NTCP and Beyond: Opening the Door to Unveil Hepatitis B Virus Entry



{kind=link}
Abstract
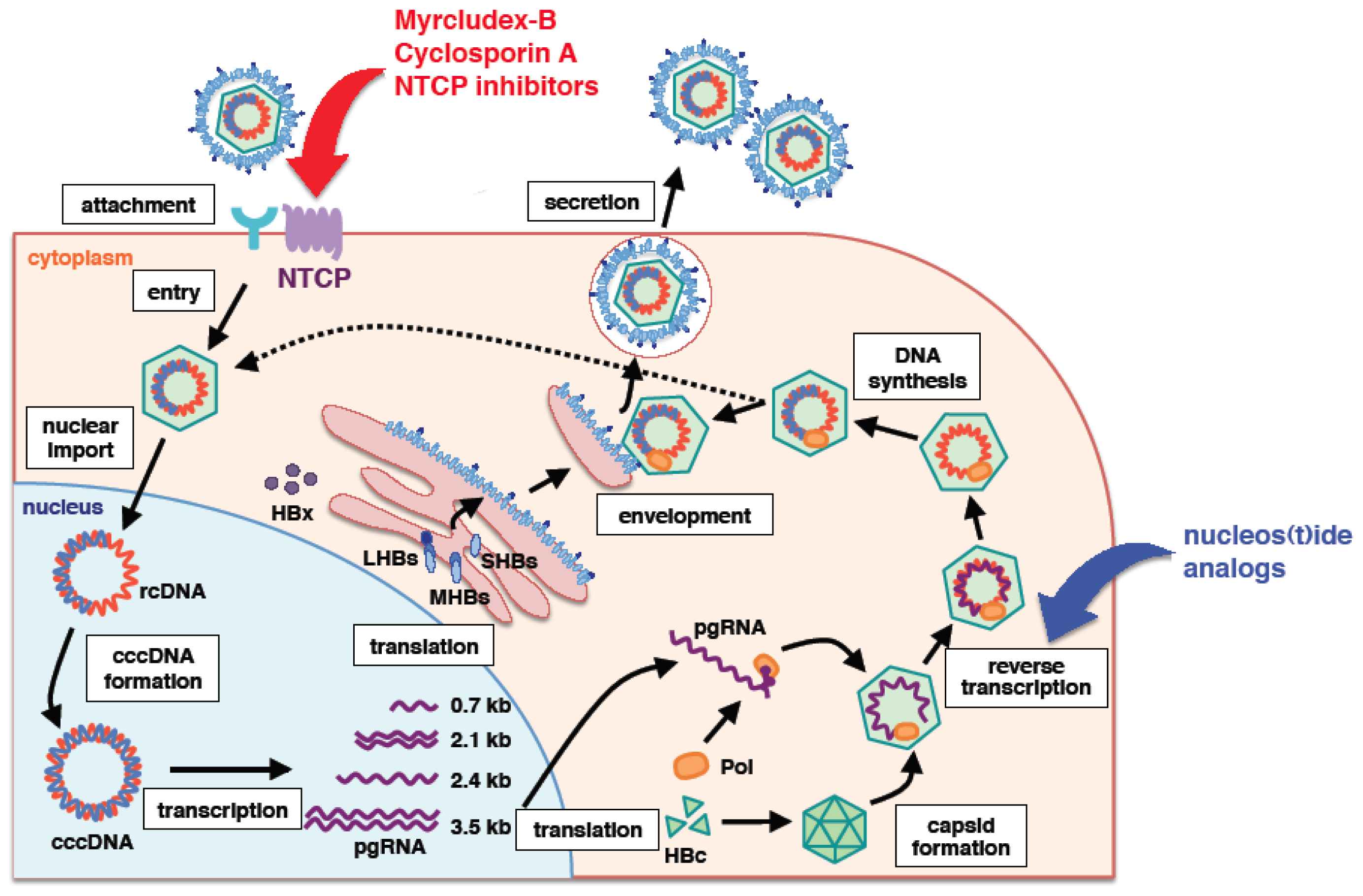
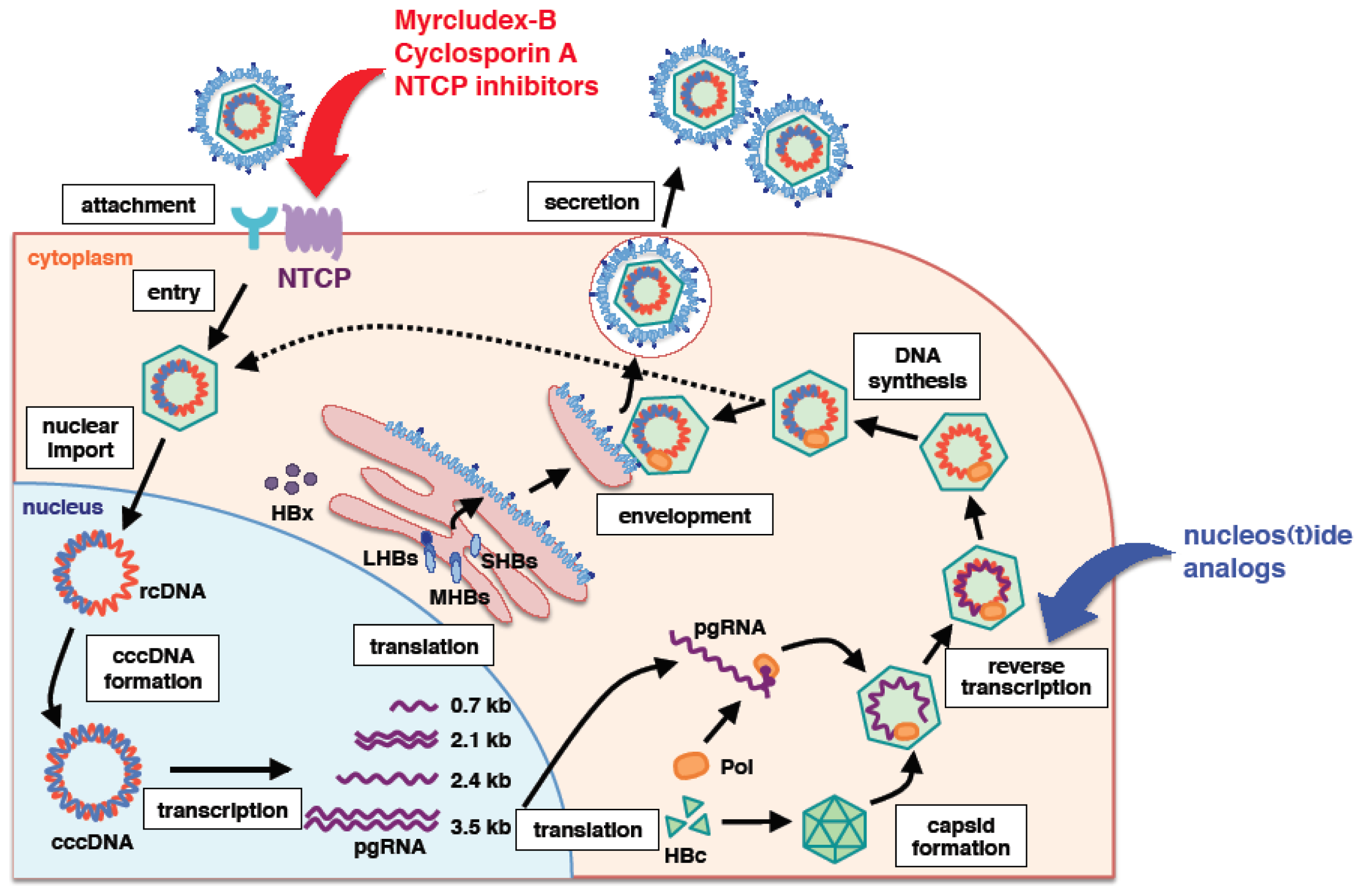
:1. Introduction
2. PreS1 Region of HBV Surface Protein Is Essential for Viral Entry
3. Sodium Taurocholate Co-Transporting Polypeptide (NTCP) as a Bona Fide HBV Receptor
4. Other Factors Essential for HBV Infection?
5. General Characteristic Features of NTCP
6. NTCP as a Target for Anti-HBV Agents
7. Conclusions
Acknowledgments
Conflicts of Interest
- Author ContributionsAll of the authors wrote the paper.
