Interactive Association of Drugs Binding to Human Serum Albumin

Abstract
:1. Introduction
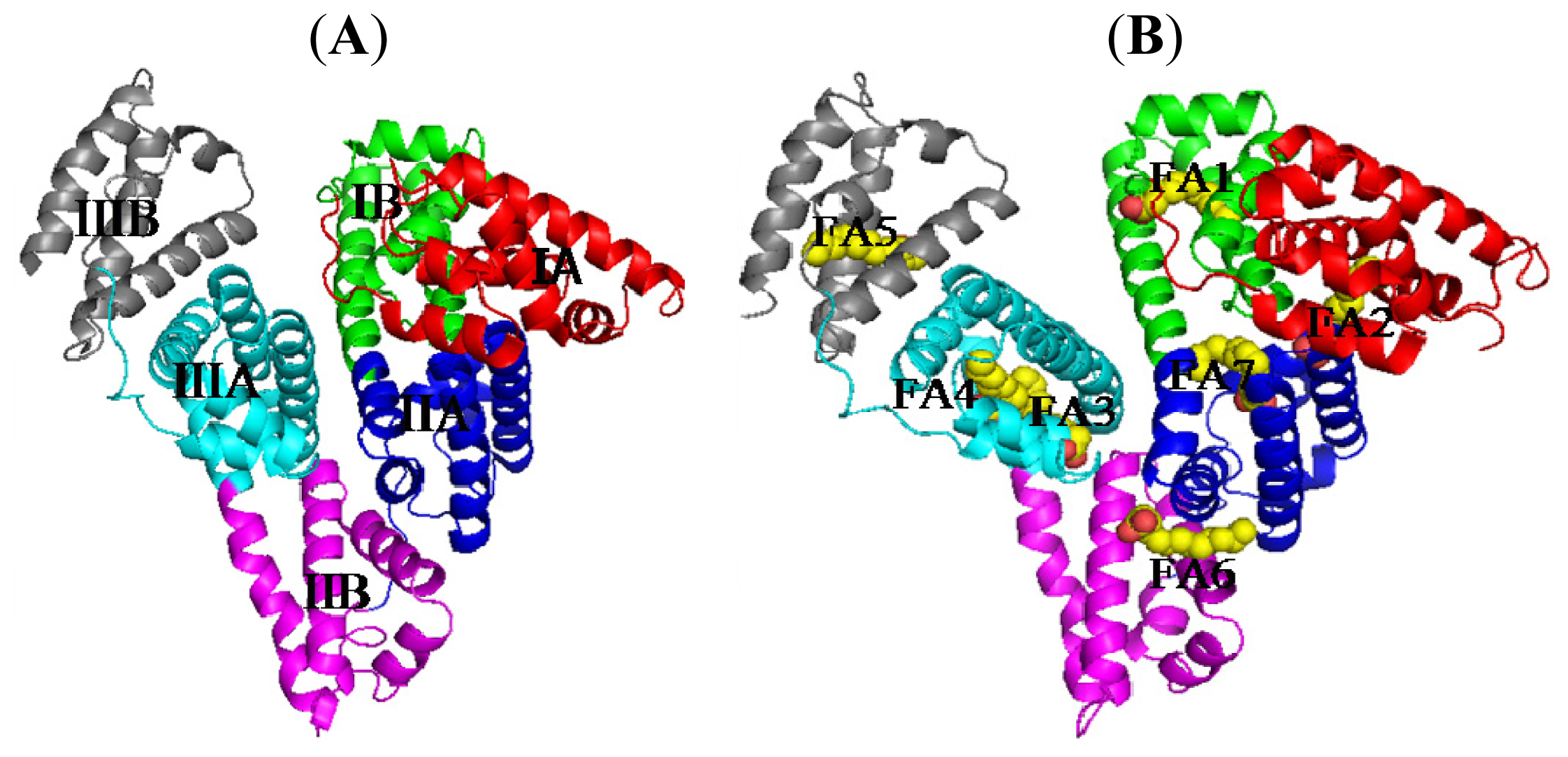
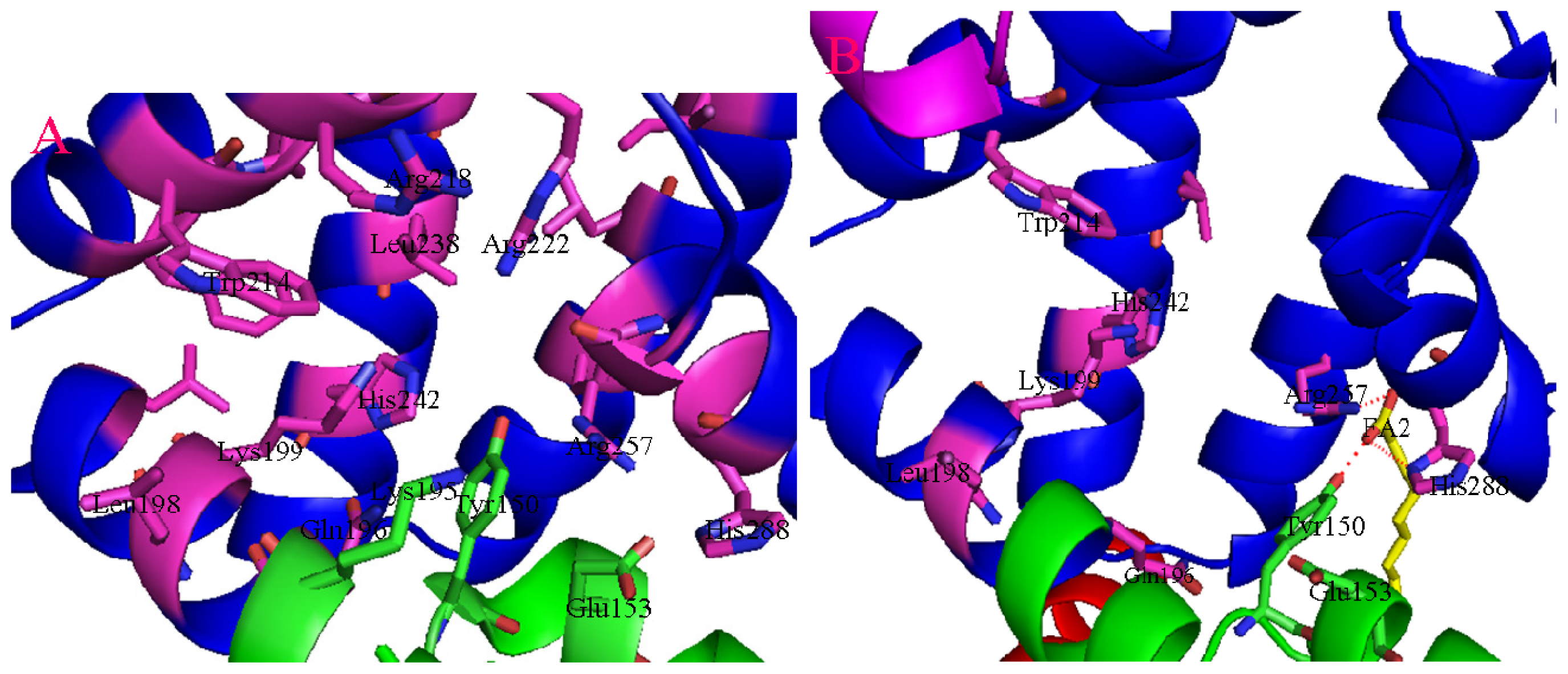
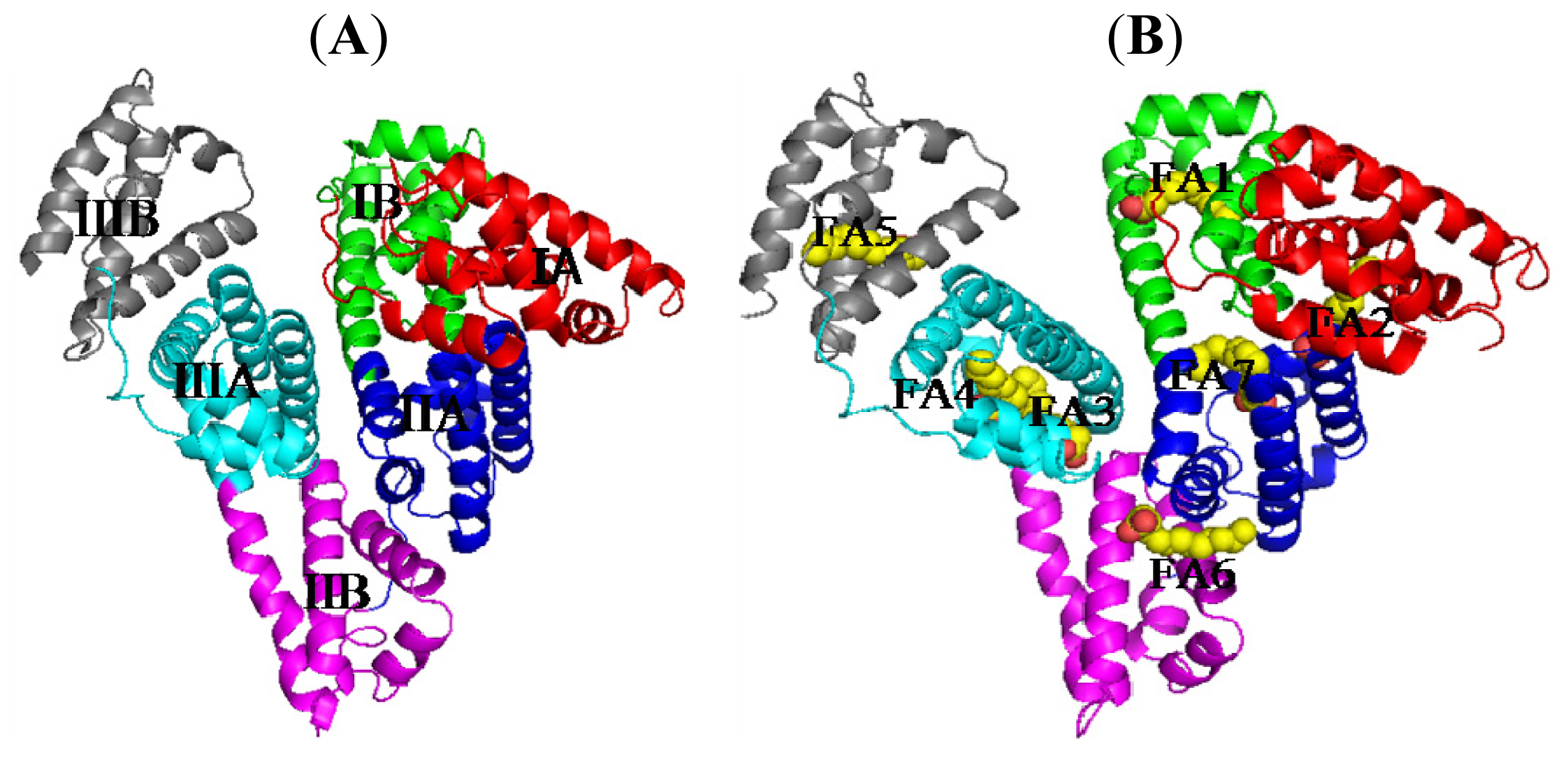
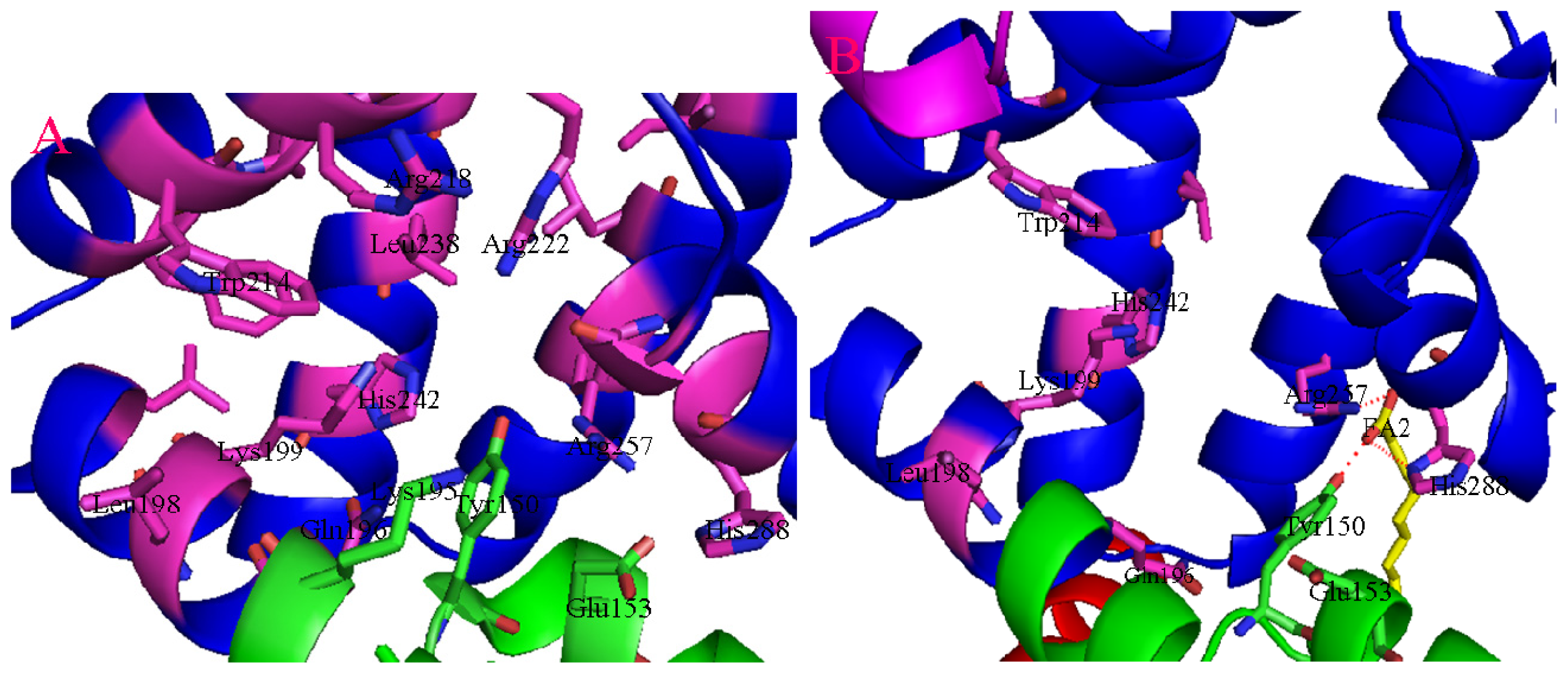
2. Binding Site of Drugs in IIA Subdomain of HSA
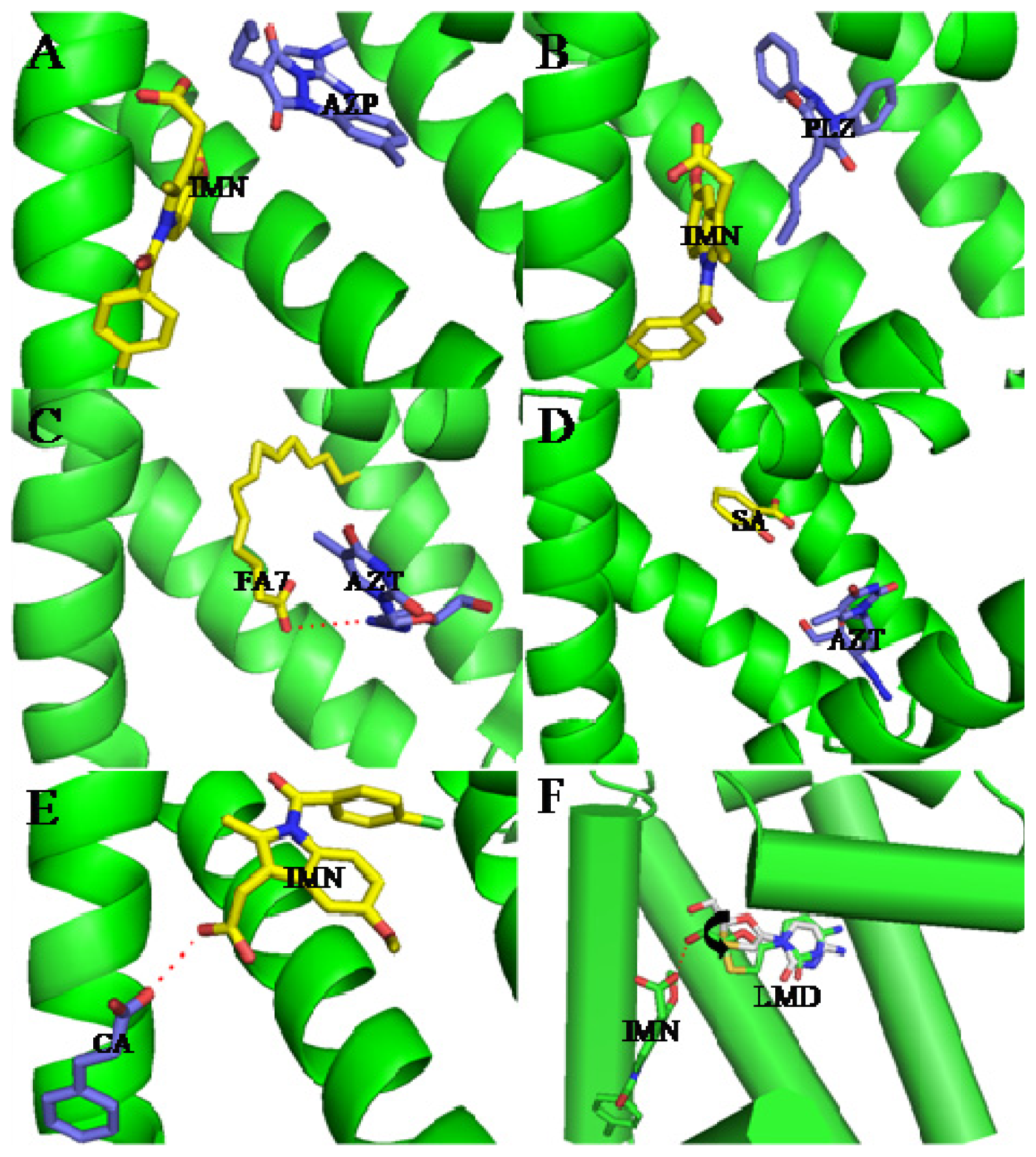
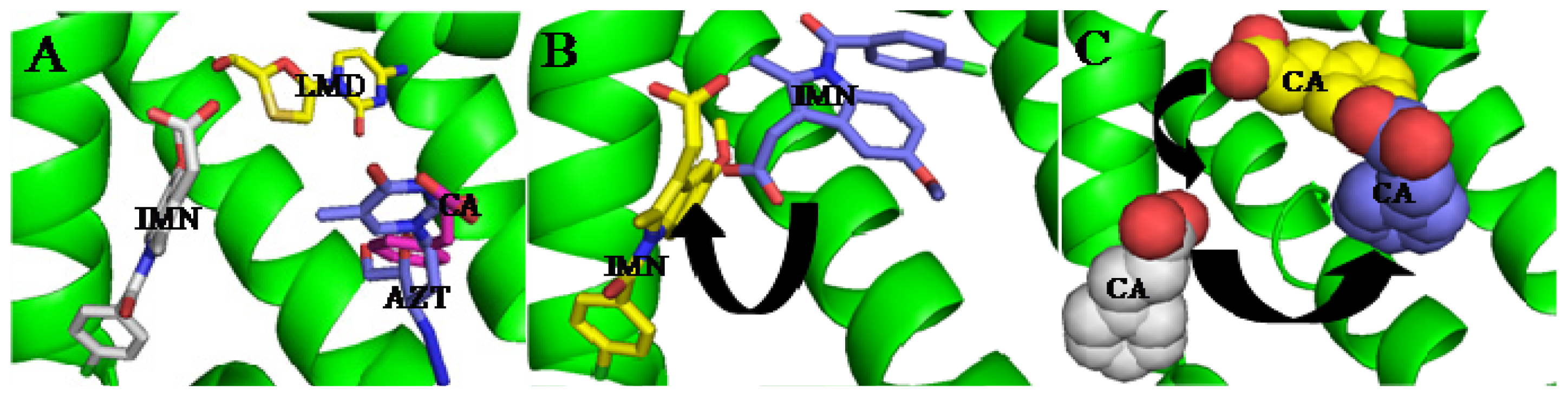
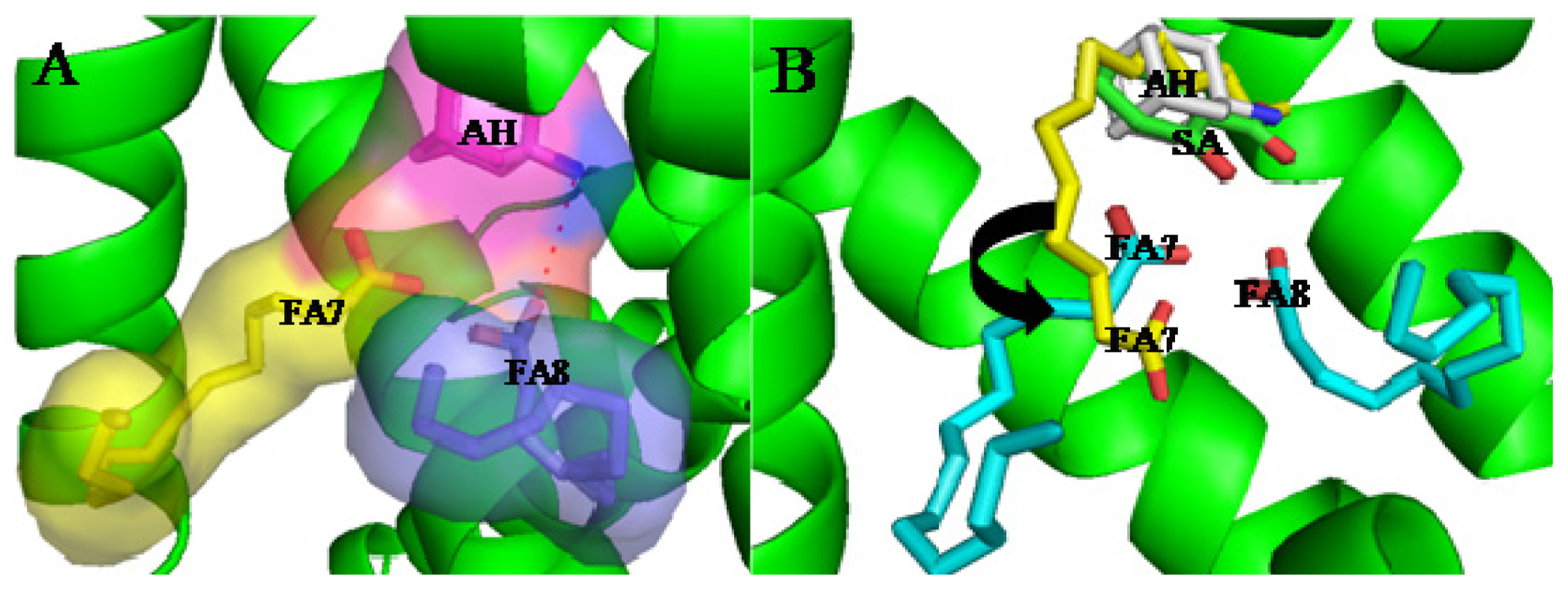
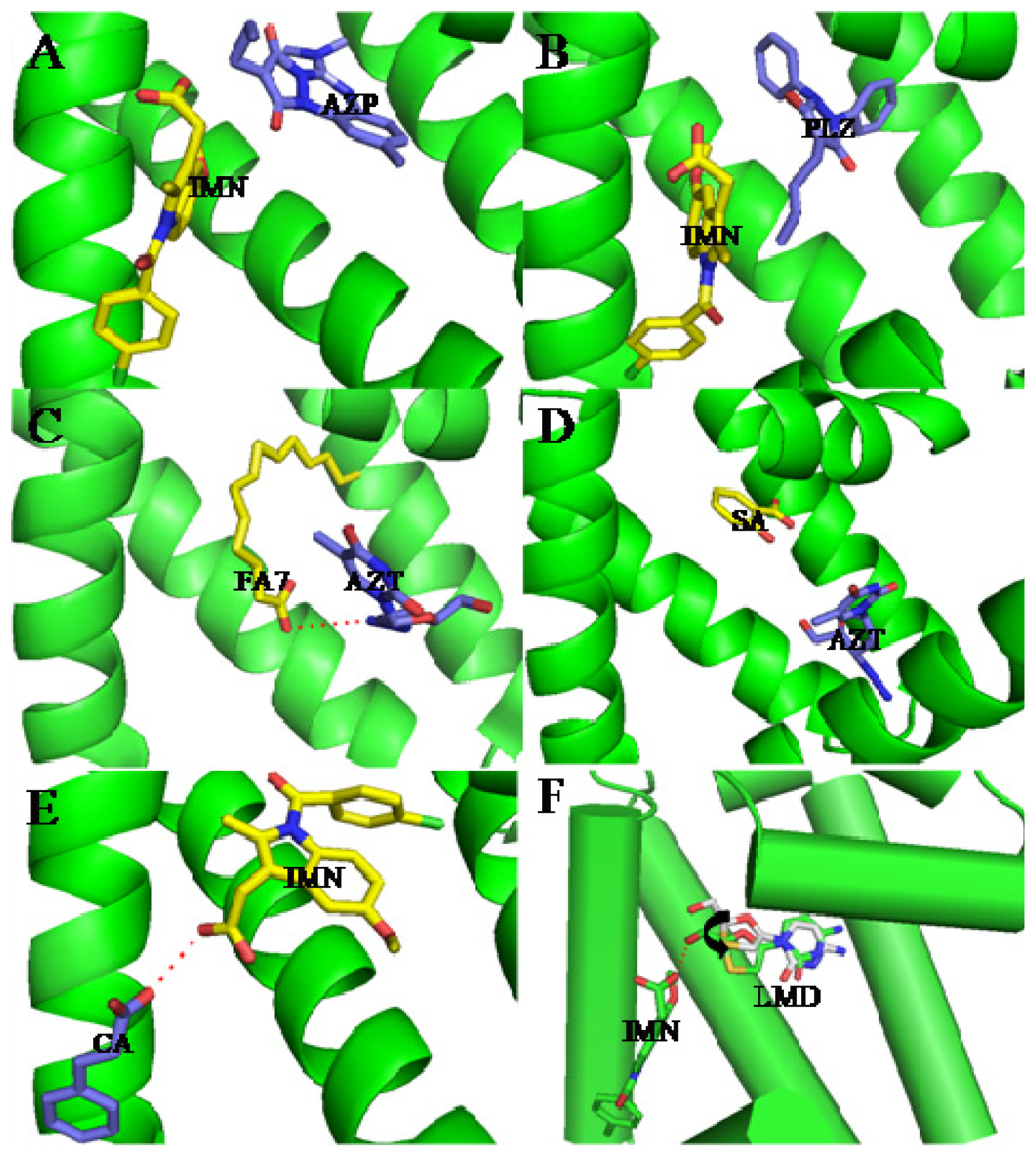
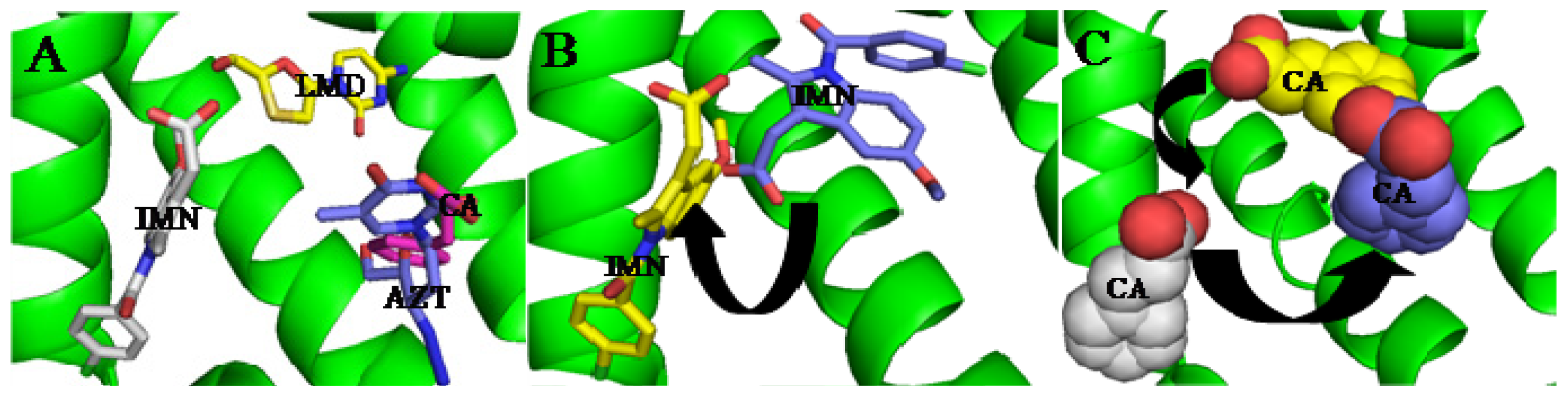
3. Interactive Association of Drug–Drug with HSA
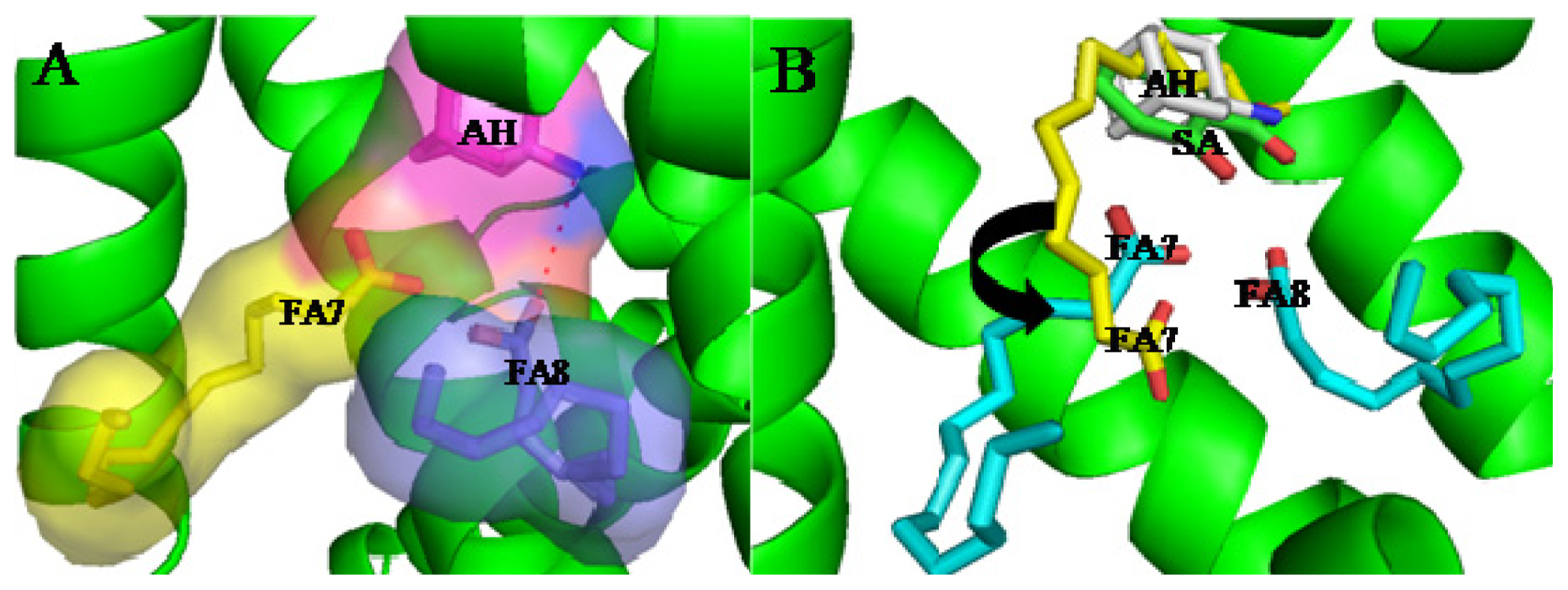
4. Interactive Association of Drug–Drug–Drug with HSA
5. Conclusions
Acknowledgments
Conflicts of Interest
References
- Peters, T. All about Albumin: Biochemistry, Genetics, and Medical Applications; Academic Press: San Diego, CA, USA, 1996. [Google Scholar]
- Fanali, G.; di Masi, A.; Trezza, V.; Marino, M.; Fasano, M.; Ascenzi, P. Human serum albumin: From bench to bedside. Mol. Asp. Med. 2012, 33, 209–290. [Google Scholar]
- Fasano, M.; Curry, S.; Terreno, E.; Galliano, M.; Fanali, G.; Narciso, P.; Notari, S.; Ascenzi, P. The extraordinary ligand binding properties of human serum albumin. IUBMB Life 2005, 57, 787–796. [Google Scholar]
- Ha, C.E.; Bhagavan, N.V. Novel insights into the pleiotropic effects of human serum albumin in health and disease. Biochim. Biophys. Acta 2013, 1830, 5486–5493. [Google Scholar]
- Kragh-Hansen, U.; Chuang, V.T.; Otagiri, M. Practical aspects of the ligand-binding and enzymatic properties of human serum albumin. Biol. Pharm. Bull. 2002, 25, 695–704. [Google Scholar]
- Bal, W.; Sokołowska, M.; Kurowska, E.; Faller, P. Binding of transition metal ions to albumin: Sites affinities and rates. Biochim. Biophys. Acta 2013, 1830, 5444–5455. [Google Scholar]
- Yamasaki, K.; Chuang, V.T.; Maruyama, T.; Otagiri, M. Albumin–drug interaction and its clinical implication. Biochim. Biophys. Acta 2013, 1830, 5435–5443. [Google Scholar]
- Otagiri, M. A molecular functional study on the interactions of drugs with plasma proteins. Drug Metab. Pharmacokinet. 2005, 20, 309–323. [Google Scholar]
- Ascenzi, P.; Bocedi, A.; Notari, S.; Fanali, G.; Fesce, R.; Fasano, M. Allosteric modulation of drug binding to human serum albumin. Mini Rev. Med. Chem. 2006, 6, 483–489. [Google Scholar]
- Bertucci, C.; Domenici, E. Reversible and covalent binding of drugs to human serum albumin: Methodological approaches and physiological relevance. Curr. Med. Chem. 2002, 9, 1463–1481. [Google Scholar]
- Colmenarejo, G. In silico prediction of drug-binding strengths to human serum albumin. Med. Res. Rev. 2003, 23, 275–301. [Google Scholar]
- Kawakami, A.; Kubota, K.; Yamada, N.; Tagami, U.; Takehana, K.; Sonaka, I.; Suzuki, E.; Hirayama, K. Identification and characterization of oxidized human serum albumin. FEBS J. 2006, 273, 3346–3357. [Google Scholar]
- Sleep, D.; Cameron, J.; Evans, L.R. Albumin as a versatile platform for drug half-life extension. Biochim. Biophys. Acta 2013, 1830, 5526–5534. [Google Scholar]
- Sugio, S.; Kashima, A.; Mochizuki, S.; Noda, M.; Kobayashi, K. Crystal structure of human serum albumin at 25 Å resolution. Protein Eng. 1999, 12, 439–446. [Google Scholar]
