Evodiamine Induces Apoptosis and Enhances TRAIL-Induced Apoptosis in Human Bladder Cancer Cells through mTOR/S6K1-Mediated Downregulation of Mcl-1

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
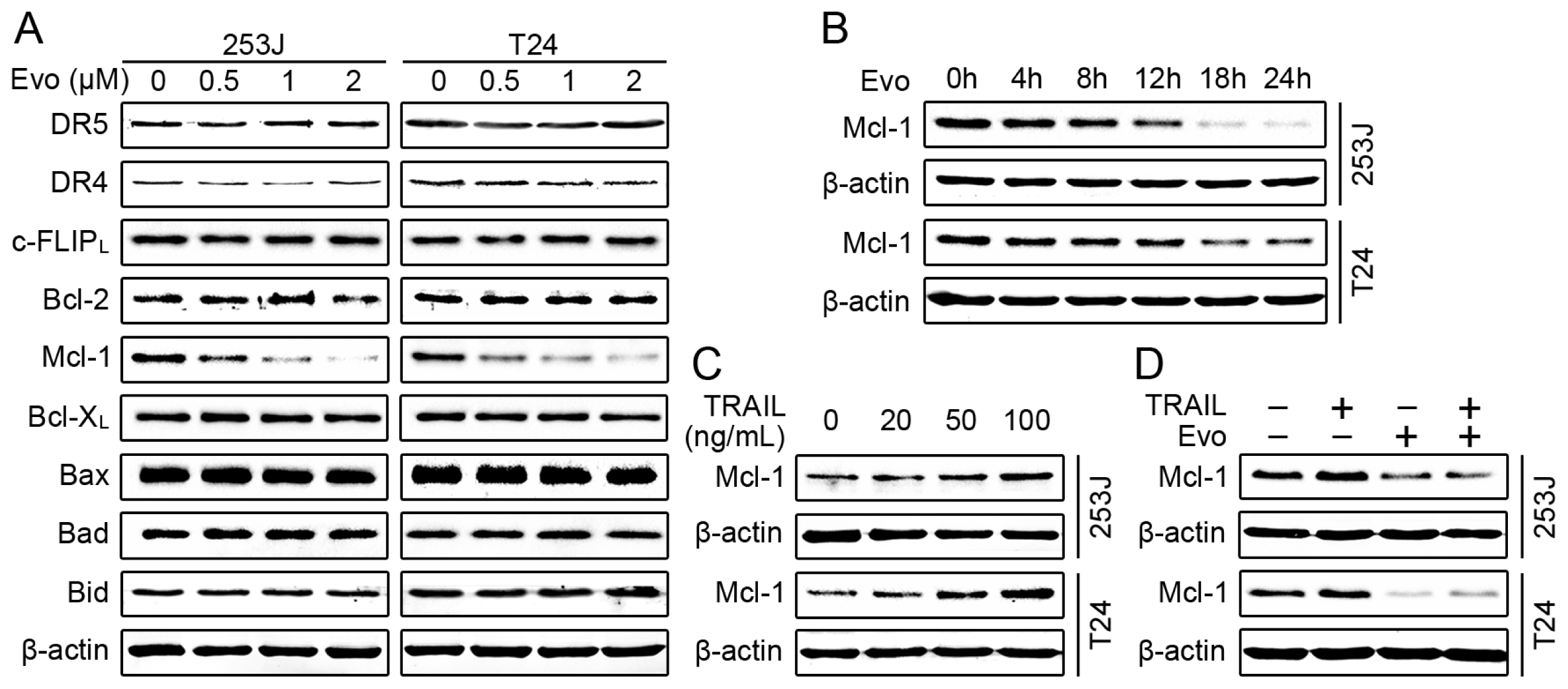
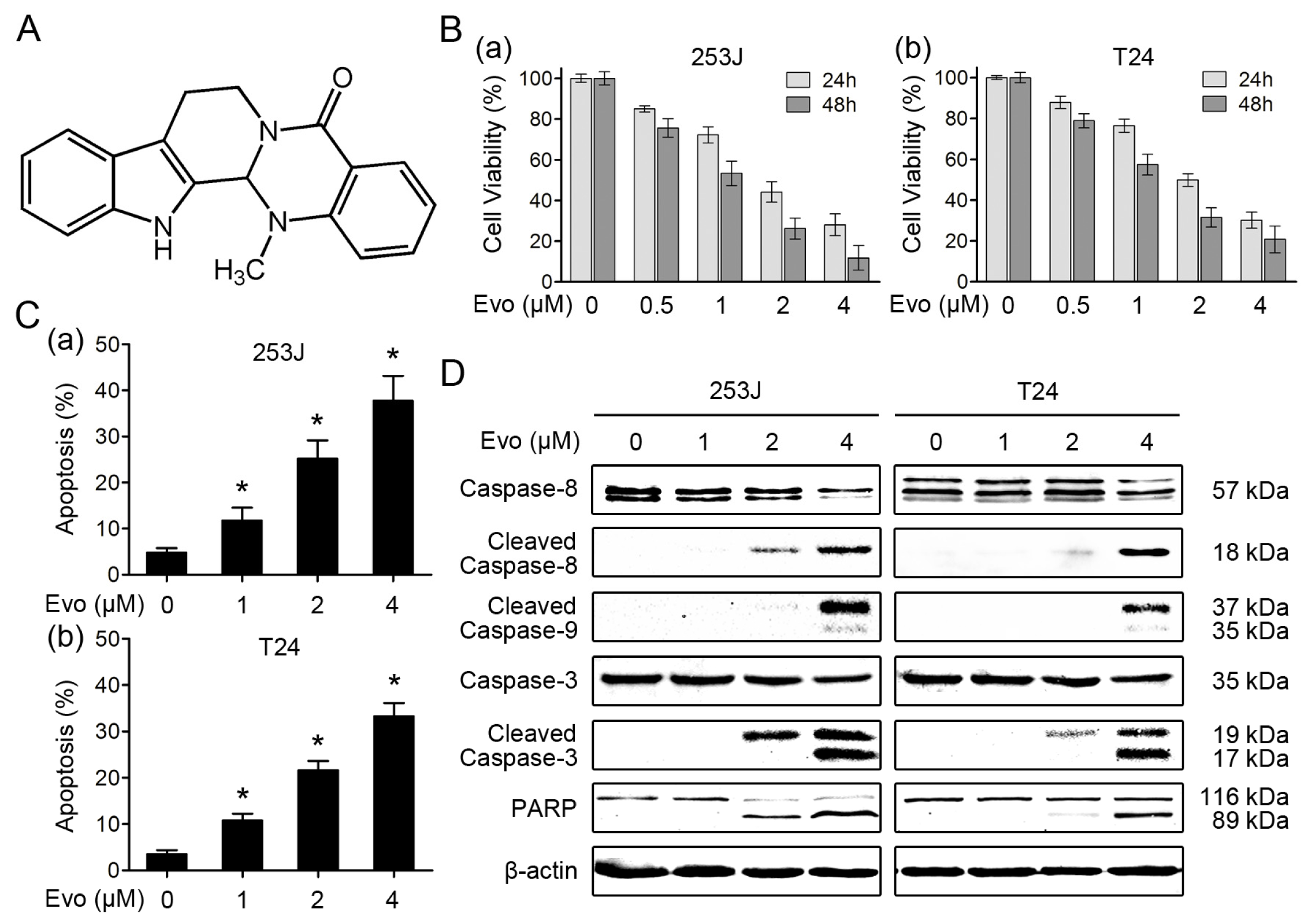
2.1. Evodiamine Induces Apoptosis in Human Bladder Cancer Cells
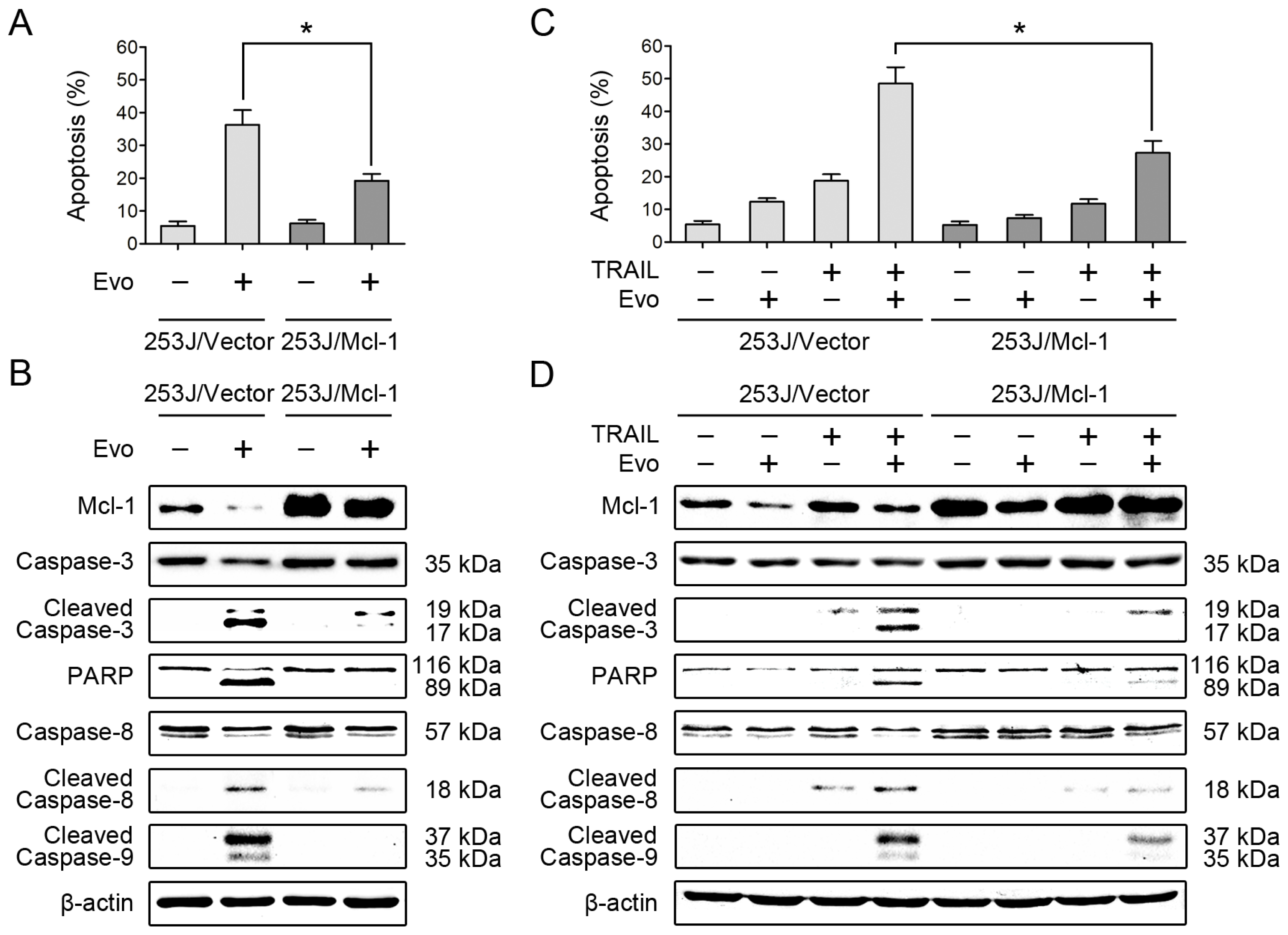
2.2. Evodiamine Enhances TRAIL-Induced Apoptosis in Human Bladder Cancer Cells
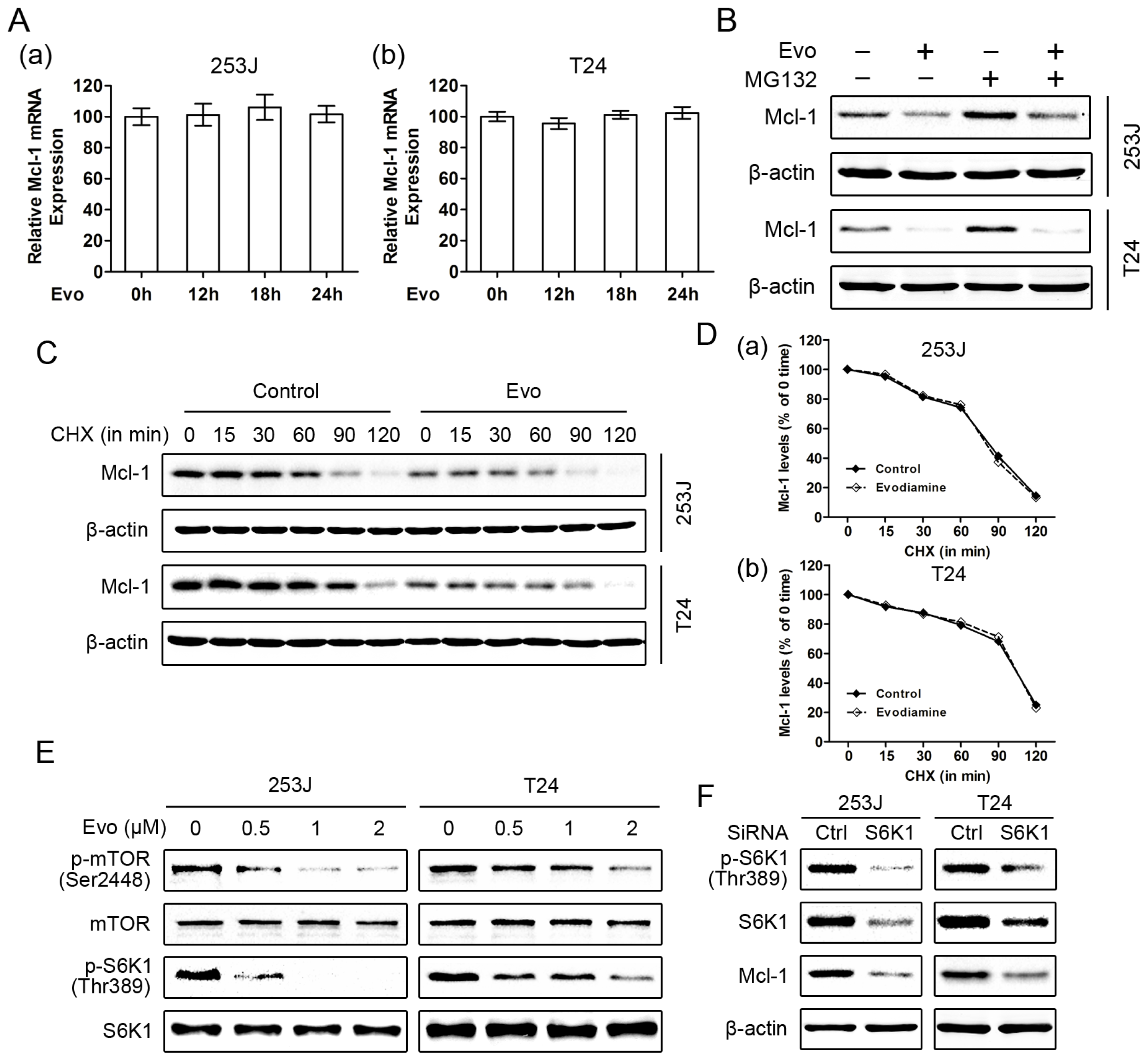
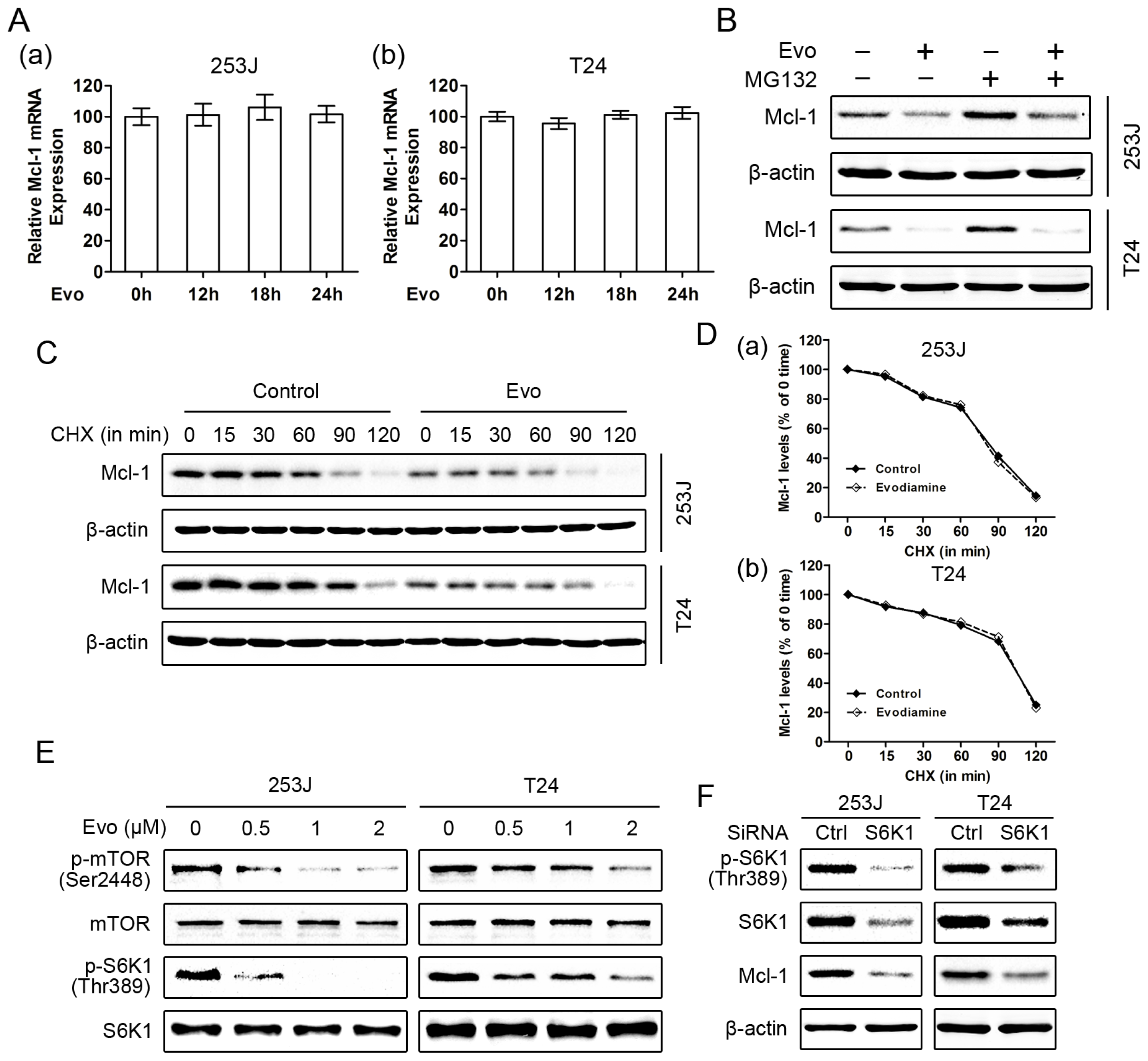
2.3. Evodiamine Reduces the Levels of Mcl-1 and Blocks TRAIL-Induced Mcl-1 Upregulation
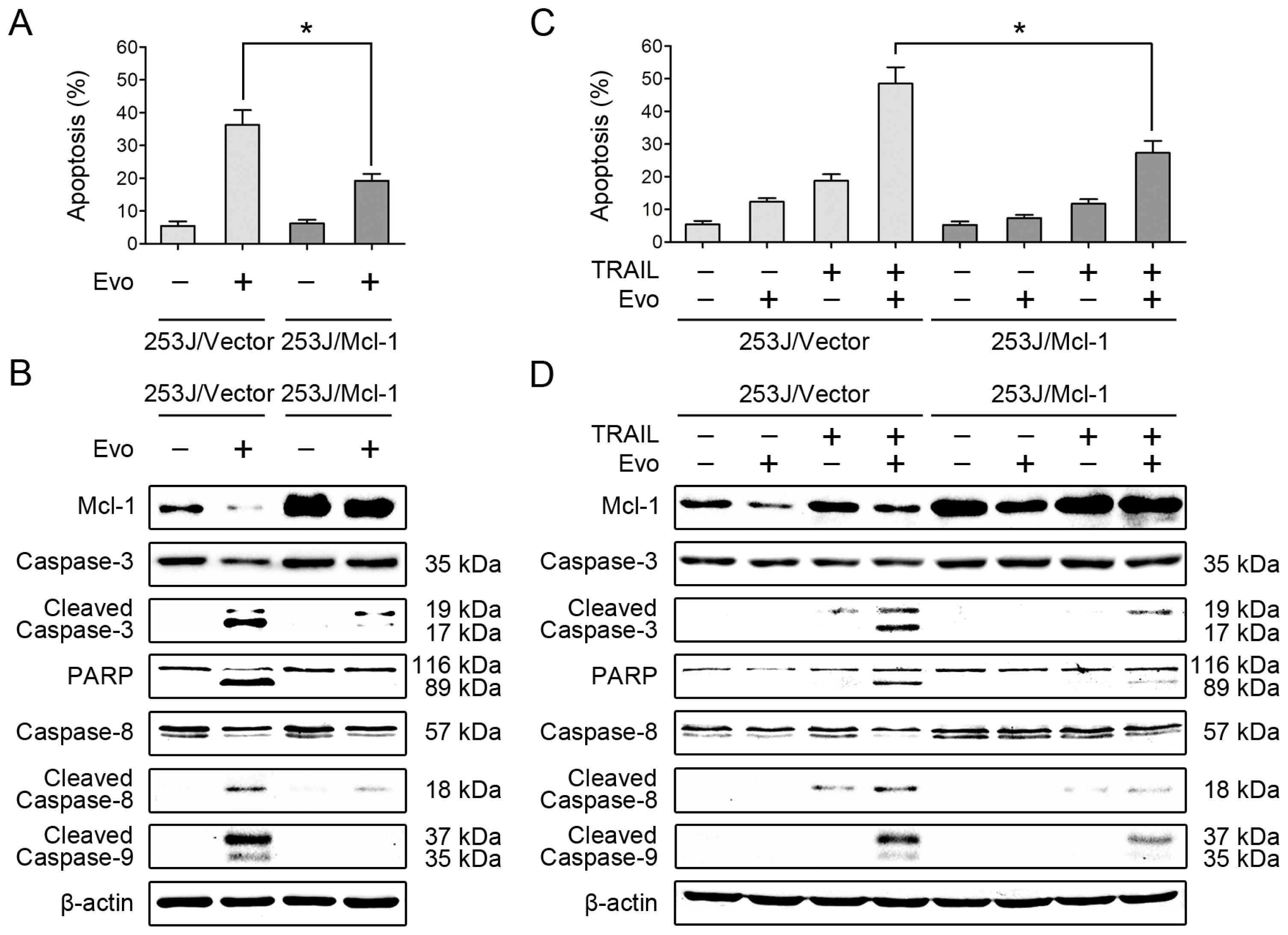
2.4. Overexpression of Mcl-1 Attenuates the Effects of Evodiamine Alone or in Combination with TRAIL on Induction of Apoptosis
2.5. Evodiamine Downregulates Mcl-1 Expression through the mTOR/S6K1 Pathway
3. Discussion
4. Experimental Section
4.1. Reagents and Antibodies
4.2. Cell Lines and Culture Conditions
4.3. Cell Viability Assay
4.4. Quantitative Detection of Apoptosis
4.5. RNA Isolation and Quantitative Real-Time PCR
4.6. Plasmids and Establishment of Stable Cell Lines Overexpressing Mcl-1
4.7. SiRNA and Transfection
4.8. Western Blot Analysis
4.9. Statistical Analysis
5. Conclusions
Acknowledgments
Conflicts of Interest
References
- Siegel, R.; Naishadham, D.; Jemal, A. Cancer statistics, 2012. CA Cancer J. Clin 2012, 62, 10–29. [Google Scholar]
- Babjuk, M.; Burger, M.; Zigeuner, R.; Shariat, S.F.; van Rhijn, B.W.; Comperat, E.; Sylvester, R.J.; Kaasinen, E.; Bohle, A.; Palou, R.J.; et al. EAU guidelines on non-muscle-invasive urothelial carcinoma of the bladder: Update 2013. Eur. Urol 2013, 64, 639–653. [Google Scholar]
