Structural Data on the Periplasmic Aldehyde Oxidoreductase PaoABC from Escherichia coli: SAXS and Preliminary X-ray Crystallography Analysis

Abstract
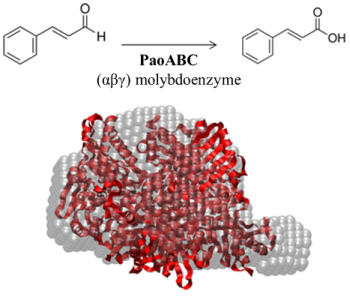
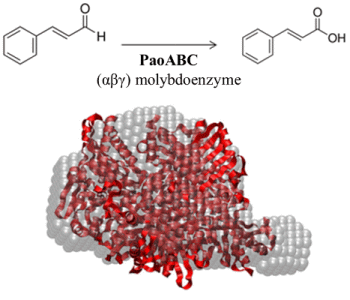
:
1. Introduction
2. Results and Discussion
2.1. Crystallization and Data Processing
2.2. Structure Determination
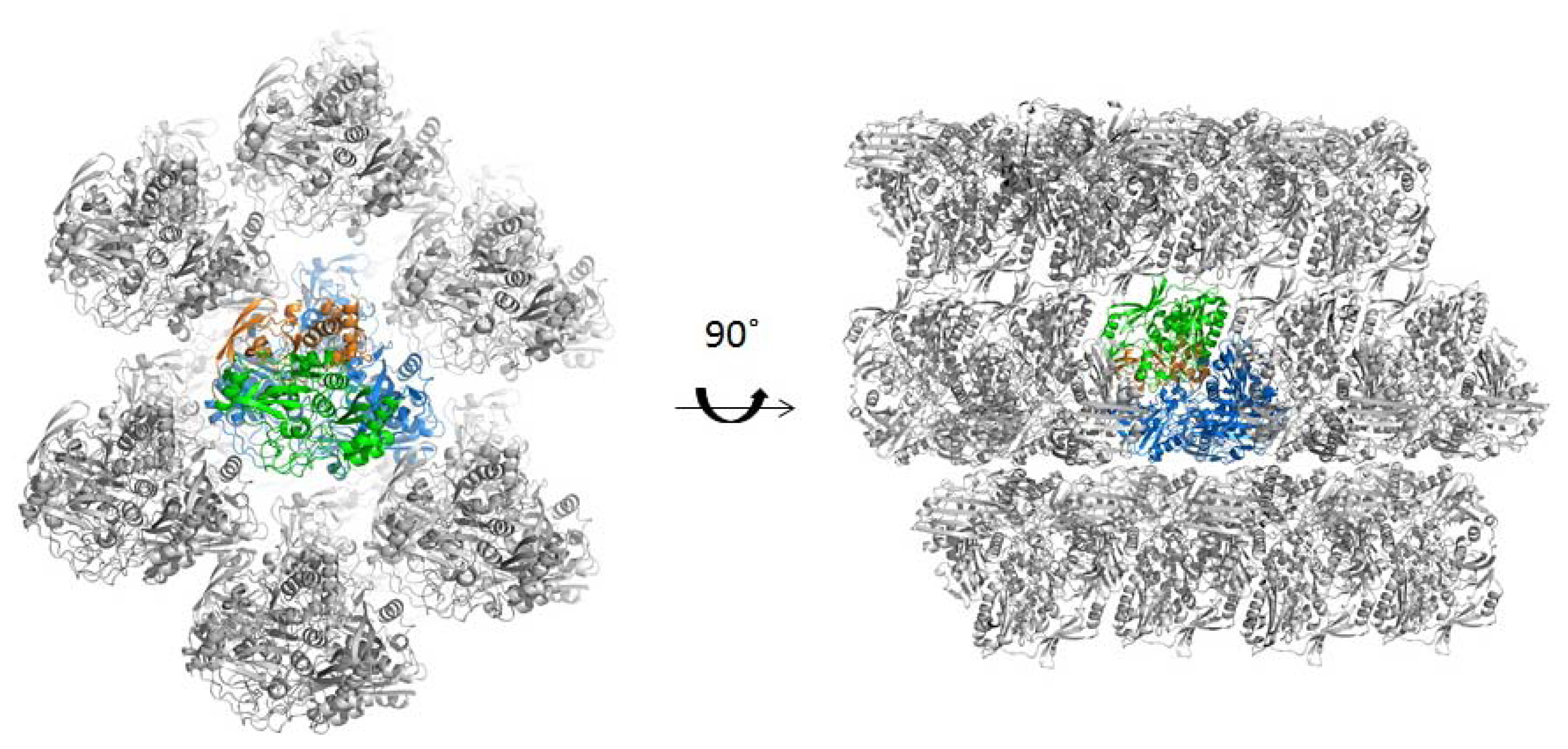
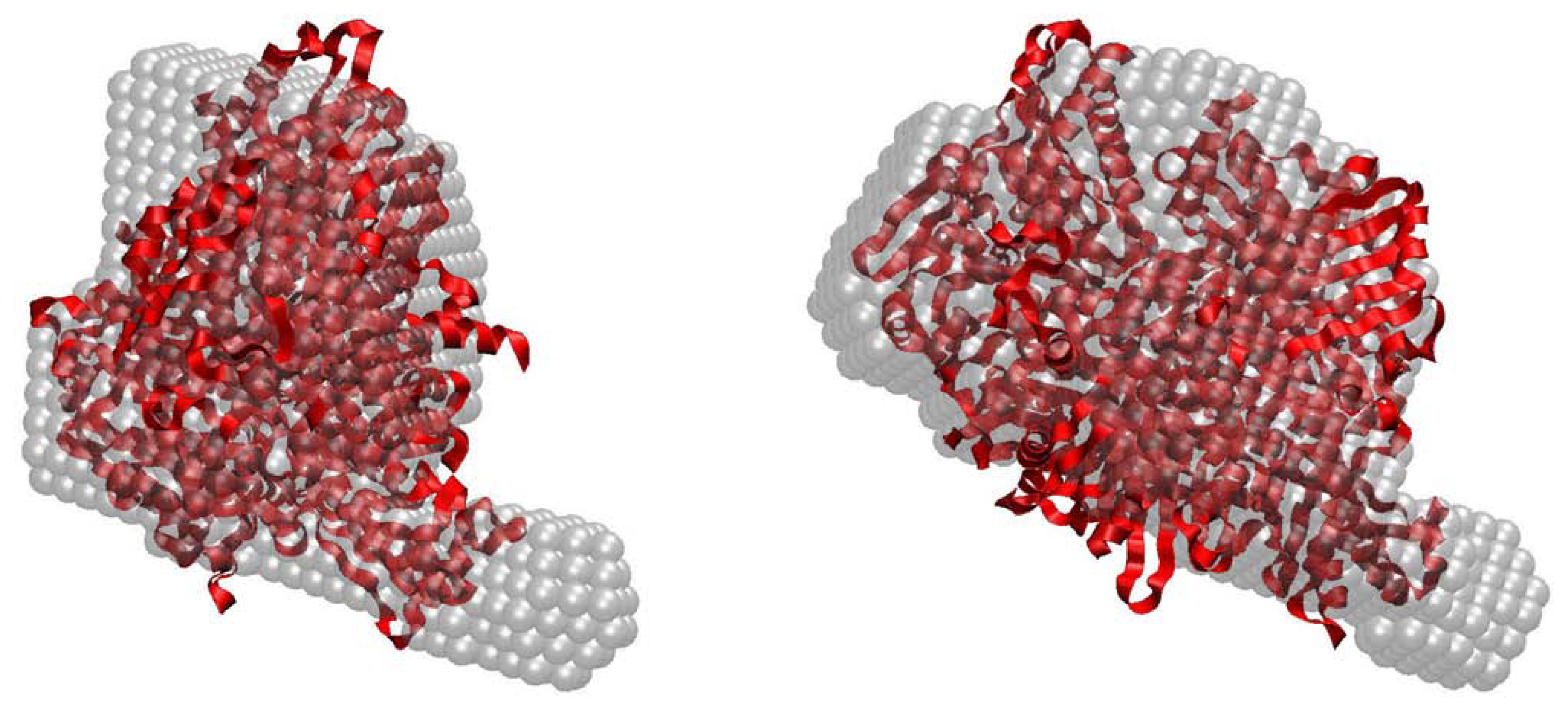
2.3. Quaternary Structure Analysis of PaoABC
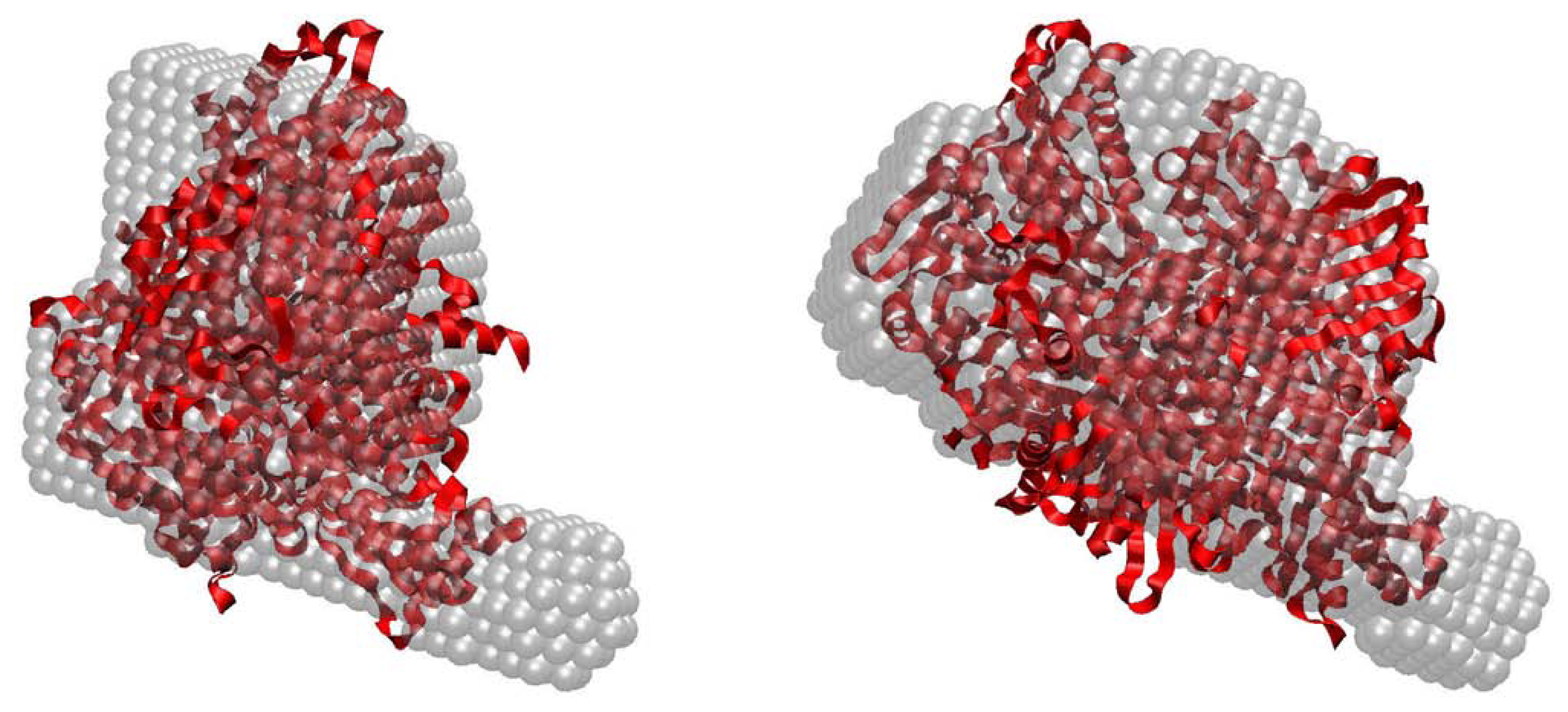
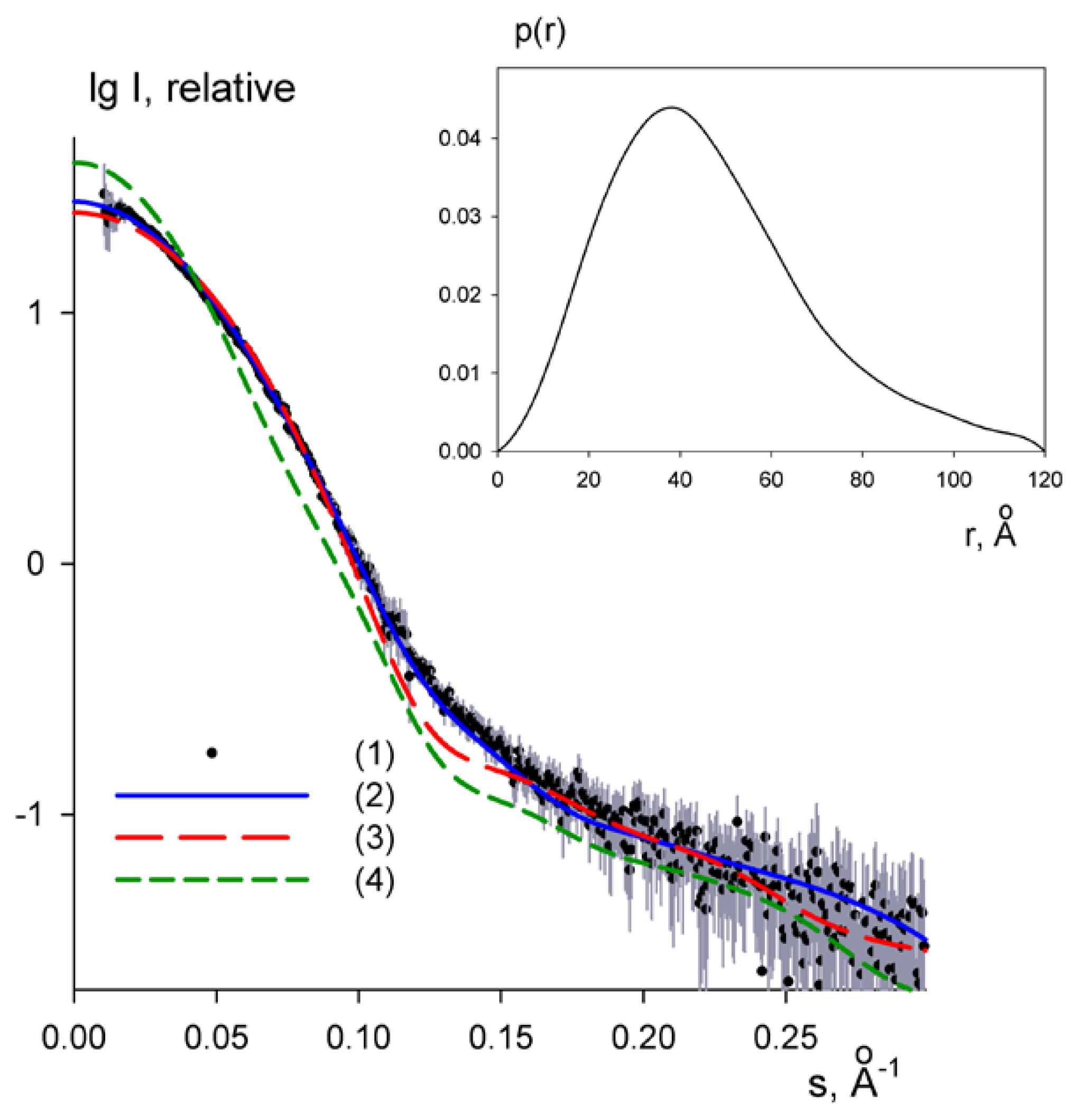
2.4. Small-Angle X-ray Scattering of PaoABC
3. Experimental Section
3.1. Crystallization
3.2. Data Collection and Processing
3.3. Structure Solution
3.4. SAXS Assays
4. Conclusions
Acknowledgments
Conflicts of Interest
References
- Hille, R. The mononuclear molybdenum enzymes. Chem. Rev 1996, 96, 2757–2816. [Google Scholar]
- Rajagopalan, K.V.; Johnson, J.L. The pterin molybdenum cofactors. J. Biol. Chem 1992, 267, 10199–10202. [Google Scholar]
- Schindelin, H.; Kisker, C.; Hilton, J.; Rajagopalan, K.V.; Rees, D.C. Crystal structure of DMSO reductase: Redox-linked changes in molybdopterin coordination. Science 1996, 272, 1615–1621. [Google Scholar]
- Kisker, C.; Schindelin, H.; Rees, D.C. Molybdenum-cofactor-containing enzymes: Structure and mechanism. Annu. Rev. Biochem 1997, 66, 233–267. [Google Scholar]
- Neumann, M.; Mittelstädt, G.; Seduk, F.; Iobbi-Nivol, C.; Leimkühler, S. MocA is a specific cytidylyltransferase involved in molybdopterin cytosine dinucleotide biosynthesis in Escherichia coli. J. Biol. Chem 2009, 284, 21891–21898. [Google Scholar]