- Rowland, M.; Tozer, T.N. Clinical Pharmacokinetics and Pharmacodynamics: Concepts and Applications, 4th ed.; Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2010. [Google Scholar]
- Koch-Weser, J.; Sellers, E.M. Binding of drugs to serum albumin (first of two parts). N. Engl. J. Med. 1976, 294, 311–316. [Google Scholar]
- Smith, D.A.; Di, L.; Kerns, E.H. The effect of plasma protein binding on in vivo efficacy: Misconceptions in drug discovery. Nat. Rev. Drug Discov. 2010, 9, 929–939. [Google Scholar]
- Vallner, J.J. Binding of drugs by albumin and plasma protein. J. Pharm. Sci. 1977, 66, 447–465. [Google Scholar]
- Otagiri, M. Study on binding of drug to serum protein. Yakugaku Zasshi 2009, 129, 413–425. [Google Scholar]
- Meyer, M.C.; Guttman, D.E. The binding of drugs by plasma proteins. J. Pharm. Sci. 1968, 57, 895–918. [Google Scholar]
- Tillement, J.P.; Duché, J.C.; Barré, J. Drug binding to blood proteins: Characteristics roles and pathophysiological changes. Bull. Acad. Natl. Med. 2006, 190, 935–946. [Google Scholar]
- Jusko, W.J.; Gretch, M. Plasma and tissue protein binding of drugs in pharmacokinetics. Drug Metab. Rev. 1976, 5, 43–140. [Google Scholar]
- Frostell-Karlsson, A.; Remaeus, A.; Roos, H.; Andersson, K.; Borg, P.; Hämäläinen, M.; Karlsson, R. Biosensor analysis of the interaction between immobilized human serum albumin and drug compounds for prediction of human serum albumin binding levels. J. Med. Chem. 2000, 43, 1986–1992. [Google Scholar]
- Ascoli, G.A.; Bertucci, C.; Salvadori, P. Ligand binding to a human serum albumin stationary phase: Use of same-drug competition to discriminate pharmacologically relevant interactions. Biomed. Chromatogr. 1998, 12, 248–254. [Google Scholar]
- Watanabe, H.; Tanase, S.; Nakajou, K.; Maruyama, T.; Kragh-Hansen, U.; Otagiri, M. Role of arg-410 and tyr-411 in human serum albumin for ligand binding and esterase-like activity. Biochem. J. 2000, 349, 813–819. [Google Scholar]
- Ahmad, B.; Parveen, S.; Khan, R.H. Effect of albumin conformation on the binding of ciprofloxacin to human serum albumin: A novel approach directly assigning binding site. Biomacromolecules 2006, 7, 1350–1356. [Google Scholar]
- Sinha, S.S.; Mitra, R.K.; Pal, S.K. Temperature-dependent simultaneous ligand binding in human serum albumin. J. Phys. Chem. B 2008, 112, 4884–4891. [Google Scholar]
- Lucas, L.H.; Price, K.E.; Larive, C.K. Epitope mapping and competitive binding of HSA drug site II ligands by NMR diffusion measurements. J. Am. Chem. Soc. 2004, 126, 14258–14266. [Google Scholar]
- Colmenarejo, G.; Alvarez-Pedraglio, A.; Lavandera, J.L. Cheminformatic models to predict binding affinities to human serum albumin. J. Med. Chem. 2001, 44, 4370–4378. [Google Scholar]
- Mahesha, H.G.; Singh, S.A.; Srinivasan, N.; Rao, A.G. A spectroscopic study of the interaction of isoflavones with human serum albumin. FEBS J. 2006, 273, 451–467. [Google Scholar]
- Garg, A.; Manidhar, D.M.; Gokara, M.; Malleda, C.; Reddy, C.S.; Subramanyam, R. Elucidation of the binding mechanism of coumarin derivatives with human serum albumin. PLoS One 2013, 8, e63805. [Google Scholar]