- Leopardo, D.; Cecere, S.C.; di Napoli, M.; Cavaliere, C.; Pisano, C.; Striano, S.; Marra, L.; Menna, L.; Claudio, L.; Perdona, S.; et al. Intravesical chemo-immunotherapy in non muscle invasive bladder cancer. Eur. Rev. Med. Pharmacol. Sci 2013, 17, 2145–2158. [Google Scholar]
- Herr, H.W.; Morales, A. History of bacillus Calmette-Guerin and bladder cancer: An immunotherapy success story. J. Urol 2008, 179, 53–56. [Google Scholar]
- Shang, P.F.; Kwong, J.; Wang, Z.P.; Tian, J.; Jiang, L.; Yang, K.; Yue, Z.J.; Tian, J.Q. Intravesical Bacillus Calmette-Guerin versus epirubicin for Ta and T1 bladder cancer. Cochrane Database Syst. Rev 2011. [Google Scholar] [CrossRef]
- Schaefer, U.; Voloshanenko, O.; Willen, D.; Walczak, H. TRAIL: A multifunctional cytokine. Front. Biosci 2007, 12, 3813–3824. [Google Scholar]
- Herbst, R.S.; Eckhardt, S.G.; Kurzrock, R.; Ebbinghaus, S.; O’Dwyer, P.J.; Gordon, M.S.; Novotny, W.; Goldwasser, M.A.; Tohnya, T.M.; Lum, B.L.; et al. Phase I dose-escalation study of recombinant human Apo2L/TRAIL, a dual proapoptotic receptor agonist, in patients with advanced cancer. J. Clin. Oncol 2010, 28, 2839–2846. [Google Scholar]
- Ludwig, A.T.; Moore, J.M.; Luo, Y.; Chen, X.; Saltsgaver, N.A.; O’Donnell, M.A.; Griffith, T.S. Tumor necrosis factor-related apoptosis-inducing ligand: A novel mechanism for Bacillus Calmette-Guerin-induced antitumor activity. Cancer Res 2004, 64, 3386–3390. [Google Scholar]
- Moibi, J.A.; Mak, A.L.; Sun, B.; Moore, R.B. Urothelial cancer cell response to combination therapy of gemcitabine and TRAIL. Int. J. Oncol 2011, 39, 61–71. [Google Scholar]
