Dynamin-Related Protein 1 Inhibitors Protect against Ischemic Toxicity through Attenuating Mitochondrial Ca2+ Uptake from Endoplasmic Reticulum Store in PC12 Cells

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
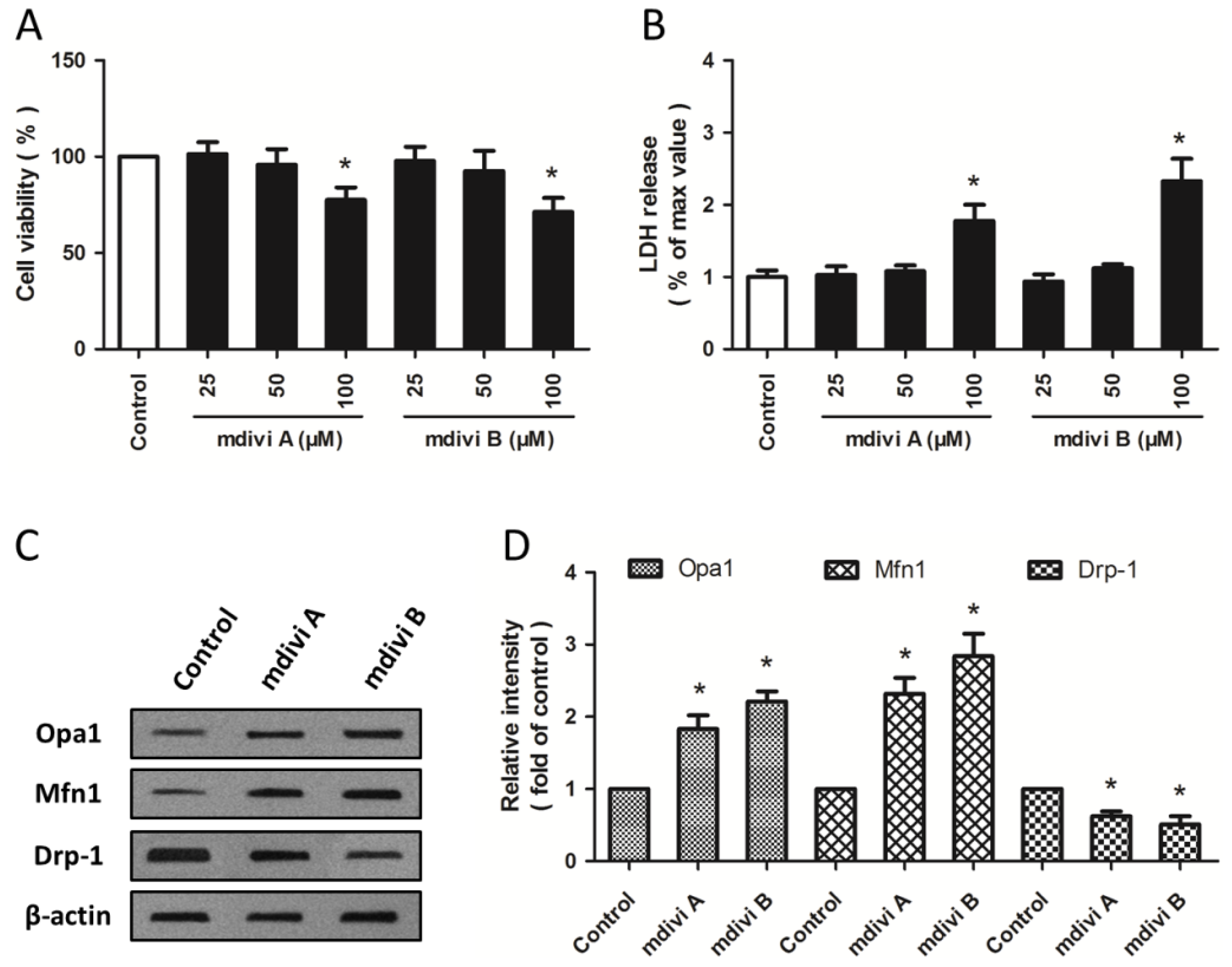
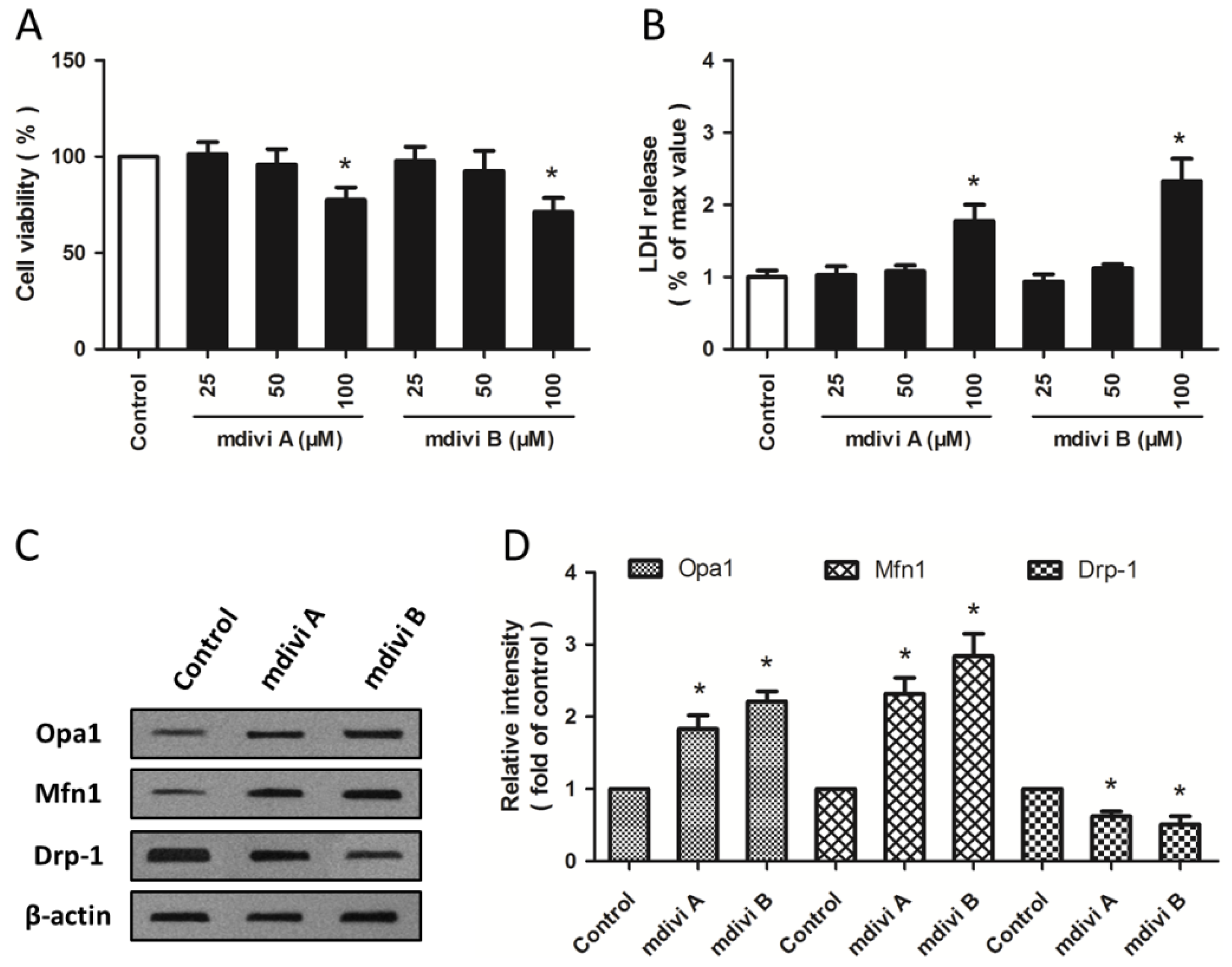
2.1. Effects of Drp-1 Inhibitors on Mitochondrial Dynamic Proteins
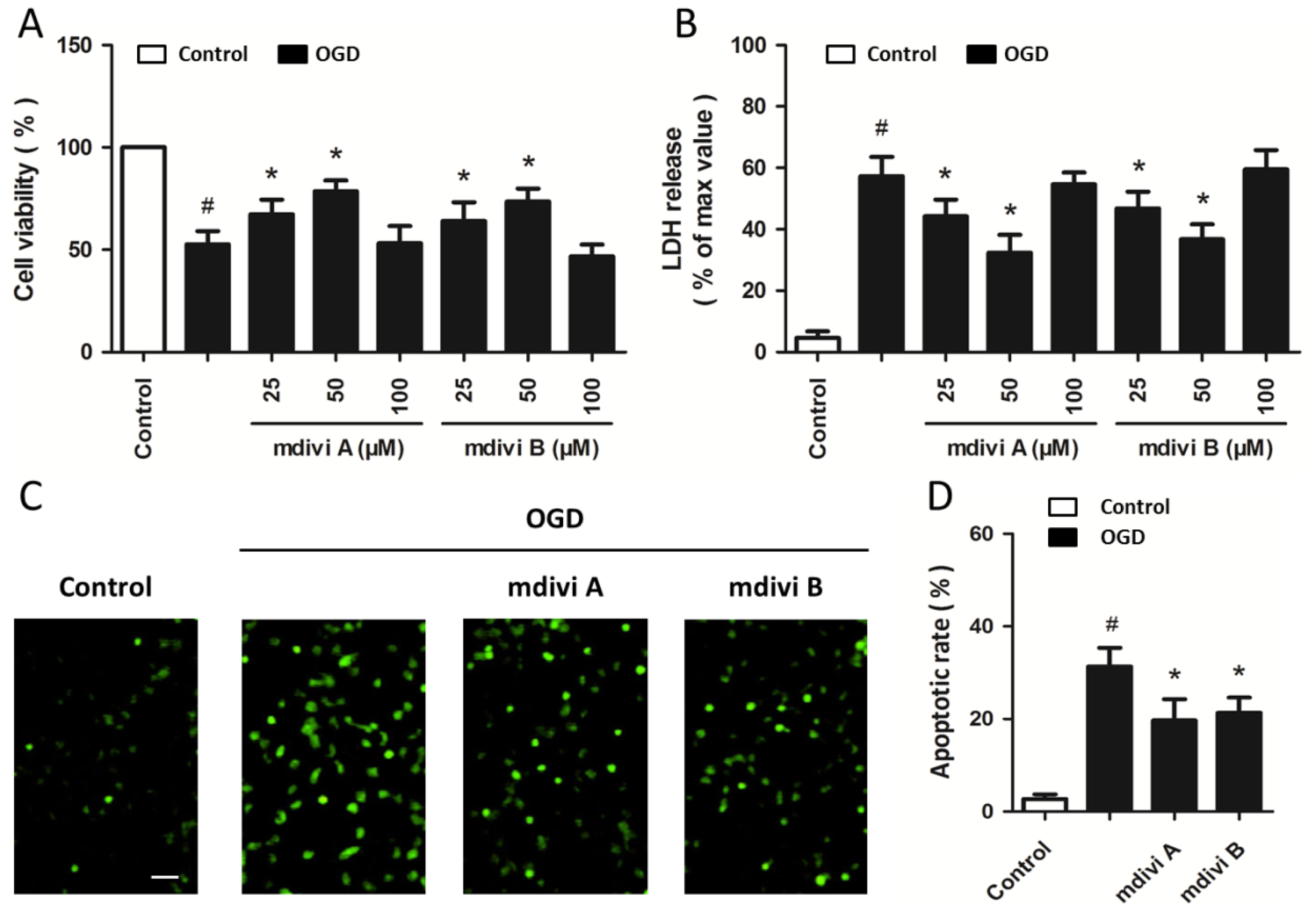
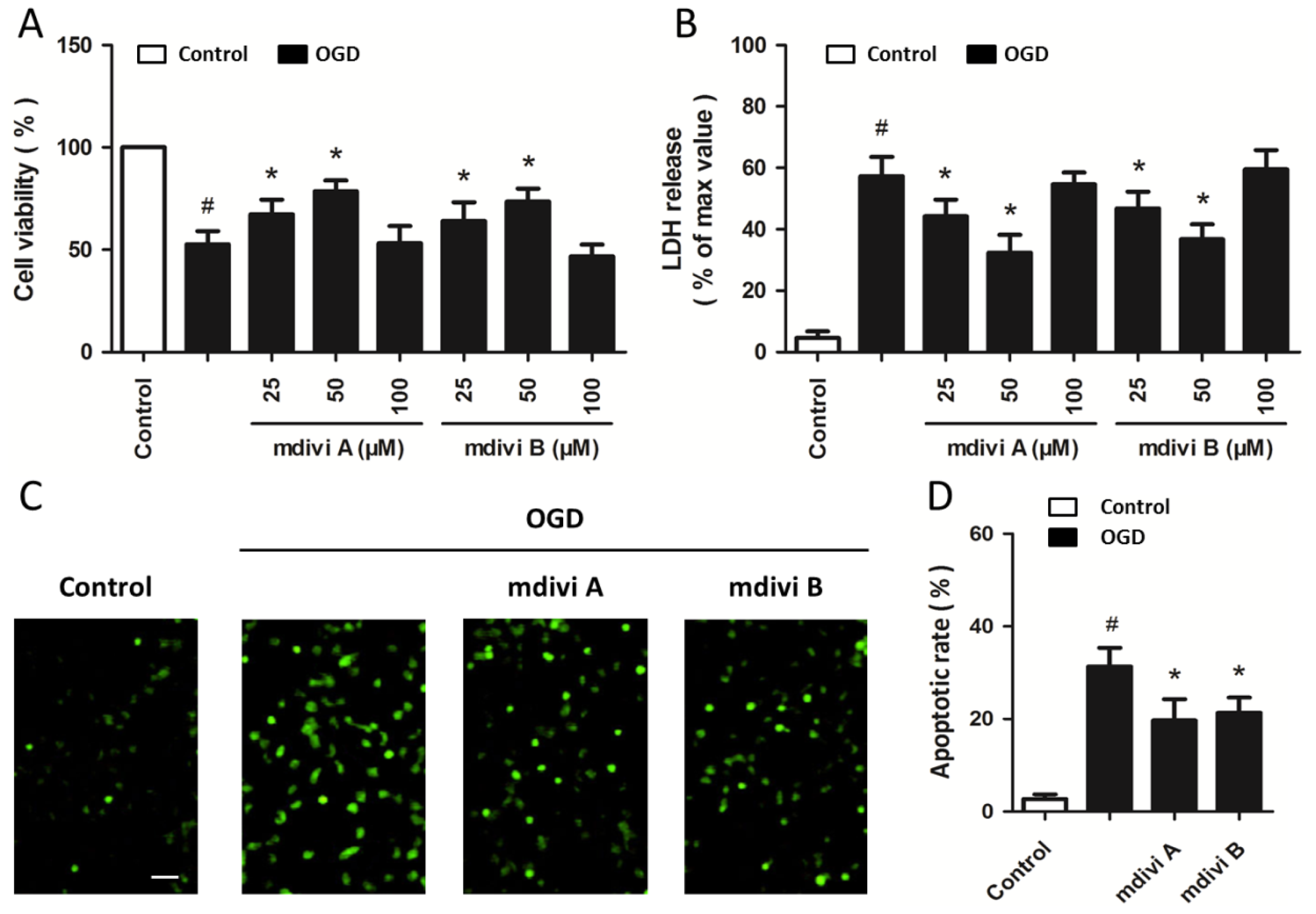
2.2. Drp-1 Inhibitors Reduce Ischemic Toxicity in PC12 Cells
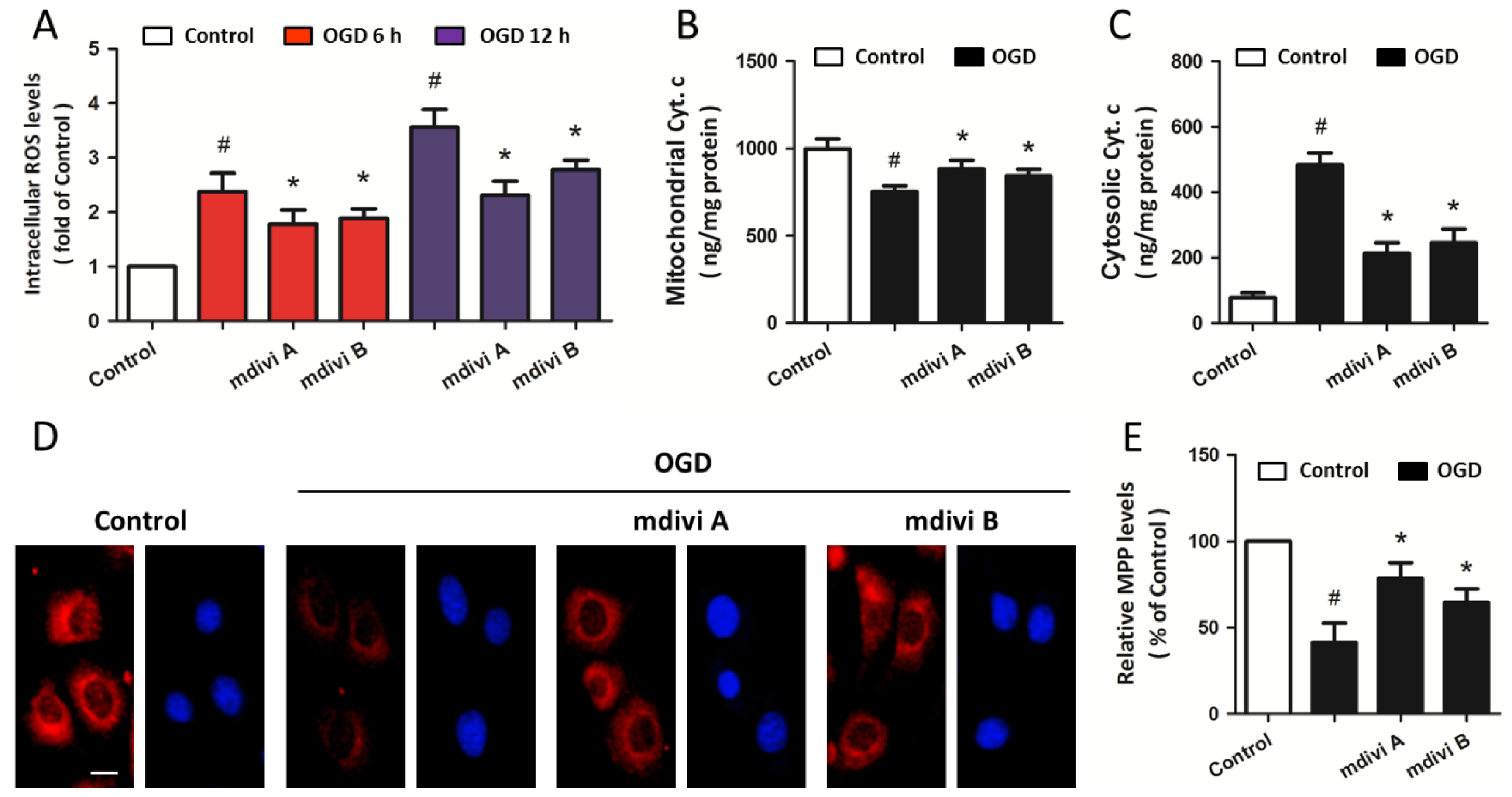
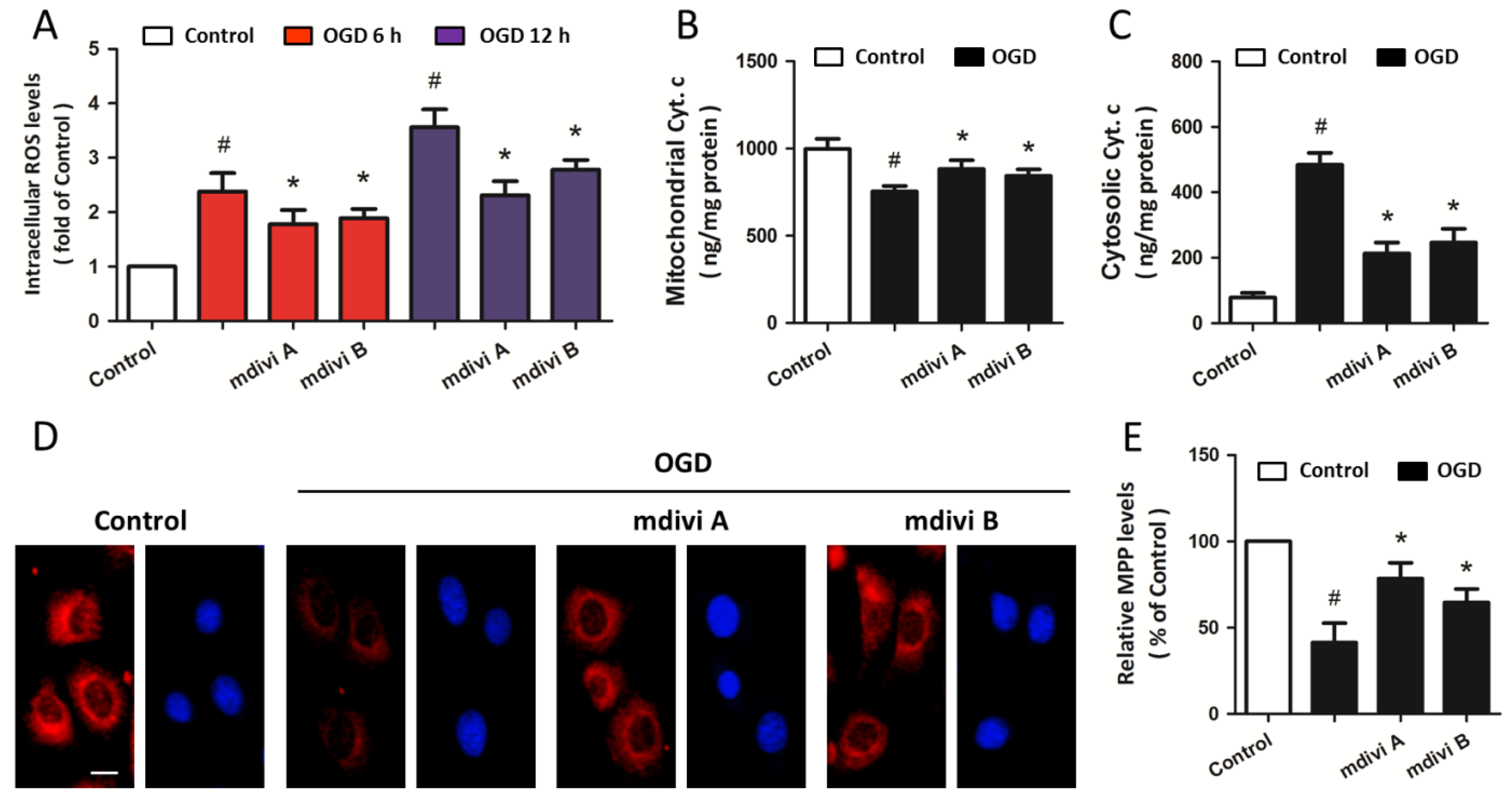
2.3. Drp-1 Inhibitors Attenuate Mitochondrial Dysfunction
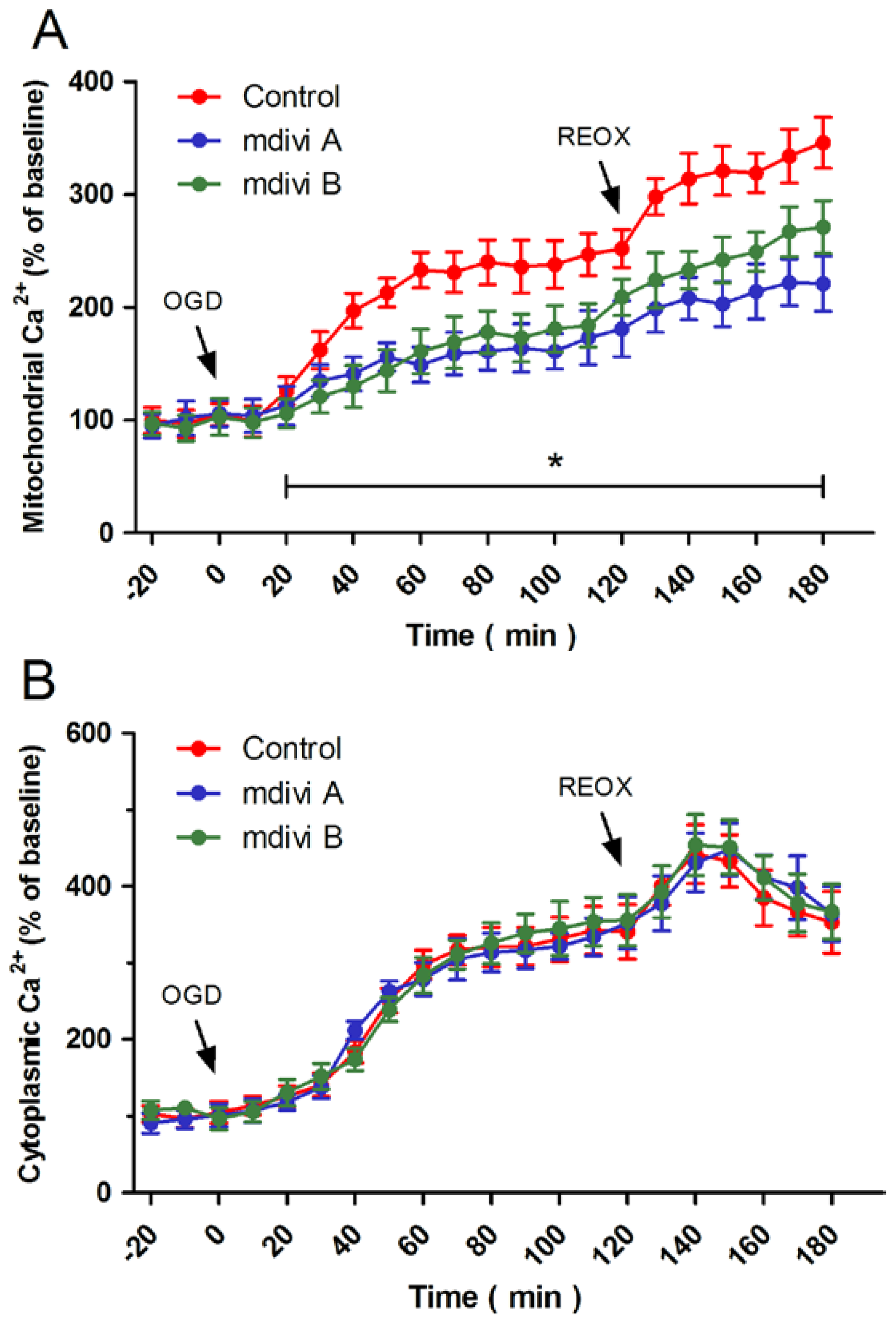
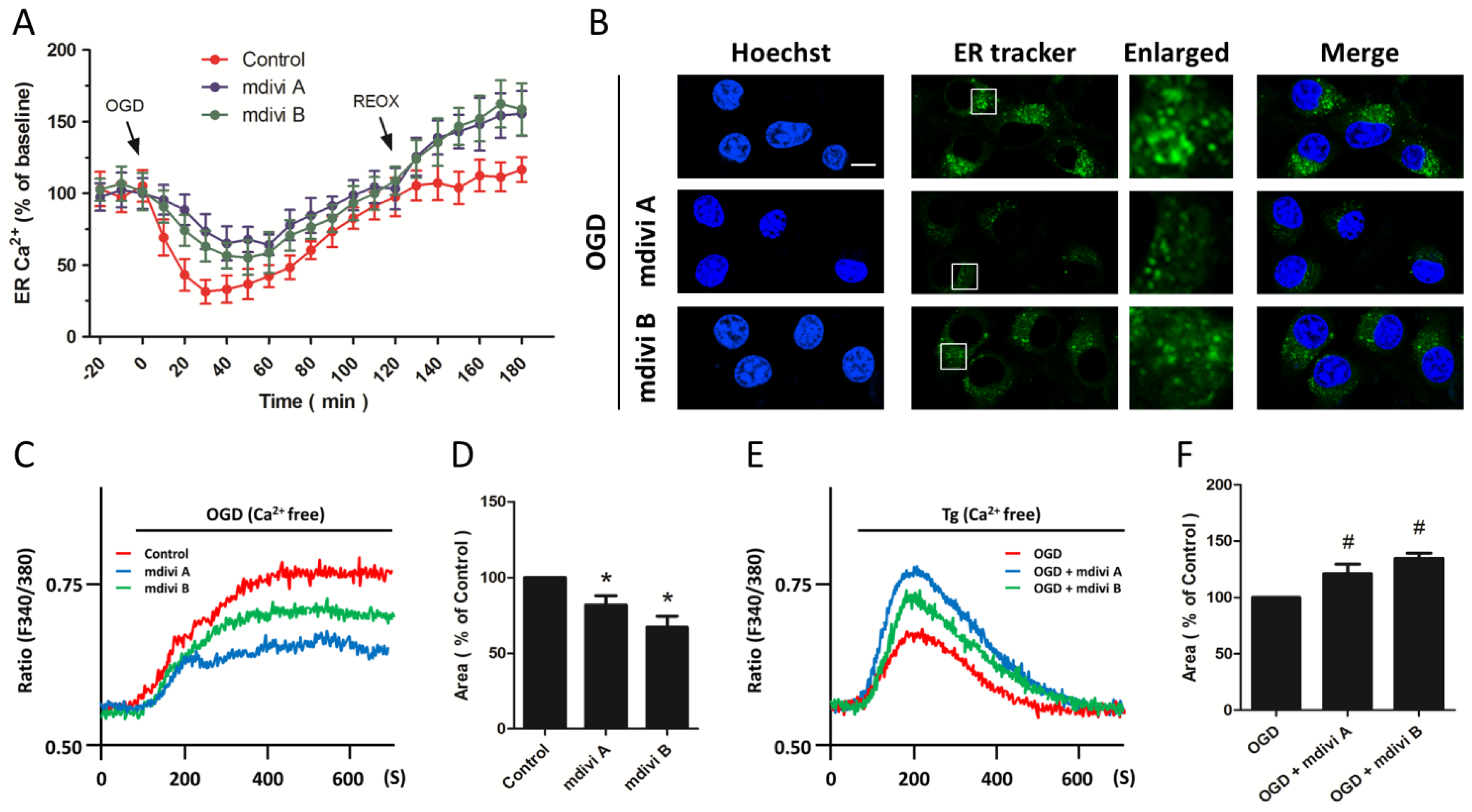
2.4. Effects of Drp-1 Inhibitors on Mitochondrial Ca2+ Uptake
2.5. Effects of Drp-1 Inhibitors on ER Ca2+ Release
2.6. Effects of Drp-1 Inhibitors on ER Ca2+ Release
3. Discussion
4. Experimental Section
4.1. Cell Culture
4.2. OGD
4.3. Cell Viability Assay
4.4. LDH Release Assay
4.5. TUNEL Staining
4.6. Measurement of Intra-Cellular ROS