- Abou-Zied, O.K.; AlShihi, O.I. Characterization of subdomain IIA binding site of human serum albumin in its native unfolded and refolded states using small molecular probes. J. Am. Chem. Soc. 2008, 130, 10793–10801. [Google Scholar]
- Liang, H.; Huang, J.; Tu, C.Q.; Zhang, M.; Zhou, Y.Q.; Shen, P.W. The subsequent effect of interaction between Co2+ and human serum albumin or bovine serum albumin. J. Inorg. Biochem. 2001, 85, 167–171. [Google Scholar]
- Shen, X.C.; Liang, H.; Guo, J.H.; Song, C.; He, X.W.; Yuan, Y.Z. Studies on the interaction between Ag+ and human serum albumin. J. Inorg. Biochem. 2003, 95, 124–130. [Google Scholar]
- Stewart, A.J.; Blindauer, C.A.; Berezenko, S.; Sleep, D.; Sadler, P.J. Interdomain zinc site on human albumin. Proc. Natl. Acad. Sci. USA 2003, 100, 3701–3706. [Google Scholar]
- Blindauer, C.A.; Harvey, I.; Bunyan, K.E.; Stewart, A.J.; Sleep, D.; Harrison, D.J.; Berezenko, S.; Sadler, P.J. Structure properties and engineering of the major zinc binding site on human albumin. J. Biol. Chem. 2009, 284, 23116–23124. [Google Scholar]
- Carter, D.C.; He, X.M.; Munson, S.H.; Twigg, P.D.; Gernert, K.M.; Broom, M.B.; Miller, T.Y. Three-dimensional structure of human serum albumin. Science 1989, 244, 1195–1198. [Google Scholar]
- Carter, D.C.; He, X.M. Structure of human serum albumin. Science 1990, 249, 302–303. [Google Scholar]
- Carter, D.C.; Ho, J.X. Structure of serum albumin. Adv. Protein Chem. 1994, 45, 153–203. [Google Scholar]
- He, X.M.; Carter, D.C. Atomic structure and chemistry of human serum albumin. Nature 1992, 358, 209–215. [Google Scholar]
- Curry, S.; Mandelkow, H.; Brick, P.; Franks, N. Crystal structure of human serum albumin complexed with fatty acid reveals an asymmetric distribution of binding sites. Nat. Struct. Biol. 1998, 5, 827–835. [Google Scholar]
- Wardell, M.; Wang, Z.; Ho, J.X.; Robert, J.; Ruker, F.; Ruble, J.; Carter, D.C. The atomic structure of human methemalbumin at 19 Å. Biochem. Biophys. Res. Commun. 2002, 291, 813–819. [Google Scholar]
- Wang, Z.M.; Ho, J.X.; Ruble, J.R.; Rose, J.; Rüker, F.; Ellenburg, M.; Murphy, R.; Click, J.; Soistman, E.; Wilkerson, L.; et al. Structural studies of several clinically important oncology drugs in complex with human serum albumin. Biochim. Biophys. Acta 2013, 1830, 5356–5374. [Google Scholar]
- Lejon, S.; Frick, I.M.; Björck, L.; Wikström, M.; Svensson, S. Crystal structure and biological implications of a bacterial albumin binding module in complex with human serum albumin. J. Biol. Chem. 2004, 279, 42924–42928. [Google Scholar]
- Lejon, S.; Cramer, J.F.; Nordberg, P. Structural basis for the binding of naproxen to human serum albumin in the presence of fatty acids and the GA module. Acta Crystallogr. Sect. F 2008, 64, 64–69. [Google Scholar]
- Bhattacharya, A.A.; Curry, S.; Franks, N.P. Binding of the general anesthetics propofol and halothane to human serum albumin High resolution crystal structures. J. Biol. Chem. 2000, 275, 38731–38738. [Google Scholar]
- Zunszain, P.A.; Ghuman, J.; Komatsu, T.; Tsuchida, E.; Curry, S. Crystal structural analysis of human serum albumin complexed with hemin and fatty acid. BMC Struct. Biol. 2003, 3, 6. [Google Scholar]