- Neumann, M.; Seduk, F.; Iobbi-Nivol, C.; Leimkühler, S. Molybdopterin dinucleotide biosynthesis in Escherichia coli: Identification of amino acid residues of molybdopterin dinucleotide transferases that determine specificity for binding of guanine or cytosine nucleotides. J. Biol. Chem 2011, 286, 1400–1408. [Google Scholar]
- Iobbi-Nivol, C.; Leimkühler, S. Molybdenum enzymes, their maturation and molybdenum cofactor biosynthesis in Escherichia coli. Biochim. Biophys. Acta 2013, 1827, 1086–1101. [Google Scholar]
- Meyer, O.; Rajagopalan, K.V. Molybdopterin in carbon monoxide oxidase from carboxydotrophic bacteria. J. Bacteriol 1984, 157, 643–648. [Google Scholar]
- Freudenberg, W.; König, K.; Andreesen, J.R. Nicotine dehydrogenase from Arthrobacter oxidans: Containing hydroxylase. FEMS Microbiol. Lett 1988, 52, 13–17. [Google Scholar]
- Romao, M.J.; Archer, M.; Moura, I.; Moura, J.J.; LeGall, J.; Engh, R.; Schneider, M.; Hof, P.; Huber, R. Crystal structure of the xanthine oxidase-related aldehyde oxido-reductase from D. gigas. Science 1995, 270, 1170–1176. [Google Scholar]
- Calzi, M.L.; Raviolo, C.; Ghibaudi, E.; de Gioia, L.; Salmona, M.; Cazzaniga, G.; Kurosaki, M.; Terao, M.; Garattini, E. Purification, cDNA cloning, and tissue distribution of bovine liver aldehyde oxidase. J. Biol. Chem 1995, 270, 31037–31045. [Google Scholar]
- Yasuhara, A.; Akiba-Goto, M.; Fujishiro, K.; Uchida, H.; Uwajima, T.; Aisaka, K. Production of aldehyde oxidases by microorganisms and their enzymatic properties. J. Biosci. Bioeng 2002, 94, 124–129. [Google Scholar]
- Kim, S.W.; Luykx, D.M.; de Vries, S.; Duine, J.A. A second molybdoprotein aldehyde dehydrogenase from Amycolatopsis methanolica NCIB 11946. Arch. Biochem. Biophys 1996, 325, 1–7. [Google Scholar]