References
- Ott, J.J.; Stevens, G.A.; Groeger, J.; Wiersma, S.T. Global epidemiology of hepatitis B virus infection: New estimates of age-specific HBsAg seroprevalence and endemicity. Vaccine 2012, 30, 2212–2219. [Google Scholar]
- Zoulim, F.; Locarnini, S. Optimal management of chronic hepatitis B patients with treatment failure and antiviral drug resistance. Liver Int 2013, 33, 116–124. [Google Scholar]
- Zoulim, F.; Perrillo, R.; Hepatitis, B. Reflections on the current approach to antiviral therapy. J. Hepatol 2008, 48, S2–S19. [Google Scholar]
- Marcellin, P.; Bonino, F.; Yurdaydin, C.; Hadziyannis, S.; Moucari, R.; Kapprell, H.P.; Rothe, V.; Popescu, M.; Brunetto, M.R. Hepatitis B surface antigen levels: Association with 5-year response to peginterferon alfa-2a in hepatitis B e-antigen-negative patients. Hepatol. Int 2013, 7, 88–97. [Google Scholar]
- Belloni, L.; Allweiss, L.; Guerrieri, F.; Pediconi, N.; Volz, T.; Pollicino, T.; Petersen, J.; Raimondo, G.; Dandri, M.; Levrero, M. IFN-alpha inhibits HBV transcription and replication in cell culture and in humanized mice by targeting the epigenetic regulation of the nuclear cccDNA minichromosome. J. Clin. Invest 2012, 122, 529–537. [Google Scholar]
- Haqqani, A.A.; Tilton, J.C. Entry inhibitors and their use in the treatment of HIV-1 infection. Antiviral Res 2013, 98, 158–170. [Google Scholar]
- Mercer, J.; Helenius, A. Gulping rather than sipping: Macropinocytosis as a way of virus entry. Curr. Opin. Microbiol 2012, 15, 490–499. [Google Scholar]
- Zeisel, M.B.; Fofana, I.; Fafi-Kremer, S.; Baumert, T.F. Hepatitis C virus entry into hepatocytes: Molecular mechanisms and targets for antiviral therapies. J. Hepatol 2011, 54, 566–576. [Google Scholar]
- Yan, H.; Zhong, G.; Xu, G.; He, W.; Jing, Z.; Gao, Z.; Huang, Y.; Qi, Y.; Peng, B.; Wang, H.; et al. Sodium taurocholate cotransporting polypeptide is a functional receptor for human hepatitis B and D virus. Elife 2012, 1, e00049. [Google Scholar]
- Meier, A.; Mehrle, S.; Weiss, T.S.; Mier, W.; Urban, S. Myristoylated PreS1-domain of the hepatitis B virus L-protein mediates specific binding to differentiated hepatocytes. Hepatology 2013, 58, 31–42. [Google Scholar]
- Glebe, D.; Urban, S. Viral and cellular determinants involved in hepadnaviral entry. World J. Gastroenterol 2007, 13, 22–38. [Google Scholar]
- Lamas Longarela, O.; Schmidt, T.T.; Schoneweis, K.; Romeo, R.; Wedemeyer, H.; Urban, S.; Schulze, A. Proteoglycans act as cellular hepatitis delta virus attachment receptors. PLoS One 2013, 8, e58340. [Google Scholar]
- Leistner, C.M.; Gruen-Bernhard, S.; Glebe, D. Role of glycosaminoglycans for binding and infection of hepatitis B virus. Cell. Microbiol 2008, 10, 122–133. [Google Scholar]
- Schulze, A.; Gripon, P.; Urban, S. Hepatitis B virus infection initiates with a large surface protein-dependent binding to heparan sulfate proteoglycans. Hepatology 2007, 46, 1759–1768. [Google Scholar]
- Cooper, A.; Shaul, Y. Clathrin-mediated endocytosis and lysosomal cleavage of hepatitis B virus capsid-like core particles. J. Biol. Chem 2006, 281, 16563–16569. [Google Scholar]
- Gao, Z.; Li, M.; He, W.; Li, W. Hepatitis B virus may enter HepG2 cells complemented with human NTCP via macropinocytosis. Proceedings of the 2013 International Meeting on Molecular Biology of Hepatitis B Viruses, Shanghai, China, 20–23 October 2013; p. O-2.