- Ryan, A.J.; Chung, C.W.; Curry, S. Crystallographic analysis reveals the structural basis of the high-affinity binding of iophenoxic acid to human serum albumin. BMC Struct. Biol. 2011, 11, 18. [Google Scholar]
- Bhattacharya, A.A.; Grüne, T.; Curry, S. Crystallographic analysis reveals common modes of binding of medium and long-chain fatty acids to human serum albumin. J. Mol. Biol. 2000, 303, 721–732. [Google Scholar]
- Petitpas, I.; Grüne, T.; Bhattacharya, A.A.; Curry, S. Crystal structures of human serum albumin complexed with monounsaturated and polyunsaturated fatty acids. J. Mol. Biol. 2001, 314, 955–960. [Google Scholar]
- Ryan, A.J.; Ghuman, J.; Zunszain, P.A.; Chung, C.W.; Curry, S. Structural basis of binding of fluorescent site-specific dansylated amino acids to human serum albumin. J. Struct. Biol. 2011, 174, 84–91. [Google Scholar]
- Petitpas, I.; Bhattacharya, A.A.; Twine, S.; East, M.; Curry, S. Crystal structure analysis of warfarin binding to human serum albumin: Anatomy of drug site I. J. Biol. Chem. 2001, 276, 22804–22809. [Google Scholar]
- Ghuman, J.; Zunszain, P.A.; Petitpas, I.; Bhattacharya, A.A.; Otagiri, M.; Curry, S. Structural basis of the drug-binding specificity of human serum albumin. J. Mol. Biol. 2005, 353, 38–52. [Google Scholar]
- Zunszain, P.A.; Ghuman, J.; McDonagh, A.F.; Curry, S. Crystallographic analysis of human serum albumin complexed with 4Z15E-bilirubin-IXα. J. Mol. Biol. 2008, 381, 394–406. [Google Scholar]
- Petitpas, I.; Petersen, C.E.; Ha, C.E.; Bhattacharya, A.A.; Zunszain, P.A.; Ghuman, J.; Bhagavan, N.V.; Curry, S. Structural basis of albumin–thyroxine interactions and familial dysalbuminemic hyperthyroxinemia. Proc. Natl. Acad. Sci. USA 2003, 100, 6440–6445. [Google Scholar]
- Zhu, L.; Yang, F.; Chen, L.; Meehan, E.J.; Huang, M. A new drug binding subsite on human serum albumin and drug–drug interaction studied by X-ray crystallography. J. Struct. Biol. 2008, 162, 40–49. [Google Scholar]
- Yang, F.; Bian, C.; Zhu, L.; Zhao, G.; Huang, Z.; Huang, M. Effect of human serum albumin on drug metabolism: Structural evidence of esterase activity of human serum albumin. J. Struct. Biol. 2007, 157, 348–355. [Google Scholar]
- Guo, S.; Shi, X.; Yang, F.; Chen, L.; Meehan, E.J.; Bian, C.; Huang, M. Structural basis of transport of lysophospholipids by human serum albumin. Biochem. J. 2009, 423, 23–30. [Google Scholar]
- Wang, Y.; Yu, H.; Shi, X.; Luo, Z.; Lin, D.; Huang, M. Structural mechanism of ring-opening reaction of glucose by human serum albumin. J. Biol. Chem. 2013, 288, 15980–15987. [Google Scholar]
- Luo, Z.; Shi, X.; Hu, Q.; Zhao, B.; Huang, M. Structural evidence of perfluorooctane sulfonate transport by human serum albumin. Chem. Res. Toxicol. 2012, 25, 990–992. [Google Scholar]
- Wang, Y.; Luo, Z.; Shi, X.; Wang, H.; Nie, L.; Huang, M. A fluorescent fatty acid probe DAUDA selectively displaces two myristates bound in human serum albumin. Protein Sci. 2011, 20, 2095–2101. [Google Scholar]
- Li, M.; Lee, P.; Zhang, Y.; Ma, Z.; Yang, F.; Zhou, Z.; Wu, X.; Liang, H. X-ray crystallographic and fluorometric analysis of the interactions of Rhein to human serum albumin. Chem. Biol. Drug Des. 2014, 83, 167–173. [Google Scholar]