4.7. Quantification of Cytochrome c Release
4.8. Measurement of Mitochondrial Membrane Potential (MMP)
4.9. Measurement of Mitochondrial Calcium
4.10. Calcium Imaging
4.11. ER Tracker Staining
4.12. Measurement of ER Calcium Release
4.13. Statistical Analysis
5. Conclusions
Acknowledgments
Conflicts of Interest
References
- Strong, K.; Mathers, C.; Bonita, R. Preventing stroke: Saving lives around the world. Lancet Neurol 2007, 6, 182–187. [Google Scholar]
- Arundine, M.; Tymianski, M. Molecular mechanisms of glutamate-dependent neurodegeneration in ischemia and traumatic brain injury. Cell. Mol. Life Sci 2004, 61, 657–668. [Google Scholar]
- Chen, T.; Liu, W.; Chao, X.; Qu, Y.; Zhang, L.; Luo, P.; Xie, K.; Huo, J.; Fei, Z. Neuroprotective effect of osthole against oxygen and glucose deprivation in rat cortical neurons: Involvement of mitogen-activated protein kinase pathway. Neuroscience 2011, 183, 203–211. [Google Scholar]
- White, B.C.; Sullivan, J.M.; DeGracia, D.J.; O’Neil, B.J.; Neumar, R.W.; Grossman, L.I.; Rafols, J.A.; Krause, G.S. Brain ischemia and reperfusion: molecular mechanisms of neuronal injury. J. Neurol. Sci 2000, 179, 1–33. [Google Scholar]