- Shimada, O.; Wu, X.; Jin, X.; Nouh, M.A.; Fiscella, M.; Albert, V.; Matsuda, T.; Kakehi, Y. Human agonistic antibody to tumor necrosis factor-related apoptosis-inducing ligand receptor 2 induces cytotoxicity and apoptosis in prostate cancer and bladder cancer cells. Urology 2007, 69, 395–401. [Google Scholar]
- Zhang, L.; Fang, B. Mechanisms of resistance to TRAIL-induced apoptosis in cancer. Cancer Gene Ther 2005, 12, 228–237. [Google Scholar]
- Kauntz, H.; Bousserouel, S.; Gosse, F.; Raul, F. The flavonolignan silibinin potentiates TRAIL-induced apoptosis in human colon adenocarcinoma and in derived TRAIL-resistant metastatic cells. Apoptosis 2012, 17, 797–809. [Google Scholar]
- Park, S.; Cho, D.H.; Andera, L.; Suh, N.; Kim, I. Curcumin enhances TRAIL-induced apoptosis of breast cancer cells by regulating apoptosis-related proteins. Mol. Cell. Biochem 2013, 383, 39–48. [Google Scholar]
- Yu, H.; Jin, H.; Gong, W.; Wang, Z.; Liang, H. Pharmacological actions of multi-target-directed evodiamine. Molecules 2013, 18, 1826–1843. [Google Scholar]
- Wang, K.L.; Hsia, S.M.; Yeh, J.Y.; Cheng, S.C.; Wang, P.S.; Wang, S.W. Anti-Proliferative Effects of Evodiamine on Human Breast Cancer Cells. PLoS One 2013, 8, e67297. [Google Scholar]
- Huang, D.M.; Guh, J.H.; Huang, Y.T.; Chueh, S.C.; Chiang, P.C.; Teng, C.M. Induction of mitotic arrest and apoptosis in human prostate cancer pc-3 cells by evodiamine. J. Urol 2005, 173, 256–261. [Google Scholar]
- Ogasawara, M.; Matsubara, T.; Suzuki, H. Inhibitory effects of evodiamine on in vitro invasion and experimental lung metastasis of murine colon cancer cells. Biol. Pharm. Bull 2001, 24, 917–920. [Google Scholar]