- Yang, F.; Ma, Z.Y.; Zhang, Y.; Li, G.Q.; Li, M.; Qin, J.K.; Lockridge, O.; Liang, H. Human serum albumin-based design of a diflunisal prodrug. Eur. J. Pharm. Biopharm. 2013, 84, 549–554. [Google Scholar]
- Li, M.; McAuley, E.; Zhang, Y.; Kong, L.; Yang, F.; Zhou, Z.; Wu, X.; Liang, H. Comparison of binding characterization of two antiviral drugs to human serum albumin. Chem. Biol. Drug Des. 2013. [Google Scholar] [CrossRef]
- Yamaguchi, S.; Aldini, G.; Ito, S.; Morishita, N.; Shibata, T.; Vistoli, G.; Carini, M.; Uchida, K. Δ12-prostaglandin J2 as a product and ligand of human serum albumin: Formation of an unusual covalent adduct at His146. J. Am. Chem. Soc. 2010, 132, 824–832. [Google Scholar]
- Buttar, D.; Colclough, N.; Gerhardt, S.; MacFaul, P.A.; Phillips, S.D.; Plowright, A.; Whittamore, P.; Tam, K.; Maskos, K.; Steinbacher, S.; et al. A combined spectroscopic and crystallographic approach to probing drug–human serum albumin interactions. Bioorg. Med. Chem. 2010, 18, 7486–7496. [Google Scholar]
- Hein, K.L.; Kragh-Hansen, U.; Morth, J.P.; Jeppesen, M.D.; Otzen, D.; Møller, J.V.; Nissen, P. Crystallographic analysis reveals a unique lidocaine binding site on human serum albumin. J. Struct. Biol. 2010, 171, 353–360. [Google Scholar]
- Mao, H.; Hajduk, P.J.; Craig, R.; Bell, R.; Borre, T.; Fesik, S.W. Rational design of diflunisal analogues with reduced affinity for human serum albumin. J. Am. Chem. Soc. 2001, 123, 10429–10435. [Google Scholar]
- Mahesh, G.; Rajagopal, S.; et al. Molecular interaction studies of trimethoxy flavone with human serum albumin. PLoS One 2010, 1(5), e8834. [Google Scholar]
- Oltersdorf, T.; Elmore, S.W.; Shoemaker, A.R.; Armstrong, R.C.; Augeri, D.J.; Belli, B.A.; Bruncko, M.; Deckwerth, T.L.; Dinges, J.; Hajduk, P.J.; et al. An inhibitor of Bcl-2 family proteins induces regression of solid tumours. Nature 2005, 435, 677–681. [Google Scholar]
- Vallianatou, T.; Lambrinidis, G.; Tsantili-Kakoulidou, A. In silico prediction of human serum albumin binding for drug leads. Expert Opin. Drug Discov. 2013, 8, 583–595. [Google Scholar]
- Tesseromatis, C.; Alevizou, A. The role of the protein-binding on the mode of drug action as well the interactions with other drugs. Eur. J. Drug Metab. Pharmacokinet. 2008, 33, 225–230. [Google Scholar]
- Yang, F.; Lee, P.; Ma, Z.; Ma, L.; Yang, G.; Wu, X.; Liang, H. Regulation of amantadine hydrochloride binding with IIA subdomain of human serum albumin by fatty acid chains. J. Pharm. Sci. 2013, 102, 84–92. [Google Scholar]
- Yang, F.; Yue, J.; Ma, L.; Ma, Z.; Li, M.; Wu, X.; Liang, H. Interactive associations of drug–drug and drug–drug–drug with IIA subdomain of human serum albumin. Mol. Pharm. 2012, 9, 3259–3265. [Google Scholar]
- Curry, S. Lessons from the crystallographic analysis of small molecule binding to human serum albumin. Drug Metab. Pharmacokinet. 2009, 24, 342–357. [Google Scholar]
- Curry, S. X-ray Crystallography of Albumin. In Human Serum Albumin—New Insights on Its Structural Dynamics, Functional Impacts and Pharmaceutical Applications; Otagiri, M., Ed.; Sojo University Publishing Center: Kumamoto, Japan, 2011; pp. 1–29. [Google Scholar]
- Sudlow, G.; Birkett, D.J.; Wade, D.N. The characterization of two specific drug binding sites on human serum albumin. Mol. Pharmacol. 1975, 11, 824–832. [Google Scholar]