- Poels, P.A.; Groen, B.W.; Duine, J.A. NAD(P)+-independent aldehyde dehydrogenase from Pseudomonas testosteroni: A novel type of molybdenum-containing hydroxylase. Eur. J. Biochem 1987, 166, 575–579. [Google Scholar]
- Leimkühler, S.; Kern, M.; Solomon, P.S.; McEwan, A.G.; Schwarz, G.; Mendel, R.R.; Klipp, W. Xanthine dehydrogenase from the phototrophic purple bacterium Rhodobacter capsulatus is more similar to its eukaryotic counterparts than to prokaryotic molybdenum enzymes. Mol. Microbiol 1998, 27, 853–869. [Google Scholar]
- Parschat, K.; Canne, C.; Huttermann, J.; Kappl, R.; Fetzner, S. Xanthine dehydrogenase from Pseudomonas putida 86: Specificity, oxidation-reduction potentials of its redox-active centers, and first EPR characterization. Biochim. Biophys. Acta 2001, 1544, 151–165. [Google Scholar]
- Hettrich, D.; Lingens, F. Microbial metabolism of quinoline and related compounds. VIII. Xanthine dehydrogenase from a quinoline utilizing Pseudomonas putida strain. Biol. Chem. Hoppe Seyler 1991, 372, 203–211. [Google Scholar]
- Smith, S.T.; Rajagopalan, K.V.; Handler, P. Purification and properties of xanthine dehydroganase from Micrococcus lactilyticus. J. Biol. Chem 1967, 242, 4108–4117. [Google Scholar]
- Gremer, L.; Meyer, O. Characterization of xanthine dehydrogenase from the anaerobic bacterium Veillonella atypica and identification of a molybdopterin-cytosine-dinucleotide-containing molybdenum cofactor. Eur. J. Biochem 1996, 238, 862–866. [Google Scholar]
- Noriega, C.; Hassett, D.J.; Rowe, J.J. The mobA gene is required for assimilatory and respiratory nitrate reduction but not xanthine dehydrogenase activity in Pseudomonas aeruginosa. Curr. Microbiol 2005, 51, 419–424. [Google Scholar]