- Sanderson, T.H.; Reynolds, C.A.; Kumar, R.; Przyklenk, K.; Huttemann, M. Molecular mechanisms of ischemia-reperfusion injury in brain: Pivotal role of the mitochondrial membrane potential in reactive oxygen species generation. Mol. Neurobiol 2013, 47, 9–23. [Google Scholar]
- Reddy, P.H.; Reddy, T.P.; Manczak, M.; Calkins, M.J.; Shirendeb, U.; Mao, P. Dynamin-related protein 1 and mitochondrial fragmentation in neurodegenerative diseases. Brain Res. Rev 2011, 67, 103–118. [Google Scholar]
- Ishihara, N.; Nomura, M.; Jofuku, A.; Kato, H.; Suzuki, S.O.; Masuda, K.; Otera, H.; Nakanishi, Y.; Nonaka, I.; Goto, Y.; et al. Mitochondrial fission factor Drp1 is essential for embryonic development and synapse formation in mice. Nat. Cell Biol 2009, 11, 958–966. [Google Scholar]
- Otera, H.; Ishihara, N.; Mihara, K. New insights into the function and regulation of mitochondrial fission. Biochim. Biophys. Acta 2013, 1833, 1256–1268. [Google Scholar]
- Ingerman, E.; Perkins, E.M.; Marino, M.; Mears, J.A.; McCaffery, J.M.; Hinshaw, J.E.; Nunnari, J. Dnm1 forms spirals that are structurally tailored to fit mitochondria. J. Cell Biol 2005, 170, 1021–1027. [Google Scholar]
- Bossy-Wetzel, E.; Barsoum, M.J.; Godzik, A.; Schwarzenbacher, R.; Lipton, S.A. Mitochondrial fission in apoptosis, neurodegeneration and aging. Curr. Opin. Cell Biol 2003, 15, 706–716. [Google Scholar]
- Inoue-Yamauchi, A.; Oda, H. Depletion of mitochondrial fission factor DRP1 causes increased apoptosis in human colon cancer cells. Biochem. Biophys. Res. Commun 2012, 421, 81–85. [Google Scholar]
- Rehman, J.; Zhang, H.J.; Toth, P.T.; Zhang, Y.; Marsboom, G.; Hong, Z.; Salgia, R.; Husain, A.N.; Wietholt, C.; Archer, S.L. Inhibition of mitochondrial fission prevents cell cycle progression in lung cancer. FASEB J 2012, 26, 2175–2186. [Google Scholar]
- Wakabayashi, J.; Zhang, Z.; Wakabayashi, N.; Tamura, Y.; Fukaya, M.; Kensler, T.W.; Iijima, M.; Sesaki, H. The dynamin-related GTPase Drp1 is required for embryonic and brain development in mice. J. Cell Biol 2009, 186, 805–816. [Google Scholar]