- Sudlow, G.; Birkett, D.J.; Wade, D.N. Further characterization of specific drug binding sites on human serum albumin. Mol. Pharmacol. 1976, 12, 1052–1061. [Google Scholar]
- Bos, O.J.; Remijn, J.P.; Fischer, M.J.; Wilting, J.; Janssen, L.H. Location and characterization of the warfarin binding site of human serum albumin—A comparative study of two large fragments. Biochem. Pharmacol. 1988, 37, 3905–3909. [Google Scholar]
- Bos, O.J.; Fischer, M.J.; Wilting, J.; Janssen, L.H. Drug-binding and other physicochemical properties of a large tryptic and a large peptic fragment of human serum albumin. Biochim. Biophys. Acta 1988, 953, 37–47. [Google Scholar]
- Simard, J.R.; Zunszain, P.A.; Hamilton, J.A.; Curry, S. Location of high and low affinity fatty acid binding sites on human serum albumin revealed by NMR drug-competition analysis. J. Mol. Biol. 2006, 361, 336–351. [Google Scholar]
- Simard, J.R.; Zunszain, P.A.; Ha, C.E.; Yang, J.S.; Bhagavan, N.V.; Petitpas, I.; Curry, S.; Hamilton, J.A. Locating high-affinity fatty acid-binding sites on albumin by X-ray crystallography and NMR spectroscopy. Proc. Natl. Acad. Sci. USA 2005, 102, 17958–17963. [Google Scholar]
- Meijer, D.K.; van der Sluijs, P. The influence of binding to albumin and α1-acid glycoprotein on the clearance of drugs by the liver. Pharm. Weekbl. Sci. 1987, 9, 65–74. [Google Scholar]
- Kuwahara, H.; Nishida, Y.; Yokota, T. Blood–brain barrier and Alzheimer’s disease. Brain Nerve 2013, 65, 145–151. [Google Scholar]
- Sharma, H.S.; Castellani, R.J.; Smith, M.A.; Sharma, A. The blood–brain barrier in Alzheimer’s disease: Novel therapeutic targets and nanodrug delivery. Int. Rev. Neurobiol. 2012, 102, 47–90. [Google Scholar]
- Banks, W.A. Drug delivery to the brain in Alzheimer’s disease: Consideration of the blood–brain barrier. Adv. Drug Deliv. Rev. 2012, 64, 629–639. [Google Scholar]
- Chopra, A. Transferrin-Coated Gadolinium-Labeled Human Serum Albumin Nanoparticles. In Molecular Imaging and Contrast Agent Database (MICAD); National Center for Biotechnology Information: Bethesda, MD, USA, 2013. [Google Scholar]
- Kang, Y.S.; Pardridge, W.M. Brain delivery of biotin bound to a conjugate of neutral avidin and cationized human albumin. Pharm. Res. 1994, 11, 1257–1264. [Google Scholar]
- Bickel, U.; Yoshikawa, T.; Pardridge, W.M. Delivery of peptides and proteins through the blood–brain barrier. Adv. Drug Deliv. Rev. 2001, 46, 247–279. [Google Scholar]
- Patel, M.M.; Goyal, B.R.; Bhadada, S.V.; Bhatt, J.S.; Amin, A.F. Getting into the brain: Approaches to enhance brain drug delivery. CNS Drugs 2009, 23, 35–58. [Google Scholar]
- Pardridge, W.M. Drug targeting to the brain. Pharm. Res. 2007, 24, 1733–1744. [Google Scholar]
- Chuang, V.T.; Kragh-Hansen, U.; Otagiri, M. Pharmaceutical strategies utilizing recombinant human serum albumin. Pharm. Res. 2002, 19, 569–577. [Google Scholar]
- Schönfeld, D.L.; Ravelli, R.B.; Mueller, U.; Skerra, A. The 18-A crystal structure of α1-acid glycoprotein (orosomucoid) solved by UV RIP reveals the broad drug-binding activity of this human plasma lipocalin. J. Mol. Biol. 2008, 384, 393–405. [Google Scholar]