- Huang, H.C.; Chen, C.C.; Chang, W.C.; Tao, M.H.; Huang, C. Entry of hepatitis B virus into immortalized human primary hepatocytes by clathrin-dependent endocytosis. J. Virol 2012, 86, 9443–9453. [Google Scholar]
- Macovei, A.; Radulescu, C.; Lazar, C.; Petrescu, S.; Durantel, D.; Dwek, R.A.; Zitzmann, N.; Nichita, N.B. Hepatitis B virus requires intact caveolin-1 function for productive infection in HepaRG cells. J. Virol 2010, 84, 243–253. [Google Scholar]
- Stibbe, W.; Gerlich, W.H. Structural relationships between minor and major proteins of hepatitis B surface antigen. J. Virol 1983, 46, 626–628. [Google Scholar]
- Heermann, K.H.; Goldmann, U.; Schwartz, W.; Seyffarth, T.; Baumgarten, H.; Gerlich, W.H. Large surface proteins of hepatitis B virus containing the pre-S sequence. J. Virol 1984, 52, 396–402. [Google Scholar]
- Baumert, T.F.; Meredith, L.; Ni, Y.; Felmlee, D.J.; McKeating, J.A.; Urban, S. Entry of hepatitis B and C viruses—Recent progress and future impact. Curr. Opin. Virol 2014, 4C, 58–65. [Google Scholar]
- Abou-Jaoude, G.; Sureau, C. Entry of hepatitis delta virus requires the conserved cysteine residues of the hepatitis B virus envelope protein antigenic loop and is blocked by inhibitors of thiol-disulfide exchange. J. Virol 2007, 81, 13057–13066. [Google Scholar]
- Bremer, C.M.; Sominskaya, I.; Skrastina, D.; Pumpens, P.; El Wahed, A.A.; Beutling, U.; Frank, R.; Fritz, H.J.; Hunsmann, G.; Gerlich, W.H.; et al. N-terminal myristoylation-dependent masking of neutralizing epitopes in the preS1 attachment site of hepatitis B virus. J. Hepatol 2011, 55, 29–37. [Google Scholar]
- Iwarson, S.; Tabor, E.; Thomas, H.C.; Goodall, A.; Waters, J.; Snoy, P.; Shih, J.W.; Gerety, R.J. Neutralization of hepatitis B virus infectivity by a murine monoclonal antibody: An experimental study in the chimpanzee. J. Med. Virol 1985, 16, 89–96. [Google Scholar]
- Ni, Y.; Sonnabend, J.; Seitz, S.; Urban, S. The pre-s2 domain of the hepatitis B virus is dispensable for infectivity but serves a spacer function for l-protein-connected virus assembly. J. Virol 2010, 84, 3879–3888. [Google Scholar]
- Salisse, J.; Sureau, C. A function essential to viral entry underlies the hepatitis B virus “a” determinant. J. Virol 2009, 83, 9321–9328. [Google Scholar]
- Shearer, M.H.; Sureau, C.; Dunbar, B.; Kennedy, R.C. Structural characterization of viral neutralizing monoclonal antibodies to hepatitis B surface antigen. Mol. Immunol 1998, 35, 1149–1160. [Google Scholar]
- Barrera, A.; Guerra, B.; Notvall, L.; Lanford, R.E. Mapping of the hepatitis B virus pre-S1 domain involved in receptor recognition. J. Virol 2005, 79, 9786–9798. [Google Scholar]
- Engelke, M.; Mills, K.; Seitz, S.; Simon, P.; Gripon, P.; Schnolzer, M.; Urban, S. Characterization of a hepatitis B and hepatitis delta virus receptor binding site. Hepatology 2006, 43, 750–760. [Google Scholar]
- Glebe, D.; Urban, S.; Knoop, E.V.; Cag, N.; Krass, P.; Grun, S.; Bulavaite, A.; Sasnauskas, K.; Gerlich, W.H. Mapping of the hepatitis B virus attachment site by use of infection-inhibiting preS1 lipopeptides and tupaia hepatocytes. Gastroenterology 2005, 129, 234–245. [Google Scholar]