- Karbowski, M.; Lee, Y.J.; Gaume, B.; Jeong, S.Y.; Frank, S.; Nechushtan, A.; Santel, A.; Fuller, M.; Smith, C.L.; Youle, R.J. Spatial and temporal association of Bax with mitochondrial fission sites, Drp1, and Mfn2 during apoptosis. J. Cell Biol 2002, 159, 931–938. [Google Scholar]
- Harris, M.H.; Thompson, C.B. The role of the Bcl-2 family in the regulation of outer mitochondrial membrane permeability. Cell Death Differ 2000, 7, 1182–1191. [Google Scholar]
- Chen, H.; Chan, D.C. Mitochondrial dynamics—Fusion, fission, movement, and mitophagy— In neurodegenerative diseases. Hum. Mol. Genet 2009, 18, R169–R176. [Google Scholar]
- Chang, C.R.; Blackstone, C. Dynamic regulation of mitochondrial fission through modification of the dynamin-related protein Drp1. Ann. N. Y. Acad. Sci 2010, 1201, 34–39. [Google Scholar]
- Schild, L.; Huppelsberg, J.; Kahlert, S.; Keilhoff, G.; Reiser, G. Brain mitochondria are primed by moderate Ca2+ rise upon hypoxia/reoxygenation for functional breakdown and morphological disintegration. J. Biol. Chem 2003, 278, 25454–25460. [Google Scholar]