- Kragh-Hansen, U. Molecular aspects of ligand binding to serum albumin. Pharmacol. Rev. 1981, 33, 17–53. [Google Scholar]
- Zsila, F. Subdomain IB is the third major drug binding region of human serum albumin: Toward the three-sites model. Mol. Pharm. 2012, 10, 1668–1682. [Google Scholar]
- Liang, H.; Yang, F.; Lee, N.; Wu, X. HSA-based anti-inflammatory therapy: A new and improved approach. Future Med. Chem. 2014, 6, 119–121. [Google Scholar]
- Kratz, F. Albumin as a drug carrier: Design of prodrugs drug conjugates and nanoparticles. J. Control. Release 2008, 132, 171–183. [Google Scholar]
- Neumann, E.; Frei, E.; Funk, D.; Becker, M.D.; Schrenk, H.H.; Müller-Ladner, U.; Fiehn, C. Native albumin for targeted drug delivery. Expert Opin. Drug Deliv. 2010, 7, 915–925. [Google Scholar]
- Kratz, F.; Elsadek, B. Clinical impact of serum proteins on drug delivery. J. Control. Release 2012, 161, 429–445. [Google Scholar]
- Elsadek, B.; Kratz, F. Impact of albumin on drug delivery—New applications on the horizon. J. Control. Release 2012, 157, 4–28. [Google Scholar]
- Allen, T.M.; Cullis, P.R. Drug delivery systems: Entering the mainstream. Science 2004, 303, 1818–1822. [Google Scholar]
- Sweta, M.; Prakash, J.J.; Domb, A.J.; Kumar, N. Exploiting EPR in polymer drug conjugate delivery for tumor targeting. Curr. Pharm. Des. 2006, 12, 4785–4796. [Google Scholar]
- Kratz, F.; Beyer, U. Serum proteins as drug carriers of anticancer agents: A review. Drug Deliv. 1998, 5, 281–299. [Google Scholar]
- Dosio, F.; Brusa, P.; Crosasso, P.; Arpicco, S.; Cattel, L.G. Preparation characterization and properties in vitro and in vivo of a paclitaxel–albumin conjugate. J. Control. Release 1997, 47, 293–304. [Google Scholar]
- Kratz, F.; Mueller-Driver, R.; Hofmann, I.; Drevs, J.; Unger, C. A novel macromolecular prodrug concept exploiting endogenous serum albumin as a drug carrier for cancer chemotherapy. J. Med. Chem. 2000, 43, 1253–1256. [Google Scholar]
- Liu, M.; Lim, Z.J.; Gwee, Y.Y.; Levina, A.; Lay, P.A. Characterization of a ruthenium(III)/NAMI— A adduct with bovine serum albumin that exhibits a high anti-metastaticactivity. Angew. Chem. Int. Ed. 2010, 49, 1661–1664. [Google Scholar]
- Silva, D.; Cortez, C.M.; Silva, C.M.; Missailidis, S. A fluorescent spectroscopy and modelling analysis of anti-heparanase aptamers–serum protein interactions. J. Photochem. Photobiol. B 2013, 127, 68–77. [Google Scholar]
- Yewale, C.; Baradia, D.; Vhora, I.; Misra, A. Proteins: Emerging carrier for delivery of cancer therapeutics. Expert Opin. Drug Deliv. 2013, 10, 1429–1448. [Google Scholar]
- Son, S.; Song, S.; Lee, S.J.; Min, S.; Kim, S.A.; Yhee, J.Y.; Huh, M.S.; Chan, K.I.; Jeong, S.Y.; Byun, Y.; et al. Self-crosslinked human serum albumin nanocarriers for systemic delivery of polymerized siRNA to tumors. Biomaterials 2013, 34, 9475–9485. [Google Scholar]
- Ehrlich, G.K.; Michel, H.; Truitt, T.; Riboulet, W.; Pop-Damkov, P.; Goelzer, P.; Hainzl, D.; Qureshi, F.; Lueckel, B.; Danho, W.; et al. Preparation and characterization of albumin conjugates of a truncated peptide YY analogue for half-life extension. Bioconjug. Chem. 2013, 24, 2015–2024. [Google Scholar]