- Joshi, M.S.; Rajagopalan, K.V. Specific incorporation of molybdopterin in xanthine dehydrogenase of Pseudomonas aeruginosa. Arch. Biochem. Biophys 1994, 308, 331–334. [Google Scholar]
- Ohe, T.; Watanabe, Y. Purification and properties of xanthine dehydrogenase from Streptomyces cyanogenus. J. Biochem 1979, 86, 45–53. [Google Scholar]
- Woolfolk, C.A. Purification and properties of a novel ferricyanide-linked xanthine dehydrogenase from Pseudomonas putida 40. J. Bacteriol 1985, 163, 600–609. [Google Scholar]
- Sin, I.L. Purification and properties of xanthine dehydrogenase from Pseudomonas acidovorans. Biochim. Biophys. Acta 1975, 410, 12–20. [Google Scholar]
- Xiang, Q.; Edmondson, D.E. Purification and characterization of a prokaryotic xanthine dehydrogenase from Comamonas acidovorans. Biochemistry 1996, 35, 5441–5450. [Google Scholar]
- Hille, R. Molybdenum-containing hydroxylases. Arch. Biochem. Biophys 2005, 433, 107–116. [Google Scholar]
- Turner, N.; Barata, B.; Bray, R.C.; Deistung, J.; Le Gall, J.; Moura, J.J. The molybdenum iron-sulphur protein from Desulfovibrio gigas as a form of aldehyde oxidase. Biochem. J 1987, 243, 755–761. [Google Scholar]
- Neumann, M.; Leimkühler, S. The role of system-specific molecular chaperones in the maturation of molybdoenzymes in bacteria. Biochem. Res. Int 2011. [Google Scholar] [CrossRef]
- Neumann, M.; Mittelstädt, G.; Iobbi-Nivol, C.; Saggu, M.; Lendzian, F.; Hildebrandt, P.; Leimkühler, S. A periplasmic aldehyde oxidoreductase represents the first molybdopterin cytosine dinucleotide cofactor containing molybdo-flavoenzyme from Escherichia coli. FEBS J 2009, 276, 2762–2774. [Google Scholar]