- Zhang, C.; Fan, X.; Xu, X.; Yang, X.; Wang, X.; Liang, H.P. Evodiamine induces caspase-dependent apoptosis and S phase arrest in human colon lovo cells. Anticancer Drugs 2010, 21, 766–776. [Google Scholar]
- Wang, C.; Wang, M.W.; Tashiro, S.; Onodera, S.; Ikejima, T. Roles of SIRT1 and phosphoinositide 3-OH kinase/protein kinase C pathways in evodiamine-induced human melanoma A375-S2 cell death. J. Pharmacol. Sci 2005, 97, 494–500. [Google Scholar]
- Wang, C.; Li, S.; Wang, M.W. Evodiamine-induced human melanoma A375-S2 cell death was mediated by PI3K/Akt/caspase and Fas-L/NF-kappaB signaling pathways and augmented by ubiquitin-proteasome inhibition. Toxicol. In Vitro 2010, 24, 898–904. [Google Scholar]
- Jiang, J.; Hu, C. Evodiamine: A novel anti-cancer alkaloid from Evodia rutaecarpa. Molecules 2009, 14, 1852–1859. [Google Scholar]
- Wei, W.T.; Chen, H.; Wang, Z.H.; Ni, Z.L.; Liu, H.B.; Tong, H.F.; Guo, H.C.; Liu, D.L.; Lin, S.Z. Enhanced antitumor efficacy of gemcitabine by evodiamine on pancreatic cancer via regulating PI3K/Akt pathway. Int. J. Biol. Sci 2012, 8, 1–14. [Google Scholar]
- Liao, C.H.; Pan, S.L.; Guh, J.H.; Chang, Y.L.; Pai, H.C.; Lin, C.H.; Teng, C.M. Antitumor mechanism of evodiamine, a constituent from Chinese herb Evodiae fructus, in human multiple-drug resistant breast cancer NCI/ADR-RES cells in vitro and in vivo. Carcinogenesis 2005, 26, 968–975. [Google Scholar]
- Steele, L.P.; Georgopoulos, N.T.; Southgate, J.; Selby, P.J.; Trejdosiewicz, L.K. Differential susceptibility to TRAIL of normal versus malignant human urothelial cells. Cell Death Differ 2006, 13, 1564–1576. [Google Scholar]