- Vakhrusheva, T.V.; Gusev, A.A.; Gusev, S.A.; Vlasova, I.I. Albumin reduces thrombogenic potential of single-walled carbon nanotubes. Toxicol. Lett. 2013, 221, 137–145. [Google Scholar]
- Kragh-Hansen, U. Molecular and practical aspects of the enzymatic properties of human serum albumin and of albumin–ligand complexes. Biochim. Biophys. Acta 2013, 1830, 5535–5544. [Google Scholar]
- Ge, C.; Du, J.; Zhao, L.; Wang, L.; Liu, Y.; Li, D.; Yang, Y.; Zhou, R.; Zhao, Y.; Chai, Z.; et al. Binding of blood proteins to carbon nanotubes reduces cytotoxicity. Proc. Natl. Acad. Sci. USA 2011, 108, 16968–16973. [Google Scholar]
- Komatsu, T.; Qu, X.; Ihara, H.; Fujihara, M.; Azuma, H.; Ikeda, H. Virus trap in human serum albumin nanotube. J. Am. Chem. Soc. 2011, 133, 3246–3248. [Google Scholar]
- Qu, X.; Komatsu, T. Molecular capture in protein nanotubes. ACS Nano 2010, 4, 563–573. [Google Scholar]
- Komatsu, T.; Nakagawa, A.; Qu, X. Structural and mutagenic approach to create human serum albumin-based oxygen carrier and photosensitizer. Drug Metab. Pharmacokinet. 2009, 24, 287–299. [Google Scholar]
- Komatsu, T.; Ohmichi, N.; Zunszain, P.A.; Curry, S.; Tsuchida, E. Dioxygenation of human serum albumin having a prosthetic heme group in a tailor-made heme pocket. J. Am. Chem. Soc. 2004, 126, 14304–14305. [Google Scholar]
- Komatsu, T.; Wang, R.M.; Zunszain, P.A.; Curry, S.; Tsuchida, E. Photosensitized reduction of water to hydrogen using human serum albumin complexed with zinc–protoporphyrin IX. J. Am. Chem. Soc. 2006, 128, 16297–16301. [Google Scholar]
- Rozga, J.; Piątek, T.; Małkowski, P. Human albumin: Old new and emerging applications. Ann. Transplant. 2013, 18, 205–217. [Google Scholar]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
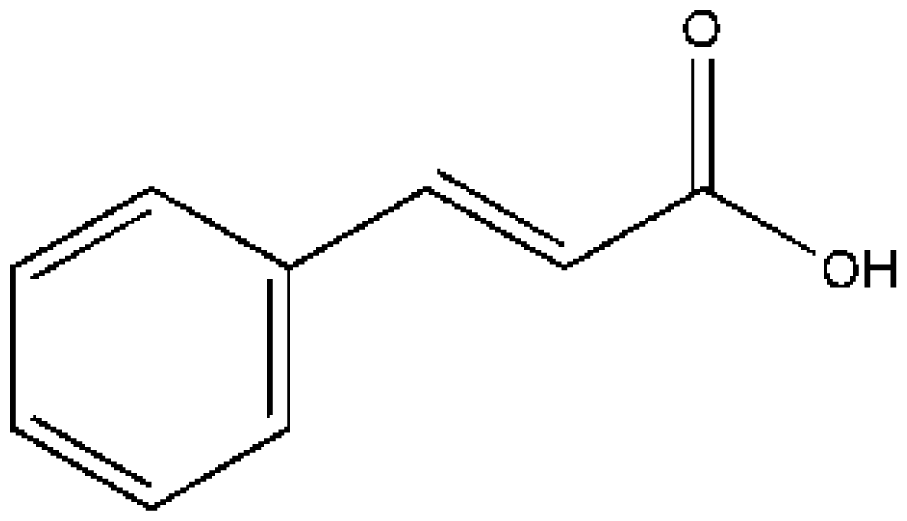
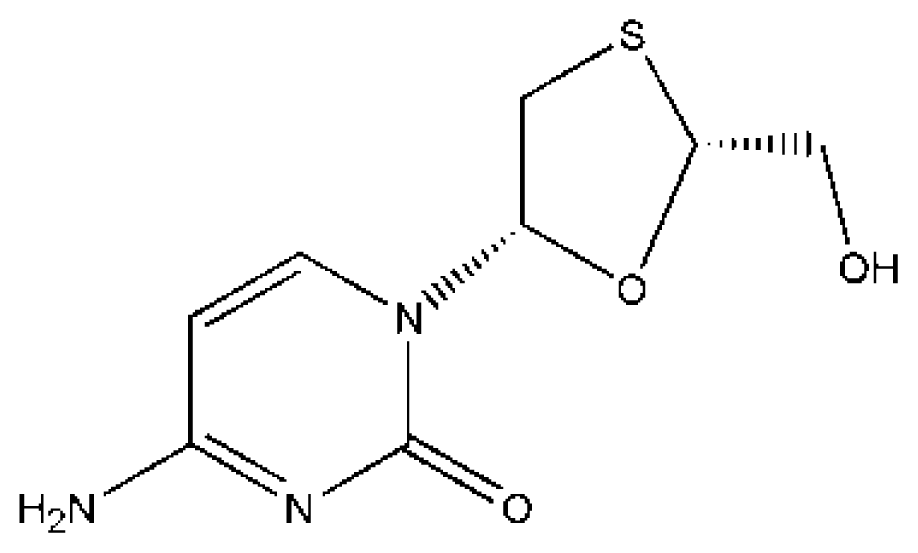
| Drugs | Ref. | HSA/HSA complex | K (×104 M−1) | ΔG (kJ/mol) |
|---|---|---|---|---|
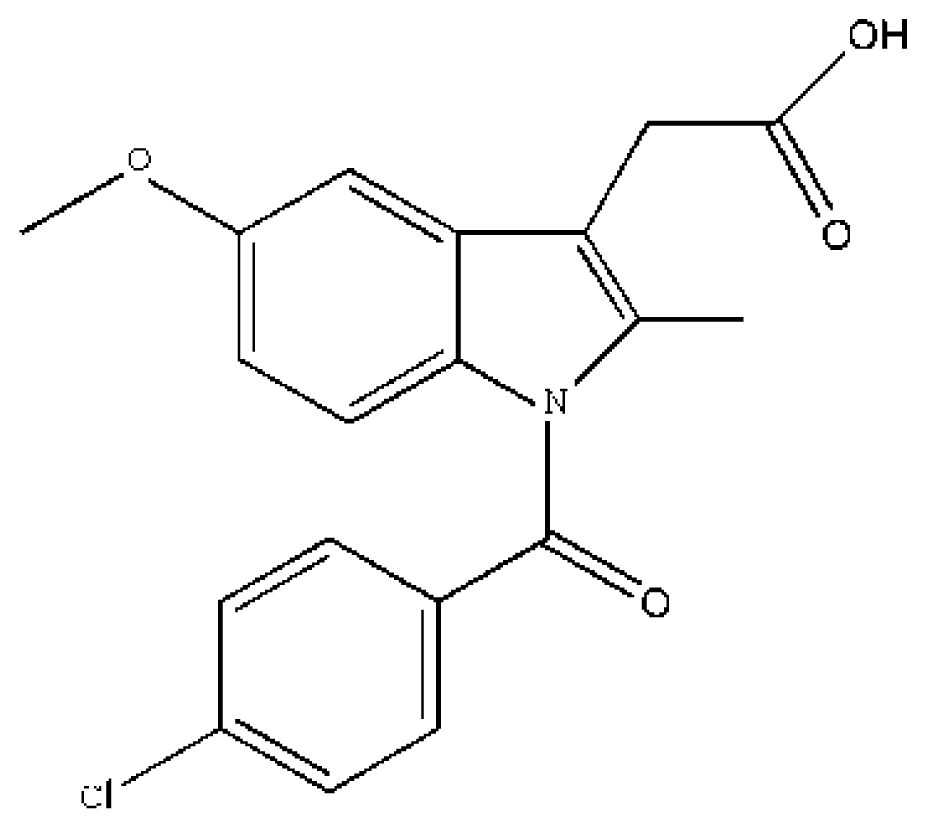
 (CA) | [74] | HSA | 5.329 ± 0.11 | −26.965 |
| HSA–IMN | 1.107 ± 0.06 | −23.071 | ||
| HSA–IMN–LMD | 3.296 ± 0.07 | −25.774 | ||
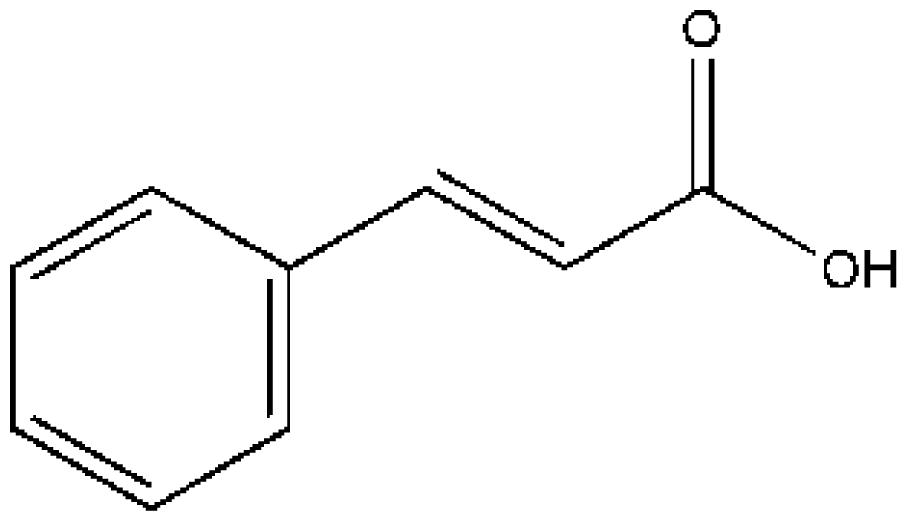
 (IMN) | [74] | HSA | 5.947 ± 0.09 | −27.237 |
| HSA–CA | 10.563 ± 0.13 | −28.660 | ||
| HSA–CA–LMD | 8.019 ± 0.11 | −27.977 | ||
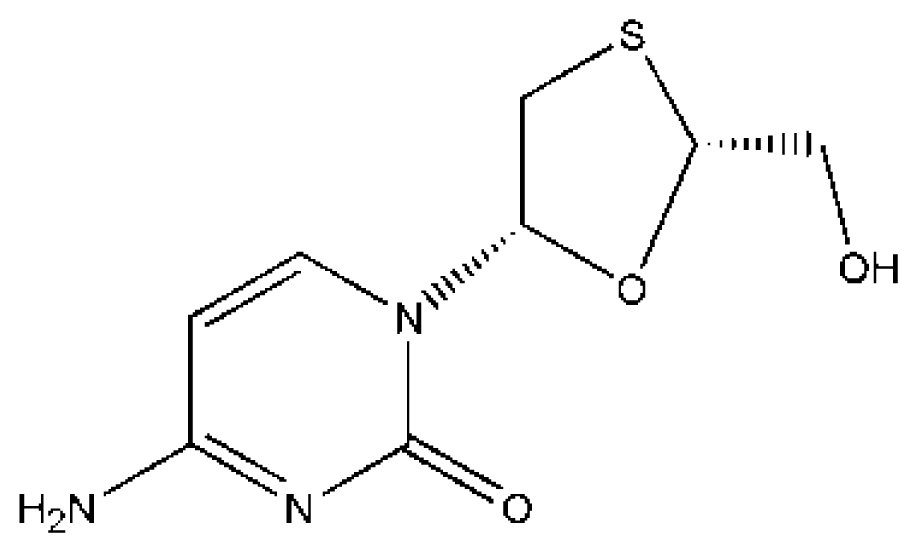 (LMD) | [64,74] | HSA | 1.688 ± 0.16 | −24.116 |
| HSA–IMN | 4.07 ± 0.12 | −27.85 | ||
| HSA–CA–IMN | 3.220 ± 0.05 | −25.717 | ||
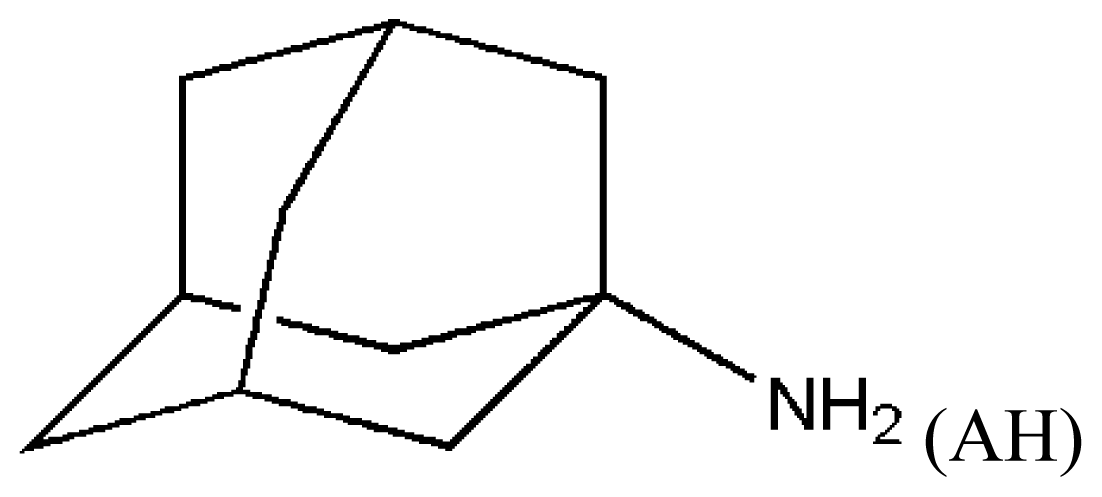
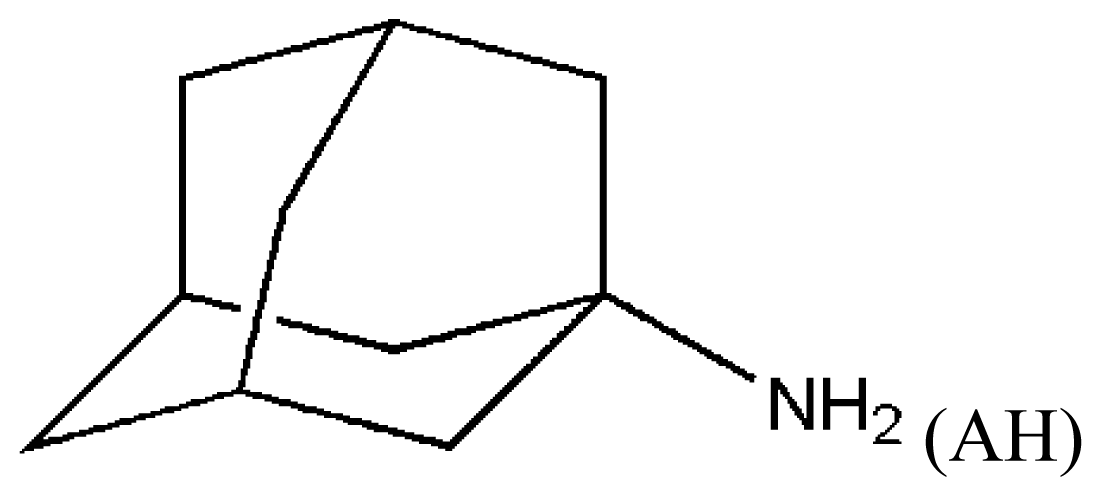 (AH) | [73] | HSA–myristate acids | 1.909 ± 0.03 | −24.421 |
| HSA–octanoic acid | 2.745 ± 0.11 | −25.321 | ||
© 2014 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Yang, F.; Zhang, Y.; Liang, H. Interactive Association of Drugs Binding to Human Serum Albumin. Int. J. Mol. Sci. 2014, 15, 3580-3595. https://doi.org/10.3390/ijms15033580
Yang F, Zhang Y, Liang H. Interactive Association of Drugs Binding to Human Serum Albumin. International Journal of Molecular Sciences. 2014; 15(3):3580-3595. https://doi.org/10.3390/ijms15033580
Chicago/Turabian StyleYang, Feng, Yao Zhang, and Hong Liang. 2014. "Interactive Association of Drugs Binding to Human Serum Albumin" International Journal of Molecular Sciences 15, no. 3: 3580-3595. https://doi.org/10.3390/ijms15033580