusion in rats. Acta Neurochir. Suppl. 2006, 96, 130–133. [Google Scholar]
- Yong, V.W. Metalloproteinases: Mediators of pathology and regeneration in the CNS. Nat. Rev. Neurosci. 2005, 6, 931–944. [Google Scholar]
- Svedin, P.; Hagberg, H.; Savman, K.; Zhu, C.; Mallard, C. Matrix metalloproteinase-9 gene knock-out protects the immature brain after cerebral hypoxia-ischemia. J. Neurosci. 2007, 27, 1511–1518. [Google Scholar]
- Zhao, B.Q.; Tejima, E.; Lo, E.H. Neurovascular proteases in brain injury hemorrhage and remodeling after stroke. Stroke 2007, 38, 748–752. [Google Scholar]
- Munkeby, B.H.; Børke, W.B.; Bjørnland, K.; Sikkeland, L.I.; Borge, G.I.; Halvorsen, B.; Saugstad, O.D. Resuscitation with 100% O2 increases cerebral injury in hypoxemic piglets. Pediatr. Res. 2004, 56, 783–790. [Google Scholar]
- Richards, J.G.; Todd, K.G.; Emara, M.; Haase, E.; Cooper, S.L.; Bigam, D.L.; Cheung, P.Y. A dose-response study of graded reoxygenation on the carotid haemodynamics matrix metalloproteinase-2 activities and amino acid concentrations in the brain of asphyxiated newborn piglets. Resuscitation 2006, 69, 319–327. [Google Scholar]
- Zhao, B.Q.; Wang, S.; Kim, H.Y.; Storrie, H.; Rosen, B.R.; Mooney, D.J.; Wang, X.; Lo, E.H. Role of matrix metalloproteinases in delayed cortical responses after stroke. Nat. Med. 2006, 12, 441–445. [Google Scholar]
- Mannello, F.; Gazzanelli, G. Tissue inhibitors of metalloproteinases and programmed cell death: Conundrums controversies and potential implications. Apoptosis 2001, 6, 479–482. [Google Scholar]
- Yong, V.W.; Power, C.; Forsyth, P.; Edwards, D.R. Metalloproteinases in biology and pathology of the nervous system. Nat. Rev. Neurosci. 2001, 2, 502–511. [Google Scholar]
- Kalb, A.; von Haefen, C.; Sifringer, M.; Tegethoff, A.; Paeschke, N.; Kostova, M.; Feldheiser, A.; Spies, C.D. Acetylcholinesterase inhibitors reduce neuroinflammation and -degeneration in the cortex and hippocampus of a surgery stress rat model. PLoS One 2013, 8, e62679. [Google Scholar]
- Kaufer, D.; Friedman, A.; Seidman, S.; Soreq, H. Acute stress facilitates long-lasting changes in cholinergic gene expression. Nature 1998, 393, 373–377. [Google Scholar]
- Meshorer, E.; Erb, C.; Gazit, R.; Pavlovsky, L.; Kaufer, D.; Friedman, A.; Glick, D.; Ben-Arie, N.; Soreq, H. Alternative splicing and neuritic mRNA translocation under long-term neuronal hypersensitivity. Science 2002, 295, 508–512. [Google Scholar]
- Zhang, X.J.; Greenberg, D.S. Acetylcholinesterase involvement in apoptosis. Front. Mol. Neurosci. 2012, 5, 40–50. [Google Scholar]
- Hwang, J.J.; Park, M.H.; Choi, S.Y.; Koh, J.Y. Activation of the Trk signaling pathway by extracellular zinc Role of metalloproteinases. J. Biol. Chem. 2005, 280, 11995–12001. [Google Scholar]
- Dikranian, K.; Ishimaru, M.J.; Tenkova, T.; Labruyere, J.; Qin, Y.Q.; Ikonomidou, C.; Olney, J.W. Apoptosis in the in vivo mammalian forebrain. Neurobiol. Dis. 2001, 8, 359–379. [Google Scholar]
- Knopman, D.S.; Morris, J.C. An update on primary drug therapies for Alzheimer disease. Arch. Neurol. 1997, 54, 1406–1409. [Google Scholar]
- Zhao, B.; Moochhala, S.M.; Tham, S.Y. Biologically active components of physostigma venenosum. J. Chromatogr. 2004, 812, 183–192. [Google Scholar]
- Lorke, D.E.; Hasan, M.Y.; Nurulain, S.M.; Shafiullah, M.; Kuca, K.; Petroianu, G.A. Pretreatment for acute exposure to diisopropylfluorophosphate: In vivo efficacy of various acetylcholinesterase inhibitors. J. Appl. Toxicol. 2011, 515–531. [Google Scholar]
- Hofer, S.; Eisenbach, C.; Lukic, I.K.; Schneider, L.; Bode, K.; Brueckmann, M.; Mautner, S.; Wente, M.N.; Encke, J.; Werner, J.; et al. Pharmacologic cholinesterase inhibition improves survival in experimental sepsis. Crit. Care Med. 2008, 36, 404–408. [Google Scholar]
- Nordberg, A. Mechanisms behind the neuroprotective actions of cholinesterase inhibitors in alzheimer disease. Alzheimer Dis. Assoc. Disord. 2006, 20, S12–S18. [Google Scholar]