- White-Gilbertson, S.J.; Kasman, L.; McKillop, J.; Tirodkar, T.; Lu, P.; Voelkel-Johnson, C. Oxidative stress sensitizes bladder cancer cells to TRAIL mediated apoptosis by down-regulating anti-apoptotic proteins. J. Urol 2009, 182, 1178–1185. [Google Scholar]
- Abdulghani, J.; El-Deiry, W.S. TRAIL receptor signaling and therapeutics. Expert Opin. Ther. Targets 2010, 14, 1091–1108. [Google Scholar]
- Kim, S.H.; Ricci, M.S.; El-Deiry, W.S. Mcl-1: A gateway to TRAIL sensitization. Cancer Res 2008, 68, 2062–2064. [Google Scholar]
- Wang, X.; Chen, W.; Zeng, W.; Bai, L.; Tesfaigzi, Y.; Belinsky, S.A.; Lin, Y. Akt-mediated eminent expression of c-FLIP and Mcl-1 confers acquired resistance to TRAIL-induced cytotoxicity to lung cancer cells. Mol. Cancer Ther 2008, 7, 1156–1163. [Google Scholar]
- Jacquemin, G.; Granci, V.; Gallouet, A.S.; Lalaoui, N.; Morle, A.; Iessi, E.; Morizot, A.; Garrido, C.; Guillaudeux, T.; Micheau, O. Quercetin-mediated Mcl-1 and survivin downregulation restores TRAIL-induced apoptosis in non-Hodgkin’s lymphoma B cells. Haematologica 2012, 97, 38–46. [Google Scholar]
- Maurer, U.; Charvet, C.; Wagman, A.S.; Dejardin, E.; Green, D.R. Glycogen synthase kinase-3 regulates mitochondrial outer membrane permeabilization and apoptosis by destabilization of MCL-1. Mol. Cell 2006, 21, 749–760. [Google Scholar]
- Opferman, J.T. Unraveling MCL-1 degradation. Cell Death Differ 2006, 13, 1260–1262. [Google Scholar]
- Mills, J.R.; Hippo, Y.; Robert, F.; Chen, S.M.; Malina, A.; Lin, C.J.; Trojahn, U.; Wendel, H.G.; Charest, A.; Bronson, R.T.; et al. mTORC1 promotes survival through translational control of Mcl-1. Proc. Natl. Acad. Sci. USA 2008, 105, 10853–10858. [Google Scholar]