- Gripon, P.; Cannie, I.; Urban, S. Efficient inhibition of hepatitis B virus infection by acylated peptides derived from the large viral surface protein. J. Virol 2005, 79, 1613–1622. [Google Scholar]
- Hong, H.J.; Ryu, C.J.; Hur, H.; Kim, S.; Oh, H.K.; Oh, M.S.; Park, S.Y. In vivo neutralization of hepatitis B virus infection by an anti-preS1 humanized antibody in chimpanzees. Virology 2004, 318, 134–141. [Google Scholar]
- Le Seyec, J.; Chouteau, P.; Cannie, I.; Guguen-Guillouzo, C.; Gripon, P. Infection process of the hepatitis B virus depends on the presence of a defined sequence in the pre-S1 domain. J. Virol 1999, 73, 2052–2057. [Google Scholar]
- Maeng, C.Y.; Ryu, C.J.; Gripon, P.; Guguen-Guillouzo, C.; Hong, H.J. Fine mapping of virus-neutralizing epitopes on hepatitis B virus PreS1. Virology 2000, 270, 9–16. [Google Scholar]
- Schulze, A.; Schieck, A.; Ni, Y.; Mier, W.; Urban, S. Fine mapping of pre-S sequence requirements for hepatitis B virus large envelope protein-mediated receptor interaction. J. Virol 2010, 84, 1989–2000. [Google Scholar]
- Petersen, J.; Dandri, M.; Mier, W.; Lutgehetmann, M.; Volz, T.; von Weizsacker, F.; Haberkorn, U.; Fischer, L.; Pollok, J.M.; Erbes, B.; et al. Prevention of hepatitis B virus infection in vivo by entry inhibitors derived from the large envelope protein. Nat. Biotechnol 2008, 26, 335–341. [Google Scholar]
- Schieck, A.; Schulze, A.; Gahler, C.; Muller, T.; Haberkorn, U.; Alexandrov, A.; Urban, S.; Mier, W. Hepatitis B virus hepatotropism is mediated by specific receptor recognition in the liver and not restricted to susceptible hosts. Hepatology 2013, 58, 43–53. [Google Scholar]
- Ni, Y.; Lempp, F.A.; Mehrle, S.; Nkongolo, S.; Kaufman, C.; Falth, M.; Stindt, J.; Koniger, C.; Nassal, M.; Kubitz, R.; et al. Hepatitis B and D viruses exploit sodium taurocholate co-transporting polypeptide for species-specific entry into hepatocytes. Gastroenterology 2014, in press. [Google Scholar]
- Zhong, G.; Yan, H.; Wang, H.; He, W.; Jing, Z.; Qi, Y.; Fu, L.; et al. Sodium taurocholate cotransporting polypeptide mediates woolly monkey hepatitis B virus infection of Tupaia hepatocytes. J. Virol 2013, 87, 7176–7184. [Google Scholar]
- Kotani, N.; Maeda, K.; Debori, Y.; Camus, S.; Li, R.; Chesne, C.; Sugiyama, Y. Expression and transport function of drug uptake transporters in differentiated HepaRG cells. Mol. Pharm 2012, 9, 3434–3441. [Google Scholar]
- Kullak-Ublick, G.A.; Beuers, U.; Paumgartner, G. Molecular and functional characterization of bile acid transport in human hepatoblastoma HepG2 cells. Hepatology 1996, 23, 1053–1060. [Google Scholar]
- Watashi, K.; Sluder, A.; Daito, T.; Matsunaga, S.; Ryo, A.; Nagamori, S.; Iwamoto, M.; Nakajima, S.; Tsukuda, S.; Borroto-Esoda, K.; et al. Cyclosporin A and its analogs inhibit hepatitis B virus entry into cultured hepatocytes through targeting a membrane transporter NTCP. Hepatology 2014, in press. [Google Scholar]
- Iwamoto, M.; Watashi, K.; Tsukuda, S.; Aly, H.H.; Fukasawa, M.; Fujimoto, A.; Suzuki, R.; Aizaki, H.; Ito, T.; Koiwai, O.; et al. Evaluation and identification of hepatitis B virus entry inhibitors using HepG2 cells overexpressing a membrane transporter NTCP. Biochem. Biophys. Res. Commun 2014, 443, 808–813. [Google Scholar]