- French, S.; Wilson, K. On the treatment of negative intensity observations. Acta Cryst. A 1978, 34, 517–525. [Google Scholar]
- Zwart, P.H.; Grosse-Kunstleve, R.W.; Adams, P.D. Xtriage and Fest: Automatic assessment of X-ray data and substructure structure factor estimation. CCP4 Newsl 2005, 43, 27–35. [Google Scholar]
- Chandra, N.; Acharya, K.R.; Moody, P.C. Analysis and characterization of data from twinned crystals. Acta Cryst. D 1999, 55, 1750–1758. [Google Scholar]
- Unciuleac, M.; Warkentin, E.; Page, C.C.; Boll, M.; Ermler, U. Structure of a xanthine oxidase-related 4-hydroxybenzoyl-CoA reductase with an additional [4Fe-4S] cluster and an inverted electron flow. Structure 2004, 12, 2249–2256. [Google Scholar]
- Bonin, I.; Martins, B.M.; Purvanov, V.; Fetzner, S.; Huber, R.; Dobbek, H. Active site geometry and substrate recognition of the molybdenum hydroxylase quinoline 2-oxidoreductase. Structure 2004, 12, 1425–1435. [Google Scholar]
- Cao, H.; Hall, J.; Hille, R. X-ray crystal structure of arsenite-inhibited xanthine oxidase: μ-sulfifo, μ-oxo double bridge between molybdenum and arsenic in the active site. J. Am. Chem. Soc 2011, 133, 12414–12417. [Google Scholar]
- Stein, N. CHAINSAW: A program for mutating pdb files used as templates in molecular replacement. J. Appl. Cryst 2008, 41, 641–643. [Google Scholar]
- McCoy, A.J.; Grosse-Kunstleve, R.W.; Adams, P.D.; Winn, M.D.; Storoni, L.C.; Read, R.J. Phaser crystallography software. J. Appl. Cryst 2007, 40, 658–674. [Google Scholar]
- Romão, M.J. Molybdenum and tungsten enzymes: A crystallographic and mechanistic overview. Dalton Trans 2009, 21, 4053–4068. [Google Scholar]