- Hong, S.E.; Kim, E.K.; Jin, H.O.; Kim, H.A.; Lee, J.K.; Koh, J.S.; Seol, H.; Kim, J.I.; Park, I.C.; Noh, W.C. S6K1 inhibition enhances tamoxifen-induced cell death in MCF-7 cells through translational inhibition of Mcl-1 and survivin. Cell Biol. Toxicol 2013, 29, 273–282. [Google Scholar]
- Viswanath, V.; Wu, Y.; Boonplueang, R.; Chen, S.; Stevenson, F.F.; Yantiri, F.; Yang, L.; Beal, M.F.; Andersen, J.K. Caspase-9 activation results in downstream caspase-8 activation and bid cleavage in 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-induced Parkinson’s disease. J. Neurosci 2001, 21, 9519–9528. [Google Scholar]
- Ferreira, K.S.; Kreutz, C.; Macnelly, S.; Neubert, K.; Haber, A.; Bogyo, M.; Timmer, J.; Borner, C. Caspase-3 feeds back on caspase-8, Bid and XIAP in type I Fas signaling in primary mouse hepatocytes. Apoptosis 2012, 17, 503–515. [Google Scholar]
- de Vries, J.F.; Wammes, L.J.; Jedema, I.; van Dreunen, L.; Nijmeijer, B.A.; Heemskerk, M.H.; Willemze, R.; Falkenburg, J.H.; Barge, R.M. Involvement of caspase-8 in chemotherapy-induced apoptosis of patient derived leukemia cell lines independent of the death receptor pathway and downstream from mitochondria. Apoptosis 2007, 12, 181–193. [Google Scholar]
- Takada, Y.; Kobayashi, Y.; Aggarwal, B.B. Evodiamine abolishes constitutive and inducible NF-kappaB activation by inhibiting IkappaBalpha kinase activation, thereby suppressing NF-kappaB-regulated antiapoptotic and metastatic gene expression, up-regulating apoptosis, and inhibiting invasion. J. Biol. Chem 2005, 280, 17203–17212. [Google Scholar]