- Yan, H.; Peng, B.; He, W.; Zhong, G.; Qi, Y.; Ren, B.; Gao, Z.; Jing, Z.; Song, M.; Xu, G.; et al. Molecular determinants of hepatitis B and D virus entry restriction in mouse sodium taurocholate cotransporting polypeptide. J. Virol 2013, 87, 7977–7991. [Google Scholar]
- Nkongolo, S.; Ni, Y.; Lempp, F.A.; Kaufman, C.; Lindner, T.; Esser-Nobis, K.; Lohmann, V.; Mier, W.; Mehrle, S.; Urban, S. Cyclosporin A inhibits Hepatitis B and Hepatitis D Virus entry by Cyclophilin-independent interference with the NTCP receptor. J. Hepatol 2014, in press. [Google Scholar]
- Taylor, J.M. Hepatitis delta virus. Virology 2006, 344, 71–76. [Google Scholar]
- Lindenbach, B.D.; Rice, C.M. The ins and outs of hepatitis C virus entry and assembly. Nat. Rev. Microbiol 2013, 11, 688–700. [Google Scholar]
- Zeisel, M.B.; Lupberger, J.; Fofana, I.; Baumert, T.F. Host-targeting agents for prevention and treatment of chronic hepatitis C—Perspectives and challenges. J. Hepatol 2013, 58, 375–384. [Google Scholar]
- Dorner, M.; Horwitz, J.A.; Robbins, J.B.; Barry, W.T.; Feng, Q.; Mu, K.; Jones, C.T.; Schoggins, J.W.; Catanese, M.T.; Burton, D.R.; et al. A genetically humanized mouse model for hepatitis C virus infection. Nature 2011, 474, 208–211. [Google Scholar]
- Kuroki, K.; Eng, F.; Ishikawa, T.; Turck, C.; Harada, F.; Ganem, D. gp180, a host cell glycoprotein that binds duck hepatitis B virus particles, is encoded by a member of the carboxypeptidase gene family. J. Biol. Chem 1995, 270, 15022–15028. [Google Scholar]
- Breiner, K.M.; Urban, S.; Schaller, H.; Carboxypeptidase, D. (gp180), a Golgi-resident protein, functions in the attachment and entry of avian hepatitis B viruses. J. Virol 1998, 72, 8098–8104. [Google Scholar]
- Urban, S.; Breiner, K.M.; Fehler, F.; Klingmuller, U.; Schaller, H. Avian hepatitis B virus infection is initiated by the interaction of a distinct pre-S subdomain with the cellular receptor gp180. J. Virol 1998, 72, 8089–8097. [Google Scholar]
- Taylor, J.M. Virus entry mediated by hepatitis B virus envelope proteins. World J. Gastroenterol 2013, 19, 6730–6734. [Google Scholar]
- Claro da Silva, T.; Polli, J.E.; Swaan, P.W. The solute carrier family 10 (SLC10): Beyond bile acid transport. Mol. Aspects Med 2013, 34, 252–269. [Google Scholar]
- Eloranta, J.J.; Jung, D.; Kullak-Ublick, G.A. The human Na+-taurocholate cotransporting polypeptide gene is activated by glucocorticoid receptor and peroxisome proliferator-activated receptor-gamma coactivator-1alpha, and suppressed by bile acids via a small heterodimer partner-dependent mechanism. Mol. Endocrinol 2006, 20, 65–79. [Google Scholar]
- Ananthanarayanan, M.; Ng, O.C.; Boyer, J.L.; Suchy, F.J. Characterization of cloned rat liver Na(+)-bile acid cotransporter using peptide and fusion protein antibodies. Am. J. Physiol 1994, 267, G637–G643. [Google Scholar]
- Doring, B.; Lutteke, T.; Geyer, J.; Petzinger, E. The SLC10 carrier family: Transport functions and molecular structure. Curr. Top. Membr 2012, 70, 105–168. [Google Scholar]
- Kosters, A.; Karpen, S.J. Bile acid transporters in health and disease. Xenobiotica 2008, 38, 1043–1071. [Google Scholar]
- Hagenbuch, B.; Meier, P.J. Molecular cloning, chromosomal localization, and functional characterization of a human liver Na+/bile acid cotransporter. J. Clin. Invest 1994, 93, 1326–1331. [Google Scholar]