- Starkov, A.A.; Chinopoulos, C.; Fiskum, G. Mitochondrial calcium and oxidative stress as mediators of ischemic brain injury. Cell Calcium 2004, 36, 257–264. [Google Scholar]
- Castilho, R.F.; Ward, M.W.; Nicholls, D.G. Oxidative stress, mitochondrial function, and acute glutamate excitotoxicity in cultured cerebellar granule cells. J. Neurochem 1999, 72, 1394–1401. [Google Scholar]
- Yoon, Y. Regulation of mitochondrial dynamics: Another process modulated by Ca2+ signals? Sci. Signal 2005, 2005, pe18. [Google Scholar]
- Rowland, A.A.; Voeltz, G.K. Endoplasmic reticulum-mitochondria contacts: Function of the junction. Nat. Rev. Mol. Cell Biol 2012, 13, 607–625. [Google Scholar]
- Rizzuto, R.; Pinton, P.; Ferrari, D.; Chami, M.; Szabadkai, G.; Magalhaes, P.J.; di Virgilio, F.; Pozzan, T. Calcium and apoptosis: Facts and hypotheses. Oncogene 2003, 22, 8619–8627. [Google Scholar]
- Montessuit, S.; Somasekharan, S.P.; Terrones, O.; Lucken-Ardjomande, S.; Herzig, S.; Schwarzenbacher, R.; Manstein, D.J.; Bossy-Wetzel, E.; Basanez, G.; Meda, P.; et al. Membrane remodeling induced by the dynamin-related protein Drp1 stimulates Bax oligomerization. Cell 2010, 142, 889–901. [Google Scholar]
- Cassidy-Stone, A.; Chipuk, J.E.; Ingerman, E.; Song, C.; Yoo, C.; Kuwana, T.; Kurth, M.J.; Shaw, J.T.; Hinshaw, J.E.; Green, D.R.; et al. Chemical inhibition of the mitochondrial division dynamin reveals its role in Bax/Bak-dependent mitochondrial outer membrane permeabilization. Dev. Cell 2008, 14, 193–204. [Google Scholar]
- Pinton, P.; Ferrari, D.; Magalhaes, P.; Schulze-Osthoff, K.; DiVirgilio, F.; Pozzan, T.; Rizzuto, R. Reduced loading of intracellular Ca2+ stores and downregulation of capacitative Ca2+ influx in Bcl-2-overexpressing cells. J. Cell Biol 2000, 148, 857–862. [Google Scholar]
- Chen, T.; Fei, F.; Jiang, X.F.; Zhang, L.; Qu, Y.; Huo, K.; Fei, Z. Down-regulation of Homer1b/c attenuates glutamate-mediated excitotoxicity through endoplasmic reticulum and mitochondria pathways in rat cortical neurons. Free Radic. Biol. Med 2012, 52, 208–217. [Google Scholar]