- Krissinel, E.; Henrick, K. Inference of macromolecular assemblies from crystalline state. J. Mol. Biol 2007, 372, 774–797. [Google Scholar]
- DeLano, W. The PyMOL Molecular Graphics System; CA DeLano Sci: San Carlos, CA, USA, 2002. [Google Scholar]
- Svergun, D.I. Restoring low resolution structure of biological macromolecules from solution scattering using simulated annealing. Biophys. J 1999, 76, 2879–2886. [Google Scholar]
- Franke, D.; Svergun, D.I. DAMMIF a program for rapid ab-initio shape determination in small-angle scattering. J. Appl. Cryst 2009, 42, 342–346. [Google Scholar]
- Volkov, V.; Svergun, D.I. Uniqueness of ab initio shape determination in small-angle scattering. J. Appl. Cryst 2003, 36, 860–864. [Google Scholar]
- Svergun, D.I.; Barberato, C.; Koch, M. CRYSOL—A program to evaluate X-ray solution scattering of biological macromolecules from atomic coordinates. J. Appl. Cryst 1995, 28, 768–773. [Google Scholar]
- Rajagopalan, K.V.; Fridovich, I.; Handler, P. Hepatic aldehyde oxidase. J. Biol. Chem 1962, 237, 922–928. [Google Scholar]
- Jancarik, J.; Kim, S.H. Sparse matrix sampling: A screening method for crystallization of proteins. J. Appl. Cryst 1991, 24, 409–411. [Google Scholar]
- Matthews, B.W. Solvent content of protein crystals. J. Mol. Biol 1968, 33, 491–497. [Google Scholar]
- Leslie, A.G.W. Recent Changes to the MOSFLM Package for Processing Film and Image Plate Data. In Jt. CCP4 + ESF-EAMCB Newsl. Protein Cryst.; CCP4: Oxon, UK, 1992. [Google Scholar]
- Kabsch, W. Evaluation of single-crystal X-ray diffraction data from a position-sensitive detector. J. Appl. Cryst 1988, 21, 916–924. [Google Scholar]
- Number 4, C.C.P. The CCP4 suite: Programs for protein crystallography. Acta Cryst. D 1994, 50, 760–763.