- Hallen, S.; Mareninova, O.; Branden, M.; Sachs, G. Organization of the membrane domain of the human liver sodium/bile acid cotransporter. Biochemistry 2002, 41, 7253–7266. [Google Scholar]
- Mareninova, O.; Shin, J.M.; Vagin, O.; Turdikulova, S.; Hallen, S.; Sachs, G. Topography of the membrane domain of the liver Na+-dependent bile acid transporter. Biochemistry 2005, 44, 13702–13712. [Google Scholar]
- Hu, N.J.; Iwata, S.; Cameron, A.D.; Drew, D. Crystal structure of a bacterial homologue of the bile acid sodium symporter ASBT. Nature 2011, 478, 408–411. [Google Scholar]
- Zhou, X.; Levin, E.J.; Pan, Y.; McCoy, J.G.; Sharma, R.; Kloss, B.; Bruni, R.; Quick, M.; Zhou, M. Structural basis of the alternating-access mechanism in a bile acid transporter. Nature 2013, 505, 569–573. [Google Scholar]
- Hallen, S.; Branden, M.; Dawson, P.A.; Sachs, G. Membrane insertion scanning of the human ileal sodium/bile acid co-transporter. Biochemistry 1999, 38, 11379–11388. [Google Scholar]
- Zhang, E.Y.; Phelps, M.A.; Banerjee, A.; Khantwal, C.M.; Chang, C.; Helsper, F.; Swaan, P.W. Topology scanning and putative three-dimensional structure of the extracellular binding domains of the apical sodium-dependent bile acid transporter (SLC10A2). Biochemistry 2004, 43, 11380–11392. [Google Scholar]
- Ho, R.H.; Leake, B.F.; Roberts, R.L.; Lee, W.; Kim, R.B. Ethnicity-dependent polymorphism in Na+-taurocholate cotransporting polypeptide (SLC10A1) reveals a domain critical for bile acid substrate recognition. J. Biol. Chem 2004, 279, 7213–7222. [Google Scholar]
- Pan, W.; Song, I.S.; Shin, H.J.; Kim, M.H.; Choi, Y.L.; Lim, S.J.; Kim, W.Y.; Lee, S.S.; Shin, J.G. Genetic polymorphisms in Na+-taurocholate co-transporting polypeptide (NTCP) and ileal apical sodium-dependent bile acid transporter (ASBT) and ethnic comparisons of functional variants of NTCP among Asian populations. Xenobiotica 2011, 41, 501–510. [Google Scholar]
- Yan, H.; Peng, B.; Liu, Y.; Xu, G.; He, W.; Ren, B.; Jing, Z.; Sui, J.; Li, W. Viral entry of Hepatitis B and D viruses and bile salts transportation share common molecular determinants on sodium taurocholate cotransporting polypeptide. J. Virol 2014, in press. [Google Scholar]
- Kilby, J.M.; Hopkins, S.; Venetta, T.M.; DiMassimo, B.; Cloud, G.A.; Lee, J.Y.; Alldredge, L.; Hunter, E.; Lambert, D.; Bolognesi, D.; et al. Potent suppression of HIV-1 replication in humans by T-20, a peptide inhibitor of gp41-mediated virus entry. Nat. Med 1998, 4, 1302–1307. [Google Scholar]
- Warner, N.; Locarnini, S. The new front-line in hepatitis B/D research: Identification and blocking of a functional receptor. Hepatology 2013, 58, 9–12. [Google Scholar]
- Watashi, K.; Shimotohno, K. Cyclophilin and viruses: Cyclophilin as a cofactor for viral infection and possible anti-viral target. Drug Target Insights 2007, 2, 9–18. [Google Scholar]
- Watashi, K.; Hijikata, M.; Hosaka, M.; Yamaji, M.; Shimotohno, K. Cyclosporin A suppresses replication of hepatitis C virus genome in cultured hepatocytes. Hepatology 2003, 38, 1282–1288. [Google Scholar]