© 2014 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Tian, Y.; Li, B.; Shi, W.-Z.; Chang, M.-Z.; Zhang, G.-J.; Di, Z.-L.; Liu, Y. Dynamin-Related Protein 1 Inhibitors Protect against Ischemic Toxicity through Attenuating Mitochondrial Ca2+ Uptake from Endoplasmic Reticulum Store in PC12 Cells. Int. J. Mol. Sci. 2014, 15, 3172-3185. https://doi.org/10.3390/ijms15023172
Tian Y, Li B, Shi W-Z, Chang M-Z, Zhang G-J, Di Z-L, Liu Y. Dynamin-Related Protein 1 Inhibitors Protect against Ischemic Toxicity through Attenuating Mitochondrial Ca2+ Uptake from Endoplasmic Reticulum Store in PC12 Cells. International Journal of Molecular Sciences. 2014; 15(2):3172-3185. https://doi.org/10.3390/ijms15023172
Chicago/Turabian StyleTian, Ye, Bin Li, Wen-Zhen Shi, Ming-Ze Chang, Ge-Juan Zhang, Zheng-Li Di, and Yong Liu. 2014. "Dynamin-Related Protein 1 Inhibitors Protect against Ischemic Toxicity through Attenuating Mitochondrial Ca2+ Uptake from Endoplasmic Reticulum Store in PC12 Cells" International Journal of Molecular Sciences 15, no. 2: 3172-3185. https://doi.org/10.3390/ijms15023172