- Takada-Takatori, Y.; Kume, T.; Izumi, Y.; Ohgi, Y.; Niidome, T.; Fujii, T.; Sugimoto, H.; Akaike, A. Roles of nicotinic receptors in acetylcholinesterase inhibitor-induced neuroprotection and nicotinic receptor up-regulation. Biol. Pharm. Bull. 2009, 32, 318–324. [Google Scholar]
- Muthuraju, S.; Maiti, P.; Solanki, P.; Sharma, A.K.; Amitabh Singh, S.B.; Prasad, D.; Ilavazhagan, G. Acetylcholinesterase inhibitors enhance cognitive functions in rats following hypobaric hypoxia. Behav. Brain Res. 2009, 203, 1–14. [Google Scholar]
- Shaked, I.; Meerson, A.; Wolf, Y.; Avni, R.; Greenberg, D.; Gilboa-Geffen, A.; Soreq, H. MicroRNA-132 potentiates cholinergic anti-inflammatory signaling by targeting acetylcholinesterase. Immunity 2009, 31, 965–973. [Google Scholar]
- Shaltiel, G.; Hanan, M.; Wolf, Y.; Barbash, S.; Kovalev, E.; Shoham, S.; Soreq, H. Hippocampal microRNA-132 mediates stress-inducible cognitive deficits through its acetylcholinesterase target. Brain Struct. Funct. 2013, 218, 59–72. [Google Scholar]
- Soreq, H.; Wolf, Y. NeurimmiRs: MicroRNAs in the neuroimmune interface. Trends Mol. Med. 2011, 17, 548–555. [Google Scholar]
- Amantea, D.; Corasaniti, M.T.; Mercuri, N.B.; Bernardi, G.; Bagetta, G. Brain regional and cellular localization of gelatinase activity in rat that have undergone transient middle cerebral artery occlusion. Neuroscience 2008, 152, 8–17. [Google Scholar]
- Chen, W.; Hartman, R.; Ayer, R.; Marcantonio, S.; Kamper, J.; Tang, J.; Zhang, J.H. Matrix metalloproteinases inhibition provides neuroprotection against hypoxia-ischemia in the developing brain. J. Neurochem. 2009, 111, 726–736. [Google Scholar]
- Sifringer, M.; Stefovska, V.; Zentner, I.; Hansen, B.; Stepulak, A.; Knaute, C.; Marzahn, J.; Ikonomidou, C. The role of matrix metalloproteinases in infant traumatic brain injury. Neurobiol. Dis. 2007, 25, 526–535. [Google Scholar]
- Uckermann, O.; Luksch, H.; Stefovska, V.; Hoehna, Y.; Marzahn, J.; Theil, M.; Pesic, M.; Gorkiewicz, T.; Gawlak, M.; Wilczynski, G.M.; et al. Matrix metalloproteinases 2 and 9 fail to influence drug-induced neuroapoptosis in developing rat brain. Neurotox. Res. 2011, 19, 638–648. [Google Scholar]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Oligonucleotide sequences 5′-3′ | ||
|---|---|---|---|
| BDNF | Forward | TCAGCAGTCAAGTGCCTTTGG | M61175 |
| reverse | CGCCGAACCCTCATAGACATG | ||
| probe | CCTCCTCTGCTCTTTCTGCTGGAGGAATACAA | ||
| TIMP-2 | Forward | GGCAACCCCATCAAGAGGAT | NM_021989 |
| reverse | GGGCCGTGTAGATAAATTCGAT | ||
| probe | AGATGTTCAAAGGACCTGAC | ||
| β-actin | Forward | GTACAACCTCCTTGCAGCTCCT | NM_031144 |
| reverse | TTGTCGACGACGACGGC | ||
| probe | CGCCACCAGTTCGCCATGGAT | ||
© 2014 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Bendix, I.; Serdar, M.; Herz, J.; Von Haefen, C.; Nasser, F.; Rohrer, B.; Endesfelder, S.; Felderhoff-Mueser, U.; Spies, C.D.; Sifringer, M. Inhibition of Acetylcholinesterase Modulates NMDA Receptor Antagonist Mediated Alterations in the Developing Brain. Int. J. Mol. Sci. 2014, 15, 3784-3798. https://doi.org/10.3390/ijms15033784
Bendix I, Serdar M, Herz J, Von Haefen C, Nasser F, Rohrer B, Endesfelder S, Felderhoff-Mueser U, Spies CD, Sifringer M. Inhibition of Acetylcholinesterase Modulates NMDA Receptor Antagonist Mediated Alterations in the Developing Brain. International Journal of Molecular Sciences. 2014; 15(3):3784-3798. https://doi.org/10.3390/ijms15033784
Chicago/Turabian StyleBendix, Ivo, Meray Serdar, Josephine Herz, Clarissa Von Haefen, Fatme Nasser, Benjamin Rohrer, Stefanie Endesfelder, Ursula Felderhoff-Mueser, Claudia D. Spies, and Marco Sifringer. 2014. "Inhibition of Acetylcholinesterase Modulates NMDA Receptor Antagonist Mediated Alterations in the Developing Brain" International Journal of Molecular Sciences 15, no. 3: 3784-3798. https://doi.org/10.3390/ijms15033784
APA StyleBendix, I., Serdar, M., Herz, J., Von Haefen, C., Nasser, F., Rohrer, B., Endesfelder, S., Felderhoff-Mueser, U., Spies, C. D., & Sifringer, M. (2014). Inhibition of Acetylcholinesterase Modulates NMDA Receptor Antagonist Mediated Alterations in the Developing Brain. International Journal of Molecular Sciences, 15(3), 3784-3798. https://doi.org/10.3390/ijms15033784