- Bienkowska-Haba, M.; Patel, H.D.; Sapp, M. Target cell cyclophilins facilitate human papillomavirus type 16 infection. PLoS Pathog 2009, 5, e1000524. [Google Scholar]
- Liu, X.; Zhao, Z.; Li, Z.; Xu, C.; Sun, L.; Chen, J.; Liu, W. Cyclosporin A inhibits the influenza virus replication through cyclophilin A-dependent and -independent pathways. PLoS One 2012, 7, e37277. [Google Scholar]
- Luban, J.; Bossolt, K.L.; Franke, E.K.; Kalpana, G.V.; Goff, S.P. Human immunodeficiency virus type 1 Gag protein binds to cyclophilins A and B. Cell 1993, 73, 1067–1078. [Google Scholar]
- Pfefferle, S.; Schopf, J.; Kogl, M.; Friedel, C.C.; Muller, M.A.; Carbajo-Lozoya, J.; Stellberger, T.; von Dall’Armi, E.; Herzog, P.; Kallies, S.; et al. The SARS-coronavirus-host interactome: Identification of cyclophilins as target for pan-coronavirus inhibitors. PLoS Pathog 2011, 7, e1002331. [Google Scholar]
- Qing, M.; Yang, F.; Zhang, B.; Zou, G.; Robida, J.M.; Yuan, Z.; Tang, H.; Shi, P.Y. Cyclosporine inhibits flavivirus replication through blocking the interaction between host cyclophilins and viral NS5 protein. Antimicrob. Agents Chemother 2009, 53, 3226–3235. [Google Scholar]
- Towers, G.J.; Hatziioannou, T.; Cowan, S.; Goff, S.P.; Luban, J.; Bieniasz, P.D. Cyclophilin A modulates the sensitivity of HIV-1 to host restriction factors. Nat. Med 2003, 9, 1138–1143. [Google Scholar]
- Bouchard, M.J.; Puro, R.J.; Wang, L.; Schneider, R.J. Activation and inhibition of cellular calcium and tyrosine kinase signaling pathways identify targets of the HBx protein involved in hepatitis B virus replication. J. Virol 2003, 77, 7713–7719. [Google Scholar]
- Watashi, K. Alisporivir, a cyclosporin derivative that selectively inhibits cyclophilin, for the treatment of HCV infection. Curr. Opin. Investig. Drugs 2010, 11, 213–224. [Google Scholar]
- Membreno, F.E.; Espinales, J.C.; Lawitz, E.J. Cyclophilin inhibitors for hepatitis C therapy. Clin. Liver Dis 2013, 17, 129–139. [Google Scholar]
- Lucifora, J.; Esser, K.; Protzer, U. Ezetimibe blocks hepatitis B virus infection after virus uptake into hepatocytes. Antiviral Res 2013, 97, 195–197. [Google Scholar]
- Dong, Z.; Ekins, S.; Polli, J.E. Structure-activity relationship for FDA approved drugs as inhibitors of the human sodium taurocholate cotransporting polypeptide (NTCP). Mol. Pharm 2013, 10, 1008–1019. [Google Scholar]
© 2014 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Watashi, K.; Urban, S.; Li, W.; Wakita, T. NTCP and Beyond: Opening the Door to Unveil Hepatitis B Virus Entry. Int. J. Mol. Sci. 2014, 15, 2892-2905. https://doi.org/10.3390/ijms15022892
Watashi K, Urban S, Li W, Wakita T. NTCP and Beyond: Opening the Door to Unveil Hepatitis B Virus Entry. International Journal of Molecular Sciences. 2014; 15(2):2892-2905. https://doi.org/10.3390/ijms15022892
Chicago/Turabian StyleWatashi, Koichi, Stephan Urban, Wenhui Li, and Takaji Wakita. 2014. "NTCP and Beyond: Opening the Door to Unveil Hepatitis B Virus Entry" International Journal of Molecular Sciences 15, no. 2: 2892-2905. https://doi.org/10.3390/ijms15022892
APA StyleWatashi, K., Urban, S., Li, W., & Wakita, T. (2014). NTCP and Beyond: Opening the Door to Unveil Hepatitis B Virus Entry. International Journal of Molecular Sciences, 15(2), 2892-2905. https://doi.org/10.3390/ijms15022892