- Hänzelmann, P.; Dobbek, H.; Gremer, L.; Huber, R.; Meyer, O. The effect of intracellular molybdenum in Hydrogenophaga pseudoflava on the crystallographic structure of the seleno-molybdo-iron-sulfur flavoenzyme carbon monoxide dehydrogenase. J. Mol. Biol 2000, 301, 1221–1235. [Google Scholar]
- Pauff, J.M.; Cao, H.; Hille, R. Substrate orientation and catalysis at molybdenum site in xanthine oxidase: Crystal structure in complex with xanthine and lumazine. J. Mol. Biol 2009, 284, 8760–8767. [Google Scholar]
- Cowtan, K.D.; Main, P. Phase combination and cross validation in iterated density-modification calculations. Acta Cryst. D 1996, 52, 43–48. [Google Scholar]
- Blanchet, C.E.; Zozulya, A.V.; Kikhney, A.G.; Franke, D.; Konarev, P.V.; Shang, W.; Klaering, R.; Robrahn, B.; Hermes, C.; Cipriani, F.; et al. Instrumental setup for high-throughput small- and wide-angle solution scattering at the X33 beamline of EMBL Hamburg. J. Appl. Cryst 2012, 45, 489–495. [Google Scholar]
- Franke, D.; Kikhney, A.G.; Svergun, D.I. Automated acquisition and analysis of small angle X-ray scattering data. Nucl. Instrum. Methods. Phys. Res. Sect. A 2012, 689, 52–59. [Google Scholar]
- Petoukhov, M.V.; Konarev, P.V.; Kikhney, A.G.; Svergun, D.I. ATSAS 2.1—Towards automated and web-supported small-angle scattering data analysis. J. Appl. Cryst 2007, 40, 223–228. [Google Scholar]
- Konarev, P.V.; Volkov, V.V.; Sokolova, A.V.; Koch, M.H.J.; Svergun, D.I. PRIMUS: A Windows PC-based system for small-angle scattering data analysis. J. Appl. Cryst 2003, 36, 1277–1282. [Google Scholar]
- Mertens, H.D.; Svergun, D.I. Structural characterization of proteins and complexes using small-angle X-ray solution scattering. J. Struct. Biol 2010, 172, 128–141. [Google Scholar]
- Porod, G. In Small Angle X-ray Scattering; Academic Press: New York, NY, USA, 1982. [Google Scholar]
- Kozin, M.B.; Svergun, D.I. Automated matching of high- and low-resolution structural models. J. Appl. Cryst 2001, 34, 33–41. [Google Scholar]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Twinned crystals (A) (293 K) | Non-twinned crystals (B) (277 K) | |
|---|---|---|
| Data collection parameters | ||
| X-ray Source | ID14-1 beam line (ESRF, Grenoble) | X06DA—PXIII beam line (SLS, Villigen) |
| Detector | ADSC Q210 CCD | PILATUS 2 M |
| Wavelength (Å) | 0.934 | 0.976 |
| Processing statistics | ||
| Unit-cell parameters (Å, °) | a = 109.50 | a = 109.42 |
| b = 78.16 | b = 78.08 | |
| c = 151.84 | c = 151.77 | |
| β = 100.15 | β = 99.77 | |
| Space group | C2 | C2 |
| Molecules per AU | 1 | 1 |
| Matthews coefficient (Å3/Da) | 2.70 | 2.73 |
| Mosaicity (°) | 0.79 | 0.64 |
| Resolution range (Å) | 30.88–1.67 (1.76–1.67) | 41.23–1.80 (1.88–1.80) |
| <I/σI> | 12.8 (5.9) | 9.91 (2.41) |
| Rmerge (%) * | 6.1 (16.8) | 7.7 (46.2) |
| Rpim (%) + | 3.9 (11.1) | 5.2 (30.9) |
| Multiplicity | 3.0 (3.0) | 2.9 (2.8) |