- Oka, N.; Tanimoto, S.; Taue, R.; Nakatsuji, H.; Kishimoto, T.; Izaki, H.; Fukumori, T.; Takahashi, M.; Nishitani, M.; Kanayama, H.O. Role of phosphatidylinositol-3 kinase/Akt pathway in bladder cancer cell apoptosis induced by tumor necrosis factor-related apoptosis-inducing ligand. Cancer Sci 2006, 97, 1093–1098. [Google Scholar]
- Ren, H.; Zhao, L.; Li, Y.; Yue, P.; Deng, X.; Owonikoko, T.K.; Chen, M.; Khuri, F.R.; Sun, S.Y. The PI3 kinase inhibitor NVP-BKM120 induces GSK3/FBXW7-dependent Mcl-1 degradation, contributing to induction of apoptosis and enhancement of TRAIL-induced apoptosis. Cancer Lett 2013, 338, 229–238. [Google Scholar]
- Murphy, A.C.; Weyhenmeyer, B.; Noonan, J.; Kilbride, S.M.; Schimansky, S.; Loh, K.P.; Kogel, D.; Letai, A.G.; Prehn, J.H.; Murphy, B.M. Modulation of Mcl-1 sensitizes glioblastoma to TRAIL-induced apoptosis. Apoptosis 2013. [Google Scholar] [CrossRef]
- Clohessy, J.G.; Zhuang, J.; de Boer, J.; Gil-Gomez, G.; Brady, H.J. Mcl-1 interacts with truncated Bid and inhibits its induction of cytochrome c release and its role in receptor-mediated apoptosis. J. Biol. Chem 2006, 281, 5750–5759. [Google Scholar]
- Akgul, C. Mcl-1 is a potential therapeutic target in multiple types of cancer. Cell. Mol. Life Sci 2009, 66, 1326–1336. [Google Scholar]
- Liu, Y.N.; Pan, S.L.; Liao, C.H.; Huang, D.Y.; Guh, J.H.; Peng, C.Y.; Chang, Y.L.; Teng, C.M. Evodiamine represses hypoxia-induced inflammatory proteins expression and hypoxia-inducible factor 1alpha accumulation in RAW264.7. Shock 2009, 32, 263–269. [Google Scholar]
- Guertin, D.A.; Sabatini, D.M. Defining the role of mTOR in cancer. Cancer Cell 2007, 12, 9–22. [Google Scholar]