| No. of observed reflections | 379108 (50315) | 324367 (40070) |
| No. of unique reflections | 124566 (16919) | 108448 (9450) |
| Completeness (%) | 85.2 (79.7) | 92.3 (80.7) |
| Data collection parameters | |
|---|---|
| Instrument | EMBL X33 beam line (DORIS-III, DESY, Hamburg) |
| Beam geometry | 2.0 × 0.6 mm2 |
| Wavelength (Å) | 1.5 |
| q range (Å−1) a | 0.01–0.5 |
| Exposure time (s) | 8 × 15 |
| Concentration range (mg/mL) | 0.3–20 |
| Temperature (K) | 293 |
| Structural parameters b | |
| I(0) (relative) [from p(r)] | 120 ± 1 |
| Rg (Å) [from p(r)] | 36 ± 1 |
| I(0) (cm−1) (from Guinier) | 120 ± 1 |
| Rg (Å) (from Guinier) | 35 ± 1 |
| Dmax (Å) | 120 ± 10 |
| Porod volume estimate (Å3) | 200,000 ± 10,000 |
| Excluded volume estimate (Å3) | 240,000 ± 10,000 |
| Molecular-mass determination | |
| I(0) (cm−1) BSA (66,000 Da) | 60 ± 1 |
| Molecular mass Mr (Da) [from I(0)] | 130,000 ± 15,000 |
| Molecular mass Mr (Da) [from Porod volume (Vp/1.6)] | 125,000 ± 10,000 |
| Molecular mass Mr (Da) [from excluded volume (Vex/2)] | 120,000 ± 10,000 |
| Calculated monomeric Mr from sequence | 136,000 |
| Software employed | |
| Primary data reduction | Automated SAXS pipeline |
| Data processing | PRIMUS |
| Ab initio analysis | DAMMIN, DAMMIF |
| Validation and averaging | SUPCOMB, DAMAVER |
| Computation of model intensities | CRYSOL |
| 3D graphics representations | VMD |
© 2014 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Otrelo-Cardoso, A.R.; Da Silva Correia, M.A.; Schwuchow, V.; Svergun, D.I.; Romão, M.J.; Leimkühler, S.; Santos-Silva, T. Structural Data on the Periplasmic Aldehyde Oxidoreductase PaoABC from Escherichia coli: SAXS and Preliminary X-ray Crystallography Analysis. Int. J. Mol. Sci. 2014, 15, 2223-2236. https://doi.org/10.3390/ijms15022223
Otrelo-Cardoso AR, Da Silva Correia MA, Schwuchow V, Svergun DI, Romão MJ, Leimkühler S, Santos-Silva T. Structural Data on the Periplasmic Aldehyde Oxidoreductase PaoABC from Escherichia coli: SAXS and Preliminary X-ray Crystallography Analysis. International Journal of Molecular Sciences. 2014; 15(2):2223-2236. https://doi.org/10.3390/ijms15022223
Chicago/Turabian StyleOtrelo-Cardoso, Ana Rita, Márcia Alexandra Da Silva Correia, Viola Schwuchow, Dmitri I. Svergun, Maria João Romão, Silke Leimkühler, and Teresa Santos-Silva. 2014. "Structural Data on the Periplasmic Aldehyde Oxidoreductase PaoABC from Escherichia coli: SAXS and Preliminary X-ray Crystallography Analysis" International Journal of Molecular Sciences 15, no. 2: 2223-2236. https://doi.org/10.3390/ijms15022223
APA StyleOtrelo-Cardoso, A. R., Da Silva Correia, M. A., Schwuchow, V., Svergun, D. I., Romão, M. J., Leimkühler, S., & Santos-Silva, T. (2014). Structural Data on the Periplasmic Aldehyde Oxidoreductase PaoABC from Escherichia coli: SAXS and Preliminary X-ray Crystallography Analysis. International Journal of Molecular Sciences, 15(2), 2223-2236. https://doi.org/10.3390/ijms15022223