- Wei, G.; Twomey, D.; Lamb, J.; Schlis, K.; Agarwal, J.; Stam, R.W.; Opferman, J.T.; Sallan, S.E.; den Boer, M.L.; Pieters, R.; et al. Gene expression-based chemical genomics identifies rapamycin as a modulator of MCL1 and glucocorticoid resistance. Cancer Cell 2006, 10, 331–342. [Google Scholar]
- Choi, H.N.; Jin, H.O.; Kim, J.H.; Hong, S.E.; Kim, H.A.; Kim, E.K.; Lee, J.K.; Park, I.C.; Noh, W.C. Inhibition of S6K1 enhances glucose deprivation-induced cell death via downregulation of anti-apoptotic proteins in MCF-7 breast cancer cells. Biochem. Biophys. Res. Commun 2013, 432, 123–128. [Google Scholar]
- Zhao, L.; Yue, P.; Khuri, F.R.; Sun, S.Y. mTOR complex 2 is involved in regulation of Cbl-dependent c-FLIP degradation and sensitivity of TRAIL-induced apoptosis. Cancer Res 2013, 73, 1946–1957. [Google Scholar]


© 2014 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Zhang, T.; Qu, S.; Shi, Q.; He, D.; Jin, X. Evodiamine Induces Apoptosis and Enhances TRAIL-Induced Apoptosis in Human Bladder Cancer Cells through mTOR/S6K1-Mediated Downregulation of Mcl-1. Int. J. Mol. Sci. 2014, 15, 3154-3171. https://doi.org/10.3390/ijms15023154
Zhang T, Qu S, Shi Q, He D, Jin X. Evodiamine Induces Apoptosis and Enhances TRAIL-Induced Apoptosis in Human Bladder Cancer Cells through mTOR/S6K1-Mediated Downregulation of Mcl-1. International Journal of Molecular Sciences. 2014; 15(2):3154-3171. https://doi.org/10.3390/ijms15023154
Chicago/Turabian StyleZhang, Tao, Shanna Qu, Qi Shi, Dalin He, and Xunbo Jin. 2014. "Evodiamine Induces Apoptosis and Enhances TRAIL-Induced Apoptosis in Human Bladder Cancer Cells through mTOR/S6K1-Mediated Downregulation of Mcl-1" International Journal of Molecular Sciences 15, no. 2: 3154-3171. https://doi.org/10.3390/ijms15023154